

Abstract
Acute infection with Trypanosoma cruzi is characterized by multiple manifestations of immunosuppression of both cellular and humoral responses. B cells isolated at the acute stage of infection have shown marked impairment in their response to polyclonal activators in vitro. The present work aims at studying the B cell compartment in the context of acute T. cruzi infection to provide evidence for B cell activation, spontaneous apoptosis and arrest of the cell cycle upon mitogenic stimulation as a mechanism underlying B cell hyporesponse. We found that B cells from acutely infected mice, which fail to respond to the mitogen LPS, showed spontaneous proliferation and production of IgM, indicating a high level of B cell activation. Furthermore, these activated B cells also exhibited an increase in Fas expression and apoptosis in cultures without an exogenous stimulus. On the other hand, B cells from early acute and chronic infected mice did not present activation or apoptosis, and were able to respond properly to the mitogen. Upon in vitro stimulation with LPS, B cells from hyporesponder mice failed to progress through the cell cycle (G0/G1 arrest), nor did they increase the levels of apoptosis. These results indicate that B cell apoptosis and cell cycle arrest could be the mechanisms that control intense B cell expansion, but at the same time could be delaying the emergence of a specific immune response against the parasite.
Keywords: Trypanosoma cruzi, B lymphocytes, immunosuppression, apoptosis, cell cycle arrest
INTRODUCTION
Acute infection of mice with Trypanosoma cruzi, the intracellular protozoan parasite responsible for Chagas’ disease, is characterized by depression of cellular and humoral immune responses [1,2]. This functional deficiency has been implicated in key events of T. cruzi pathogenesis, such as parasite persistence [3] and immunoregulatory disturbances leading to late immunological attack of the host heart and skeletal muscle or nervous system [4].
Several studies [5–7] demonstrated the importance of antibodies for survival and parasite clearance. Brener [8] proposed that trypanolytic antibodies elicited by an active infection are the major and possibly the sole immune effector mechanism controlling murine and human T. cruzi infection. Although parasite-specific antibodies are essential for controlling T. cruzi infection, it has been described that B cells from acutely T. cruzi-infected mice showed a reduced reactivity to parasite antigens in vitro[9]. Minoprio et al. [4] proposed that the humoral immunosuppression to parasite antigens observed during the acute phase of the infection might be due to the fact that parasite-specific B cells are outnumbered by polyclonally activated cells which produce non-specific antibodies that fail to bind parasite antigens. In addition, Hayes & Kierszenbaum [10] described that B cells isolated only at the acute phase of the infection show an impaired reactivity to polyclonal activators despite the significant number of B lymphocytes, indicating a B cell alteration, a suppressive phenomenon, or a combination of both. In this context, it has been reported that stimulated B lymphocytes display reduced IL-2R expression in the presence of T. cruzi trypomastigotes [11]. Other researchers have suggested that a membrane antigen of T. cruzi is involved in immunosuppression [12]. Altogether, these findings provide evidence for the coexistence of B cell immunosuppression and polyclonal activation during acute T. cruzi infection. The coexistence of these apparently contradictory immunological phenomena is consistent with the hypothesis that a continuous stimulation of lymphocytes leads them to an immune dysfunction: a loss of ability to respond to an antigen (anergy) and/or an increase in the levels of apoptosis.
Here, we document that B cell hyporesponsiveness to LPS in T. cruzi-infected mice is a result of B cell activation, spontaneous apoptosis and cell cycle arrest upon LPS stimulation.
Since different mechanisms of impaired T cell responses have been proposed [13–18] and these T cell defects may affect the B cell response in the majority of the experiments, we undertook to study the effect of mitogenic stimulation on B cells keeping them under the influence of other immune cells, as occurs in in vivo conditions. Hence, we used a B cell potent mitogen, such as LPS, and analysed its effect on CD19+ or B220+ cells of the whole splenocyte population. To simulate the in vivo conditions, we selected LPS as B cell mitogen, since LPS acts [19] in a similar way to mitogenic T. cruzi antigens [20–22]. Elucidation of the mechanisms responsible for B cell hyporesponsiveness in T. cruzi infection may provide new clues for immune intervention aimed at preventing or reversing the resulting humoral immunosuppression and further promoting the development of protective immunity against the parasite.
MATERIALS AND METHODS
Parasite and mouse strains
The Tulahuén strain of T. cruzi was used. It was maintained by weekly intraperitoneal (i.p.) inoculation in BALB/c mice. These mice (6–8 weeks old) were obtained from the Comisión Nacional de Energía Atómica (Buenos Aires, Argentina).
Infection with T. cruzi
Mice were infected intraperitoneally with 500 trypomastigotes (tp) from T. cruzi, diluted in 0·2 ml PBS. This parasite dose was selected in view of the fact that 500 tp-infected mice were able to progress to the chronic phase, so it was useful to evaluate cell behaviour at acute and chronic phases of the infection. Age-matched uninfected normal littermates were used as control mice. In some experiments we also assayed mice injected with 0·2 ml of Freund’s complete adjuvant (FCA) diluted 1:1 in PBS as control. After 8, 15 (acute phase) and 180 days (chronic phase), post-infection mice were killed by cervical dislocation and spleens were obtained by surgery.
Cell preparation
Spleens from normal or infected donors were removed at the indicated times after the infection and cell suspensions were prepared by homogenization in a tissue grinder. Erythrocytes were lysed by brief incubation in erythrocyte lysing buffer (Sigma Chemical Co.). Spleen mononuclear cells (SMC) were washed twice and resuspended in complete RPMI medium containing 10% fetal bovine serum (FBS), 50 μmβ-mercaptoethanol and 40 μ g/ml gentamycin.
For B cell purification, monocytes were removed from SMC by two rounds of plastic adherence (1 h incubation at 37°C in 10-cm Petri dishes) and T cells were depleted by magnetic cell sorting using anti-Thy1.2-coated magnetic beads (Dynal beads) following the manufacturer’s instructions. After this procedure > 95% of CD19+ cells were detected by flow cytometry.
In vitro cell cultures
SMC (2 × 105 cells/well) were cultured in triplicate in a volume of 200 μ l in flat-bottomed 96-well tissue culture plates (Corning) for 24, 48, 72, 96 and 120 h with 20 μ g/ml LPS (from Escherichia coli serotype 0127:B8; Sigma) or medium alone.
Cell proliferation was assayed by measuring the 3H-TdR incorporation (1 μ Ci/well) during the final 18 h of culture. To evaluate in vitro spontaneous proliferation, SMC were cultured for 6 h as above in the absence of exogenous stimulus following the protocol of Minoprio et al. [23]. To analyse the effects of LPS proliferation, a similar experiment was carried out in the presence of LPS (20 μ g/ml). Cells were harvested and 3H-TdR incorporation was measured by a beta scintillation counter. Results are presented as ct/min of 3H-TdR incorporation or Δct/min (Δct/min = ct/min in the presence of LPS – ct/min without stimulus) ± s.d.
In another set of experiments, SMC from normal or infected mice were cultured with or without LPS (20 μ g/ml), at 2 × 106 cells per 2 ml of complete RPMI medium in flat-bottomed 24-well plates. After culturing for different times, supernatants were collected and released IgM was determined. In these supernatants the presence of DNA fragmentation was evaluated as below. Cells were harvested and submitted to flow cytometric determination.
IgM determination
For IgM determination 96-well ELISA plates were coated with goat anti-mouse IgM overnight at 4°C, extensively washed and blocked by the addition of 1% bovine serum albumin (BSA) for 1 h at room temperature. Plates were emptied and supernatants of cultures diluted 1:10 in 1% BSA were added for 2 h at 37°C in a humidified atmosphere in air. After washing three times with PBS containing 0·05% Tween 20 (PBS–T), peroxidase-conjugated anti-mouse IgM was added and incubated for 1 h at 37°C. The reaction was developed using o-phenylenediamine. The assays were performed in duplicate and values were expressed as mean optical density at 490 nm (OD at 490 nm) ± s.d. in an ELISA reader (BioRad, Hercules, CA).
DNA fragmentation analysis
SMC were cultured as above. After 18 h of culture, supernatants were collected and further depleted of contaminating cells by centrifugation. An equal volume (600 μ l) of supernatant was obtained from each treatment and treated with 0·2 ml cell lysis buffer consisting of 0·5 (w/v) SDS, 200 μ g/ml proteinase K (Sigma), and 50 mm NaCl in TE buffer (10 mm Tris–HCl, 1 mm EDTA, pH 8·0) for 2·5 h at 42°C. The pellet was treated with 50% isopropanol/0·05 m NaCl at −20°C overnight, centrifuged, and finally the DNA was washed with 70% ethanol, air-dried and resuspended in TE buffer.
The DNA was separated on 1·5% agarose gel using a standard electrophoresis procedure. Gels were stained with 5 μ g/ml ethidium bromide (Sigma) and photographed under UV light.
Flow cytometry determinations
SMC from normal or infected mice freshly explanted or cultured for 96 h were washed three times with Hanks’ balanced salt solution (HBSS) containing 1% BSA and 0·1% NaN3 and preincubated with anti-mouse CD32/CD16 antibody for 1 h at 4°C in order to block immunoglobulin non-specific trapping through Fc receptors. Following Fc blocking, cells were incubated with PE-labelled anti-mouse CD19 (PharMingen, San Diego, CA), PE-labelled anti-mouse B220 (Sigma), and/or FITC-labelled anti-mouse Fas (PharMingen) for 30 min at 4°C using 1 μ g of antibody/106 cells. The cells were washed three times with HBSS, fixed in 2% formaldehyde and stored at 4°C in the dark until analysis.
The DNA content was determined as described by Nicoletti et al. [24]. SMC cultured during 36 h were stained with FITC-labelled anti-mouse B220 as described above and fixed in 1 ml cold 70% ethanol at 4°C. Then the propidium iodide (PI; 50 μ g/ml; Sigma) was added to stain DNA.
The number of apoptotic cells was determined by evaluating the percentage of hypodiploid nuclei in the < 2N DNA peak and the cells in G0/G1 were distinguished from those in S/G2/M cell cycle stage by the amount of DNA per cell.
Terminal deoxinucleotidyl transferase (TdT)-mediated dUTP nick-end labelling (TUNEL) assay [25] was carried out on purified B cell population using the Apoptosis Detection System, Fluorescein (Promega) according to the manufacturer’s recommended protocol for flow cytometry. Incorporation of FITC-labelled dUTP by exogenous TdT was detected by analysing green fluorescence. Negative control involved the omission of the TdT enzyme during TUNEL reaction.
Ten thousand events were acquired with a flow cytometer (Becton Dickinson) and results were analysed with WiMDI software.
Statistical analysis
All statistical analyses were performed using an unpaired Student’s t-test. P < 0·05 was considered significant.
RESULTS
Impaired reactivity to LPS in SMC from 15 days T. cruzi-infected mice
To assess the kinetics of the B cell proliferative response throughout the acute and chronic phases of T. cruzi infection, SMC from mice at 8, 15 and 180 (chronic) days post-infection were cultured with or without LPS during different time periods. Cell proliferation was assayed by measuring 3H-TdR incorporation. SMC corresponding to age-matched normal mice were used as controls
We also assessed, as control, one group of mice injected with FCA in order to imitate normal germinal centre reaction following antigenic challenge, and it was observed that the proliferative response of B cells from these FCA-injected mice was similar to normal mice (data not shown).
As observed in Fig. 1a,b, cells from 8-day-infected mice with 500 tp were able to proliferate upon LPS stimulation with a peak of proliferation reached 24 h before B cells from normal mice. In addition, B cells corresponding to 180-day (chronic)-infected mice (Fig. 1b) were able to respond to LPS to a similar extent to normal mice cells. In contrast, behaviour of B cells at day 15 post-infection (Fig. 1b) was clearly different, since their proliferative response to LPS was significantly diminished compared with controls. Figure 1b shows only 15-day age-matched control mice, since their proliferative response was similar that of 180-day age-matched control mice.
Fig. 1.
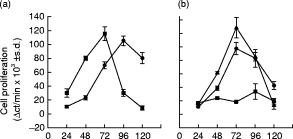
Proliferative response of spleen mononuclear cells (SMC) from normal and Trypanosoma cruzi-infected mice following stimulation with LPS. SMC (2 × 106/ml) obtained from normal (•) or 8 day-infected mice (▪) (a) or from 15- (▾) or 180 (chronic) -day-infected mice (✦) (b) were cultured with medium alone or stimulated with LPS (20 μ g/ml). (a,b) Thymidine incorporation was measured at different hours of culture and the results are presented as delta of the mean ct/min of triplicate cultures ± s.d. (see MATERIALS and METHODS). The data in (a,b) were obtained during different days and are representative of three independent experiments.
Our findings about cells from 15-day and chronic infected mice are in broad agreement with those described by Hayes & Kierszenbaum [16]. Furthermore, we also found that the ability of B cells to respond to LPS was closely related to the dose of tp inoculated and the day post-infection in which the cells were explanted. By using 10 000 and 50 000 tp, the suppression was revealed at 11 days of infection, and using 100 000 tp, it was observed at 8 days of infection (data not shown).
Lower number of B cells in SMC from infected mice that fail to respond to LPS
Since B cells from 15-day T. cruzi-infected mice are unable to respond properly to LPS, we cultured SMC from normal and 15-day infected mice for 96 h in the absence or presence of LPS and studied by FACS the percentage of viable B cells using PE-labelled anti-mouse CD19.
As expected, the percentage of CD19+ cells in cultures of SMC from normal mice (Fig. 2a) increased from 51·6% in non-stimulated cultures (Fig. 2a, left panel) up to 85·7% in cultures stimulated with LPS (Fig. 2a, right panel). However, the percentage of CD19+ cells in non-stimulated cultures of SMC from infected mice (Fig. 2b, left panel) was significantly lower than controls. Interestingly, LPS stimulation induced a more pronounced decrease of CD19+ cells from infected mice (Fig. 2b, right panel). Accordingly, SMC from infected mice presented lower absolute numbers of B cells compared with controls, and the difference was increased by LPS stimulation (data not shown).
Fig. 2.
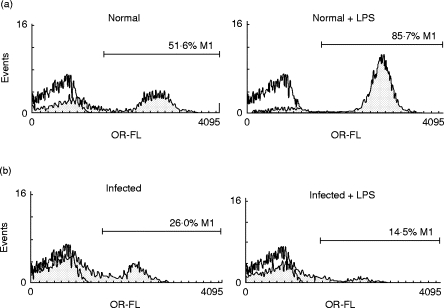
Percentage of CD19+ cells on spleen mononuclear cells (SMC) from Trypanosoma cruzi-infected mice. SMC from normal (a) or 15-day-infected mice (b) were cultured with medium alone (left panels) or LPS (20 μ g/ml) (right panels) during 96 h. The cells were then analysed for CD19+ expression by flow cytometry using PE-labelled anti-mouse CD19 antibodies. Staining with control isotype is shown by open histograms. The relative cell number is plotted against fluorescence intensity.
Two hypotheses could explain the reduced cell number: (i) an increase in the rate of cell death, or (ii) a reduction in new cells generated. In the light of these observations, the next issue we attempted to investigate was related to the molecular mechanism involved in decreased B cell number in cultures of SMC from infected mice that fail to respond to LPS.
B cells from infected mice exhibit a high rate of spontaneous apoptosis and LPS is unable to rescue them from death
In order to evaluate the first hypothesis, we measured the occurrence of apoptosis in SMC non-stimulated or stimulated with LPS after 6 h of culture. Fragmented DNA was detected in culture supernatants of SMC from 15-day-infected mice, but not in cultures of SMC from normal, 8-day and 180-day (chronic)-infected mice (Fig. 3). Simultaneously, we processed FCA-injected mice as control and did not detect DNA fragmentation at that time (data not shown). There was no increase in DNA release in any of the cell types studied upon LPS stimulation. Previous reports [26–28] have described that apoptotic cells exhibit an FSClow accompanied by an increase or no change in SSC on a flow cytometer, validating the use of light scattering properties as evidence of apoptosis. After analysing by FACS FSC versus SSC of SMC from normal and 15-day-infected mice, we observed that SMC from normal mice had raised levels of 43·5% of CD19+ B cells with FSClow in non-stimulated cultures (Fig. 4a, left panel), and this percentage decreased to 5·1% upon mitogenic stimulation (Fig. 4a, right panel). Consistently, this mitogen increased the number of CD19+ B cells with FSChigh revealing a very extensive blast transformation. On the other hand, Fig. 4b shows that cultures of SMC from 15-day-infected mice exhibited a substantial proportion of CD19+ FSClow B cells in non-stimulated cultures (90·6%), and this percentage was not modified in LPS-stimulated cultures (95·6%).
Fig. 3.
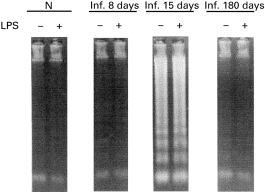
DNA fragmentation on cells obtained from Trypanosoma cruzi-infected mice. Spleen mononuclear cells (SMC) from normal 8-day, 15-day or 180-day-infected mice were cultured with medium alone or LPS (20 μ g/ml) during 6 h. DNA released into the supernatant by SMC was electrophoresed to monitor DNA fragmentation. Molecular size markers (Φλ DNA-EcoRI-Hind III digest) are shown in lane M. Data are representative of three independent experiments.
Fig. 4.
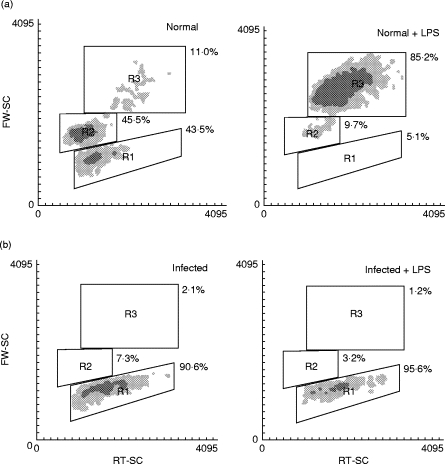
Forward scatterlow population is enriched in spleen mononuclear cells (SMC) from Trypanosoma cruzi-infected mice. SMC from normal (a) or 15-day-infected mice (b) were cultured in medium alone (left panels) or 20 μ g/ml LPS (right panels) during 96 h. Cells were stained with PE-labelled anti-mouse CD19. CD19+ cells were gated and analysed for FSC versus SSC profiles. Density plots show three distinct populations (FSClow or R1, FSCint or R2 and FSChigh or R3) based on light scattering parameters. The percentages shown indicate the number of cells within each region.
These results were in broad agreement with the percentage of B220+ cells that had a hypodiploid content of DNA after culturing SMC for 36 h (15-day-infected mice, 64%; normal mice, 36%; data not shown).
In order to assess whether increased B cell death during T. cruzi infection would be triggered by another cell type, we measured by the TUNEL technique the percentage of apoptotic cells after culturing purified B cells during 6 h. As shown in Fig. 5, cells from T. cruzi 15-day-infected mice present higher levels of apoptosis than B cells from normal mice, indicating that B cells undergo spontaneous apoptosis without any other cell type in vitro contribution. As described above, during the first 6 h of culture LPS does not modify the levels of B cell apoptosis either in normal or in infected mice. We also detected by the TUNEL technique, both in normal and in infected mice, lower apoptosis of CD19+ cells in the context of SMC in comparison with purified B cells (data not shown).
Fig. 5.
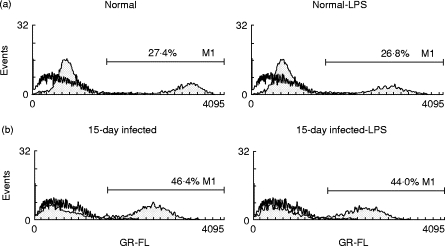
Incorporation of fluorescein-labelled dUTP into DNA strand breaks of purified B cells from Trypanosoma cruzi-infected and normal mice. B cells from normal (a) or infected mice at day 15 after infection (b) were cultured during 6 h (2 × 106 cells/well) in medium alone (left panels) or with LPS (right panels). The cells were then incubated with FITC-labelled dUTP and terminal deoxitransferase according to Promega Kit directions. The percentages of apoptotic B cells are indicated. Staining without the TdT enzyme during TUNEL reaction is shown by open histograms The data are representative of two independent experiments.
Together these results demonstrate that B cells from T. cruzi-infected mice, exhibiting a hyporesponsiveness to LPS, present an increased rate of spontaneous apoptosis, and moreover, LPS neither rescues cells from death nor increases B cell apoptosis. These results provide a clue for explaining the low number of B cells in cultures of SMC from infected mice.
Fas expression is increased among B cells during acute T. cruzi infection
To gain more insight into the mechanisms involved in spontaneous B cell apoptosis during acute T. cruzi infection, the expression of Fas molecule on B220+ B cells was studied by FACS. It was found that the percentage of Fas+ B cells was increased in 15 day-T. cruzi-infected mice in comparison with uninfected controls (Fig. 6). Moreover, statistical analysis revealed that the mean of Fas expression on B cells from infected mice was 28·4 in comparison with 17·0 in B cells from normal mice (data not shown), indicating a higher expression of Fas protein on B cell from infected mice in comparison with normal mice. These results indicate that T. cruzi infection increases the expression of the Fas pro-apoptotic molecule on B lymphocytes.
Fig. 6.
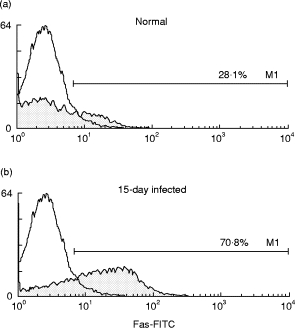
Flow cytometric analysis of Fas expression on B220+ spleen cells from Trypanosoma cruzi-infected and normal mice. Spleen mononuclear cells (SMC) from normal (a) or infected mice at day 15 after infection (b) were stained with PE-labelled anti-mouse B220+ and FITC-labelled anti-mouse Fas. B220+ cells were gated and the percentages of Fas+ cells are indicated. Staining with control isotype is shown by open histograms. The data are representative of three independent experiments.
LPS stimulation reduces spontaneous proliferation of B cells from infected mice and arrests them in the G0 /G1 cell cycle stage
Since mitogenic stimulation did not increase B cell death, the next question was why cultures of SMC from infected mice stimulated with LPS exhibited lower number of B cells than non-stimulated ones? To address this question the second hypothesis was analysed: reduction in new B cell generation upon LPS stimulus. Hence, spontaneous proliferation was evaluated by measuring the incorporation of 3H-TdR during the first 6 h of culture with or without LPS (Table 1). It was observed that SMC from 15-day-infected mice showed, in absence of exogenous stimulus, higher levels of spontaneous proliferation in comparison with normal mice (Table 1) and FCA-injected mice (data not shown). In addition, activated B cells from 15-day-infected mice were able to differentiate into immunoglobulin secretory cells, since they released spontaneously high levels of IgM after 72 h of culture compared with B cells from normal mice (Table 1). Interestingly, cells from 15-day-infected mice cultured with LPS showed lower levels of spontaneous proliferation than cells cultured in the absence of the mitogenic stimulus. These results are in agreement with the low levels of IgM released by B cells from 15-day-infected mice after 72 h of culture in the presence of LPS compared with non-stimulated cultures (Table 1).
Table 1.
Spontaneous proliferation and IgM production of spleen mononuclear cells (SMC) from Trypanosoma cruzi-infected mice
| Dose and days post-infection | Proliferation 6 h (ct/min × 103 ± s.d.) | Proliferation 6 h + LPS (ct/min × 103 ± s.d.) | IgM 72 h (OD at 492 nm ± s.d.) | IgM 72 h + LPS (OD at 492 nm ± s.d.) |
|---|---|---|---|---|
| *Normal | 464 ± 13 | 318 ± 20 | 102 ± 5 | 644 ± 3 |
| 500 tp/8 days | 523 ± 31 | 514 ± 12 | 115 ± 9 | 455 ± 3 |
| *Normal | 1046 ± 166 | 967 ± 43 | 102 ± 5 | 644 ± 3 |
| Chronic | 1171 ± 234 | 1135 ± 223 | 110 ± 14 | 644 ± 2 |
| 500 tp/15 days | 18 861 ± 1268 | 13 432 ± 940 | 408 ± 14 | 270 ± 14 |
Data obtained from two different experiments.
SMC obtained from normal, 500 trypomastigote (tp)-infected mice at days 8, 15 or 180 (chronic) after infection were cultured (2 × 10 5/well) with or without LPS. Spontaneous proliferation was measured by thymidine incorporation during the first 6 h of culture and results are presented as the mean ct/min of triplicate cultures ± s.d. IgM released to the supernatant after 72 h of culture was assayed by a capture ELISA and expressed as optical density (OD) at 490 nm of triplicate cultures ± s.d.
It should be remarked that B cells from 8- and 180-day-infected mice that were able to respond to LPS did not present spontaneous proliferation or production of IgM. Furthermore, LPS stimulation did not modify the levels of thymidine incorporated during the first 6 h of culture (Table 1). Consistent with the lymphoproliferative response shown in Fig. 1, the levels of IgM released by B cells from 8- and 180-day-infected mice increased upon LPS stimulation.
Finally, the effects of LPS were analysed on the cell cycle of the B220+ population from normal and 15-day-infected mice after 6 h of culture. Consistent with spontaneous proliferation in infected mice, we detected 23·3% of B220+ cells in the G2/M/S cell cycle stage (Fig. 7b, left panel) compared with 2·3% of B220+ cells in control mice (Fig. 7a, left panel). B cells from normal mice cultured for 6 h with LPS progressed through the cell cycle to a similar extent as non-stimulated cells. In contrast, LPS reduced the number of B220+ cells from infected mice that entered into the S/G2/M stage. In agreement with the results presented before, we observed that cultures of SMC from infected mice exhibited a higher number of hypo-diploid B220+ cells, and LPS did not alter the percentage of apoptotic B220+ cells.
Fig. 7.
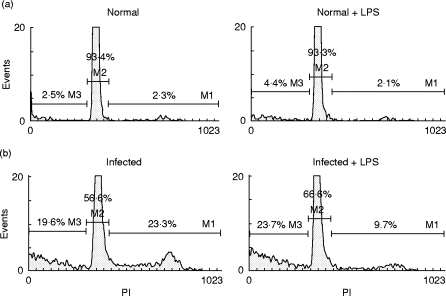
Cell cycle analysis of B220+ cells from Trypanosoma cruzi-infected and normal mice. Spleen mononuclear cells (SMC) from normal (a) or infected mice (b) were explanted at day 15 after infection and cultured during 6 h with medium alone (left panels) or with 20 μ g/ml LPS (right panels). Cells were double-stained with FITC-labelled anti-mouse B220 and propidium iodide. B220+ cells were gated and the percentages of B cells in G0/G1 (M1), S/G2/M (M2) or sub G0/G1 (M3) cell cycle stages are indicated.
These data indicate that upon LPS stimulation, B cells from infected mice are arrested in the G0/G1 cell cycle stage, avoiding spontaneous proliferation and consequently generation of novel B cells, explaining why cultures stimulated with LPS presented a lower number of B cells in comparison with non-stimulated ones.
DISCUSSION
Acute infection with T. cruzi is characterized by multiple manifestations of immunosuppression of both cellular and humoral responses. In this study, we re-validate the B cell compartment in the context of acute T. cruzi infection, providing evidence for B cell spontaneous apoptosis and arrest in the cell cycle after LPS stimulation.
B cells isolated at the acute stage of infection have shown marked impairment in their response to polyclonal activators in vitro[10]. Here, we succeeded not only in confirming these findings but also detecting that the ability of B cells from T. cruzi acutely infected mice to respond to a mitogen was closely related to the dose inoculated and the day of infection in which the cells were explanted. It was also observed that mice which failed to respond to a mitogen had B cells that proliferated and produced IgM spontaneously, indicating a high level of B cell activation. Furthermore, these activated B cells also exhibited spontaneous apoptosis in cultures without an exogenous stimulus. On the other hand, B cells from 8- and 180-day-infected mice did not present activation or apoptosis, and were able to respond properly to a mitogen. These data suggest an association between B cell activation, apoptosis and immunosuppression during acute T. cruzi infection. In some experiments we assessed FCA-injected as control mice in order to imitate normal germinal centre reaction following an antigenic challenge, and it was found that 15-day-infected mice exhibited more immunosuppression and apoptosis than FCA-injected mice. These findings may result from the higher levels of B cell activation reached after T. cruzi infection in comparison with FCA injection. The phenomenon of B cell apoptosis following intense immune stimulation is well documented [29,30], but no data of B cell apoptosis during T. cruzi infection have previously been reported. Our findings are in line with a series of experimental results during infection with several pathogens, ranging from parasites to retroviruses, where lymphocytes undergo apoptosis as a consequence of infection [31–34]. Therefore, apoptosis could be a general mechanism, which acts during virus and parasite infection, in which the immediate consequence is a state of immunosuppression. We demonstrated that during acute T. cruzi infection B cells undergo spontaneous apoptosis without contribution of any other cell type in vitro. Recently, Fas/FasL interaction has been shown to be involved in activation-induced cell death of CD4+T cells during experimental Chagas’ infection [35]. Considering that we detected an increase in Fas expression on B cell population from unresponsive mice, it is possible that the Fas/FasL pathway may be involved in B cell apoptosis, regulating the magnitude of an immune response as was previously proposed [36]. Nevertheless, further investigations should be carried out to discern whether Fas molecule is responsible for B cell apoptosis and whether an antigen-specific stimulus is able to modify it.
LPS is a potent polyclonal activator of normal murine B lymphocytes, and is able to rescue mature resting splenic B cells from spontaneous apoptosis and promote cell cycle entry [37]. Lopes et al. [38] described that CD4+ T cells activated by T. cruzi infection raised the level of apoptosis upon mitogenic stimulation, so we expected that LPS would accelerate B cell apoptosis during the first hours of culture. We observed however that during acute T. cruzi infection, stimulation with LPS: (i) did not increase the levels of B cell apoptosis; (ii) failed to reduce apoptosis levels of B cells, raising the possibility that LPS was unable to provide a survival signal for B cells from infected mice and/or that apoptosis was so advanced that survival signals failed to rescue cells from death; and (iii) arrested B cells in the G0/G1 stage, preventing proliferation of B cells from infected mice and reducing the number of B220+ cells in the S/G2/M stage of the cell cycle. Whether this phenomenon would be a consequence of B cell or macrophage activation requires more investigation. In any case, high activation of both B cells and macrophages has been reported during T. cruzi infection [4,39]. It has been proposed that parasite mitogens [20,21,40] are responsible for polyclonal activation of the B cell compartment during acute T. cruzi infection [41,42], and it is probable that the restimulation of these activated B cells with a mitogen leads them to an arrested stage. In this sense, it is probable that parasite mitogens first expand B cell clones and then arrest them by subsequent interactions, thus triggering and controlling polyclonal activation. Similar results were observed with T lymphocytes that were arrested in the G0/G1 stage by mitogenic stimulation in presence of T. cruzi trypomastigotes [11]. Also in mice with murine AIDS (MAIDS), mitogenic stimulation arrested T lymphocytes in the G0/G1 phase of the cell cycle [43]. Future investigations could generalize this phenomenon as an alternative mechanism to control intense immune responses of the B cell population.
The biological relevance of this study is supported by the fact that B cell apoptosis and cell cycle arrest could be mechanisms that control intense lymphocyte expansion during acute T. cruzi infection, but at the same time could be delaying the emergence of a specific immune response against the parasite. It would be relevant to investigate whether these mechanisms of controlling intense immune responses discriminate between parasite-specific and non-specific B cells. This issue is under current investigation in our laboratory.
Acknowledgments
We are especially grateful to Dr G. Rabinovich for useful discussion and critical advice. We also thank Dr M. Giordano from Academia Nacional de Medicina, Buenos Aires and Dr S. Sappia and Dr A. Galeano from Mater Dei, Buenos Aires, for their kind assistance in FACS analysis. This work was supported by grants from the ‘Consejo de Investigaciones Científicas y Técnicas’ (CONICET), Fundación Antorchas and ‘Consejo de Investigaciones Científicas y Técnológicas de la Provincia de Córdoba’ (CONICOR). E.Z., C.M. and C.L.M. thank CONICET and F.L.D. thanks SECYT for the fellowships granted. J.L.B. and A.G. are members of the ‘Carrera del Investigador Científico del CONICET’.
References
- 1.Ramos CE, Lamoy M, Feoly M, Rodriguez M, Perez M, Ortiz-Ortiz L. T. cruzi: immunosuppressed response to different antigens in the infected mouse. Exp Parasitol. 1978;45:190–7. doi: 10.1016/0014-4894(78)90059-0. [DOI] [PubMed] [Google Scholar]
- 2.Cunninghan DS, Kuhn RE, Rowland EC. Suppression of humoral responses during T. cruzi infection in mice. Infect Immun. 1978;22:155–62. doi: 10.1128/iai.22.1.155-160.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kierszenbaum F. On evasion of Trypanosoma cruzi from the host immune response. Lymphoproliferative responses to trypanosomal antigens during acute and chronic experimental Chagas’ disease. Immunology. 1981;44:641–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Minoprio P, Itohara S, Heusser C, Tonegawa S, Countinho A. Immunobiology of murine T. cruzi infection: the predominance of parasite nonspecific responses and the activation of TcR I T cells. Immunol Rev. 1989;112:184–206. doi: 10.1111/j.1600-065x.1989.tb00558.x. [DOI] [PubMed] [Google Scholar]
- 5.Krettli AU, Brener Z. Protective effects of specific antibodies in Trypanosoma cruzi infection. J Immunol. 1976;116:755–61. [PubMed] [Google Scholar]
- 6.Rodriguez AM, Santor F, Afchain D, Bazin H, Capron A. Trypanosoma cruzi infection in B cell deficient rats. Infect Immun. 1981;31:524–30. doi: 10.1128/iai.31.2.524-529.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kierszenbaum F, Howard JG. Mechanisms of resistance against Trypanosoma cruzi infection: the importance of antibodies and antibody forming capacity in biozzi high and low responder mice. J Immunol. 1976;116:1208–14. [PubMed] [Google Scholar]
- 8.Brener Z. Why vaccines do not work in Chagas’ disease. Parasitol Today. 1986;2:196–202. doi: 10.1016/0169-4758(86)90193-6. [DOI] [PubMed] [Google Scholar]
- 9.Tarleton RL, Kuhn RE. Measurement of parasite-specific immune responses in vitro: evidence for suppression of the antibody response to Trypanosoma cruzi. Eur J Immunol. 1985;15:845–50. doi: 10.1002/eji.1830150820. [DOI] [PubMed] [Google Scholar]
- 10.Hayes MM, Kierszenbaum F. Experimental Chagas’ disease: kinetics of lymphocyte responses and immunological control of transition from acute to chronic T. cruzi infection. Infect Immun. 1981;31:1117–24. doi: 10.1128/iai.31.3.1117-1124.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kierszenbaum F, Moretti E, Sztein MB. Molecular basis of Trypanosoma cruzi-induced immunosuppression. Altered expression by activated human lymphocytes of molecules which regulate antigen recognition and progression through the cell cycle. Biol Res. 1993;26:197–207. [PubMed] [Google Scholar]
- 12.Hernandez-Munain C, De Diego JL, Bonay P, Girones N, Fresno M. GP 50/55, a membrane antigen of Trypanosoma cruzi involved in autoimmunity and immunosuppression. Biol Res. 1993;26:209–18. [PubMed] [Google Scholar]
- 13.Reed SG, Pihl DL, Grabstein KH. Immune deficiency in chronic Trypanosoma cruzi infection: recombinant IL1 restores Th function for antibody production. J Immunol. 1989;142:2067–74. [PubMed] [Google Scholar]
- 14.La Flamme AC, Kahn SJ, Rudensky AY, Van Voorhis WC. Trypanosoma cruzi infected macrophages are defective in major histocompatibility complex class II antigen presentation. Eur J Immunol. 1997;27:3085–94. doi: 10.1002/eji.1830271202. [DOI] [PubMed] [Google Scholar]
- 15.Abrahamsonn IA, Coffman RL. Cytokine and nitric oxide regulation of the immunosuppression in T. cruzi infection. J Immunol. 1995;155:3955–63. [PubMed] [Google Scholar]
- 16.Harel-Bellan A, Joskowicz M, Fradelizi D, Eisen H. T lymphocyte function during experimental Chagas’ disease: production of and response to interleukin 2. Eur J Immunol. 1985;15:438–42. doi: 10.1002/eji.1830150505. [DOI] [PubMed] [Google Scholar]
- 17.Tarleton RL. Trypanosoma cruzi-induced suppression of IL2 production. II. Evidence for a role for suppressor cells. J Immunol. 1988;140:2769–73. [PubMed] [Google Scholar]
- 18.Lopes MF, DosReis GA. Trypanosoma cruzi-induced immunosuppression: blockade of costimulatory T cell responses in infected hosts due to defective T-cell receptor CD3 functioning. Infect Immun. 1994;62:1484–8. doi: 10.1128/iai.62.4.1484-1488.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freudenber MA, Galanos C. Bacterial lipopolysaccharide: structure, metabolism and mechanism of action. Int Rev Immunol. 1990;6:207–21. doi: 10.3109/08830189009056632. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg SS, Carneiro MN, Silva-Pereira AA, Maresguia L. Release of lipopolysaccharide (LPS) from cell surface of Trypanosoma cruzi by EDTA. Int J Parasitol. 1983;13:11–17. doi: 10.1016/s0020-7519(83)80062-9. [DOI] [PubMed] [Google Scholar]
- 21.Cordeiro Da Silva A, Espinoza AG, Taibi A, Ouassi A, Minoprio P. A 24 000 MW Trypanosoma cruzi antigen is a B-cell activator. Immunology. 1998;94:189–96. doi: 10.1046/j.1365-2567.1998.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montes CL, Zúñiga E, Minoprio P, Vottero-Cima Gruppi A. A Trypanosoma cruzi alkaline antigen induces polyclonal B-cell activation of normal murine spleen cells by T-cell-independent, BCR-directed stimulation. Scand J Immunol. 1999 doi: 10.1046/j.1365-3083.1999.00577.x. in press. [DOI] [PubMed] [Google Scholar]
- 23.Minoprio P, Eisen H, Forni L, D’Imperio Lima M, Joskowicz M, Coutinho A. Polyclonal lymphocyte response to murine Trypanosoma cruzi infection I. Quantitation of both T- and B-cell response. Scand J Immunol. 1986;24:661–8. doi: 10.1111/j.1365-3083.1986.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 24.Nicoletti Y, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–9. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 25.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishimoto H, Surh CD, Sprent J. Upregulation of surface markers on dying thymocytes. J Exp Med. 1995;181:649–55. doi: 10.1084/jem.181.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swat W, Ignatowicz L, Kisielow P. Detection of apoptosis of mature CD4 + CD8 + thymocytes by flow cytometry. 1991;137:79–87. doi: 10.1016/0022-1759(91)90396-w. [DOI] [PubMed] [Google Scholar]
- 28.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 29.Green DR, Scott DW. Activation-induced apoptosis in lymphocytes. Curr Opin Immunol. 1994;6:476–87. doi: 10.1016/0952-7915(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 30.Green DR, Bissonnette RP, Glynn JM, Shi Y. Activation-induced apoptosis in lymphoid systems. Semin Immunol. 1992;4:379–88. [PubMed] [Google Scholar]
- 31.Toure-Balde A, Sarthou JL, Aribot G, Michel P, Trape JF, Rogier C, Roussilhon C. Plasmodium falciparum induces apoptosis in human mononuclear cells. Infect Immun. 1996;64:744–50. doi: 10.1128/iai.64.3.744-750.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan YA, Matsuura T, Kasper LH. Activation-mediated CD4 + T cell unresponsiveness during acute Toxoplasma gondii infection in mice. Int Immunol. 1996;8:887–96. doi: 10.1093/intimm/8.6.887. [DOI] [PubMed] [Google Scholar]
- 33.Muro-Cacho CA, Pantaleo G, Fauci AS. Analysis of apoptosis in lymph nodes of HIV-infected persons. Intensity of apoptosis correlates with the general stage of activation of the lymphoid tissue and not with the stage of disease or viral burden. J Immunol. 1995;154:5555–66. [PubMed] [Google Scholar]
- 34.Razvi ES, Welsh RM. Programmed cell death of T lymphocytes during acute viral infection: a mechanism for virus induced immunedeficiency. J Virol. 1993;67:5754–65. doi: 10.1128/jvi.67.10.5754-5765.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopes M, Nunes M, Henriques-Pons A, Giese N, Morse H, Davidson W, Araujo-Jorge T, DosReis G. Increased susceptibility of Fas ligand deficient gld mice to Trypanosoma cruzi infection due to Th2-biased host immune response. Eur J Immunol. 1999;29:81–89. doi: 10.1002/(SICI)1521-4141(199901)29:01<81::AID-IMMU81>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 36.Hahne M, Renno T, Schroeter M, Irmler M, French L, Bornard T, MacDonald HR, Tschopp J. Activated B cells express functional Fas ligand. Eur J Immunol. 1996;26:721–4. doi: 10.1002/eji.1830260332. [DOI] [PubMed] [Google Scholar]
- 37.Jakway JP, Usinger WR, Gold MR, Mishell RI, DeFranco AL. Growth regulation of the B lymphoma cell line WeHI-231 by anti-immunoglobulin, lipopolysaccharide and other bacterial products. J Immunol. 1986;137:2225–32. [PubMed] [Google Scholar]
- 38.Lopes MF, da Veiga VF, Santos AR, Fonseca ME, DosReis GA. Activation-induced CD4 + T cell death by apoptosis in experimental Chagas’ disease. J Immunol. 1995;154:744–52. [PubMed] [Google Scholar]
- 39.Ruso M, Stanobiras N. Macrophage activation and resistance to Trypanosoma cruzi infection. Res Immunol. 1991;142:144–50. doi: 10.1016/0923-2494(91)90026-f. [DOI] [PubMed] [Google Scholar]
- 40.Montes CL, Vottero-Cima E, Gruppi A. Trypanosoma cruzi cytosolic alkaline antigens (FI) induce polyclonal activation in murine normal B cells. Scand J Immunol. 1996;44:93–100. doi: 10.1046/j.1365-3083.1996.d01-285.x. [DOI] [PubMed] [Google Scholar]
- 41.Minoprio P, Coutinho A, Joskowicz M, D’Imperio-Lima MR, Eisen H. Polyclonal lymphocyte responses to murine Trypanosoma cruzi infection. II Cytotoxic T lymphocytes. Scand J Immunol. 1986;24:669–79. doi: 10.1111/j.1365-3083.1986.tb02186.x. [DOI] [PubMed] [Google Scholar]
- 42.Ortiz-Ortiz L, Elliot L, Parks D, Rodriguez M, Weigle W. Polyclonal B lymphocyte activation during T. cruzi infection. J Immunol. 1980;124:121–5. [PubMed] [Google Scholar]
- 43.Muralidhar G, Koch S, Broome HE, Swain SL. TCR triggering of anergic CD4 T cells in murine AIDS induces apoptosis rather than cytokine synthesis and proliferation. J Immunol. 1996;157:625–35. [PubMed] [Google Scholar]