

Abstract
Antibodies to glutamic acid decarboxylase (GAD) occur frequently in patients with APECED, although clinical insulin-dependent diabetes mellitus (IDDM) is seen only in a subgroup of the patients. We studied the cellular immunity to GAD, antibodies to GAD and their association with the HLA DQB1 risk alleles for IDDM in patients with APECED. Proliferation responses to GAD were enhanced in the patients with APECED when compared with the control subjects (P = 0·004), but autoimmunity to GAD was not associated with IDDM in APECED. The levels of interferon-gamma (IFN-γ) secreted by GAD-stimulated T cells were higher in the patients than in control subjects (P = 0·001). A negative correlation (r = −0·436, P = 0·03) existed between the antibody levels and the stimulation indices (SIs) to GAD. In 14 non-diabetic patients no difference in insulin secretion was observed in intravenous glucose tolerance test (IVGTT) between the patients with and without T cell reactivity to GAD. We conclude that cellular immunity to GAD detected as T cell proliferation response to GAD or IFN-γ secretion by GAD-stimulated T cells was frequent in patients with APECED (69%) and was not restricted to the patients with clinically detectable β-cell damage.
Keywords: GAD antibodies, insulin-dependent diabetes, HLA, cytokines, T lymphocytes
INTRODUCTION
APECED, also called autoimmune polyendocrine syndrome type 1, is an autosomal recessive disease characterized by chronic mucocutaneous candidiasis, ectodermal dystrophy and multiple endocrinopathies, including in most cases hypoparathyroidism and primary adrenocortical failure [1–3]. The phenotype of the syndrome varies widely. It usually manifests in childhood but new disease components may develop throughout lifetime. A defect in a novel gene, AIRE, has recently been identified in patients with APECED [4,5]. The encoded protein is likely to be a transcription factor, that may play a role in the regulation of immune responses in APECED.
In a large series of patients with APECED the prevalence of insulin-dependent diabetes mellitus (IDDM) was 12% [1]. However, autoimmunity against islet cell antigens is more frequent [6]; antibodies against an IDDM-associated islet cell antigen, glutamic acid decarboxylase (GAD) [7], were present in 41% of patients without clinical IDDM [6]. In patients with APECED neither clinical IDDM nor the occurrence of GAD antibodies was associated with the HLA DQB1 risk alleles for IDDM, suggesting that in these patients the manifestation of IDDM may be regulated by factors other than HLA class II antigens [6]. Thus the IDDM of APECED has characteristics different from the common IDDM.
Antibodies to GAD predict common IDDM in children, although the levels of antibodies are generally lower than in patients with APECED. In vitro detectable T cell reactivity to GAD is frequently found in patients with common IDDM, their relatives and prediabetic subjects [8–11]. Since IDDM is considered a T cell-mediated disease, it has been suggested that T cell reactivity to GAD could be a better indicator for IDDM than antibodies to GAD. Cellular immune response to GAD has not yet been studied in APECED.
To study the characteristics of cellular immunity to GAD in patients with APECED, we studied T cell proliferation response to GAD and secretion of interferon-gamma (IFN-γ) by GAD-stimulated T cells. Also, we studied the relationship of T cell reactivity to GAD with antibody levels to GAD, the HLA DQB1 risk alleles for IDDM, and intravenous glucose tolerance test (IVGTT).
PATIENTS AND METHODS
Patients
All available 44 Finnish APECED patients were studied, including 27 females and 17 males, aged 10–58 years, mean (median) age 29·7 (28·7) years. They all had at least one of the following disease components: hypoparathyroidism and primary adrenocortical failure, and all had chronic mucocutaneous candidiasis. Of the 44 patients, 41 (93%) had hypoparathyroidism, 34 (77%) had primary adrenal failure, 18 (41%) had primary gonadal failure, and two (4%) had hypothyroidism. Eight (18%) of the patients had clinical IDDM. The diagnostic criteria of each disease component have been described elsewhere [1,6]. The mean (median; range) duration of IDDM was 11·2 years (11·2; 4·6–19·6). All but one patient were under 25 years at the time of IDDM diagnosis (range 4·1–45·3 years). Mean (median; range) dose of insulin in the patients was 0·68 (0·68; 0·42–0·95) U/kg per day. The diagnosis was based on classical manifestations of IDDM in seven of eight patients. Patient 8 was symptomless at the diagnosis of diabetes at 45 years of age. Three years after diagnosis his insulin dose was 0·23 U/kg per day and 4·5 years after the diagnosis (at the time of the present study) 0·42 U/kg per day. Fourteen non-diabetic patients underwent IVGTT. During a 12-month period after performing T cell assays three patients developed IDDM and are thus considered prediabetics. A control group (n = 28), including five males and 23 females, aged 23–58 years, mean (median) age being 32·8 (35·7) years, consisted of laboratory personnel and students without clinical manifestations of autoimmune disease. T cell assays in patients and control subjects were performed with fresh blood samples. The blood samples were drawn after informed consent of the patients, patients’ parents or control subjects when the patients visited the out-patient clinic of the Hospital for Children and Adolescents, University of Helsinki.
Antigens
A baculovirus expression vector pVL 1393 (Invitrogen, Leek, The Netherlands) carrying the human GAD gene was used to infect Spodoptera frugiperda (Sf9; ATTC, Rockville, MD) cells in suspension cultures [12]. The cell pellets from cultures 48–54 h post-infection were stored at −70°C. For GAD purification the protocol described earlier was used [13]. Briefly, the Sf9 cells were lysed and the supernatant was cleared by centrifugation (13 400 g for 10 min at 4°C). Immunoaffinity purification was performed using MoAb GAD-6 (Developmental Studies Hybridoma Bank, Iowa City, IA) coupled to cyanogen bromide (CNBr)-activated Sepharose (5 mg/ml gel) 4B (Pharmacia, Uppsala, Sweden). The supernatant from the infected cell lysates and the washed antibody resin were mixed and the antibody–antigen reaction was carried out in 200 mm NaHCO3 buffer at pH 9·2 for at least 16 h by rotating the mixture at 4°C. The resin was transferred to a column which was developed with 0·1 m glycine buffer pH 2·7. The effluent was neutralized with 0·1 m NaOH and the precipitated GAD was pelleted and solubilized in 100 mm NaHCO3 pH 9·2. Purity of the preparations was confirmed by 7·5% SDS–PAGE followed by staining with coomassie brilliant blue and Western blot analysis using GAD-6 or polyclonal rabbit anti-GAD as primary antibodies. The endotoxin content of the antigen preparation was below the detection level (0·062 EU/ml, which corresponds to about 2 pg/ml) of the Limulus Amebocyte Lysate test (BioWhittaker Inc., Walkersville, MD). As control antigens we used tetanus toxoid (TT) without thiomersal (National Public Health Institute, Helsinki, Finland) and in a subgroup of 10 subjects acetylcholinesterase (AChE) expressed in the baculovirus expression system and purified by immunoaffinity column using specific antibodies to AChE. As another control for possible contamination of Sf9 cell lysate in GAD we tested T cell reactivity to Sf9 cell lysate infected with baculovirus in a series of 16 individuals at a concentration of 0·1 μ g/ml and in 20 individuals at a concentration 1·0 μ g/ml. Pokeweed mitogen (PWM) was used to compare IFN-γ secretion by total peripheral blood mononuclear cell (PBMC) population from the patients and control subjects.
T cell proliferation test
PBMC were separated from heparinized blood by Ficoll–Hypaque (Pharmacia) density centrifugation. Cells (1 × 105) diluted in RPMI 1640 (Gibco, Paisley, UK) containing 5% pooled human AB+ serum (Finnish Red Cross Blood Transfusion Service, Helsinki, Finland) and 2 mm/ll-glutamine were cultured in 200 μ l volume per well in U-bottomed microwell plates (Nunc, Roskilde, Denmark). Antigens were added (20 μ l) to quadruplicate wells to provide final concentrations of 0·1, 1, and 10 μ g/ml for GAD and AChE and 8 μ g/ml for TT. After incubation for 5 days in 5% CO2 at 37°C, 1 μ Ci of tritiated thymidine (specific activity 25 Ci/mmol; Amersham, Aylesbury, UK) was added to each well. Cultures were automatically harvested 16 h later. Proliferation was expressed as a stimulation index (SI) = median ct/min incorporated in the presence of the antigen divided by median ct/min incorporated in the absence of the antigen (medium value). Because of the skew distribution of the SIs in the controls, three multiples of median SI in the controls was considered as a cut-off for positive proliferation response (corresponding SI value of 3).
ELISA for IFN-γ secretion by GAD or PWM-stimulated PBMC
Cells (3 × 106/well) with GAD at a concentration of 1 μ g/ml, TT at a concentration of 8 μ g/ml or without antigen, and 1 × 106 cells/well with or without 5 μ g/ml PWM were cultured for 72 h (PWM) or 120 h (antigens) and the supernatants were collected and stored at −70°C. IFN-γ concentration of the samples was detected by ELISA described earlier [14]. The concentrations of IFN-γ detected in the wells cultured without antigen were subtracted from the concentrations of IFN-γ detected in the antigen-stimulated wells. The detection level of the assay was 50 pg/ml.
HLA typing
HLA DQB1 genotyping was performed by dot blot hybridization of polymerase chain reaction (PCR)-amplified DNA with digoxigenin-labelled oligonucleotide probes according to the protocols of the 11th International Histocompatibility Workshop [6,15]. For the purpose of this study, only the data on the IDDM susceptibility alleles DQB1*0201 and 0302 are given, other alleles are given as x.
GAD antibody assay
GAD antibodies were determined by a radiobinding assay of Grubin et al. [16] as modified by Falorni et al. [17]. The results were expressed as an index = (sample ct/min – mean ct/min of 3 negative standard sera)/(positive standard serum ct/min – mean ct/min of 3 negative standard sera) × 100. Antibody levels exceeding an index of 5, i.e. mean + 3 s.d. of Finnish healthy children (n = 64, mean age 7·9 years), blood donors (n = 50), and non-diabetic adults (n = 182, mean age 55 years) were considered positive. All sera with an antibody index > 40 were titrated to end-point dilution still giving positive index, and the final result was expressed as the end-point result multiplied by the dilution factor as described earlier [6]. The antibody testing was performed in the serum samples taken from the patients at the time of T cell testing, although some patients overlap with the study of Tuomi et al. [6]. In the Combined Autoantibody Workshop (Orvieto, Italy, 1995) the specificity of the assay was 99%, and the sensitivity 75%.
TT antibody assay
Antibodies to TT were measured by ELISA. Maxisorb (Nunc) plates were coated overnight at 4°C with TT at a concentration of 1 μ g/ml. After washing with 0·05% Tween–PBS, residual coating was performed with 1% human serum albumin (HSA)–PBS. Plasma samples were diluted 1:800 in 0·2% HSA 0·05% Tween–PBS and incubated for 2 h in room temperature. Alkaline phosphatase-conjugated rabbit anti-human IgG (Fc) (Jackson ImmunoResearch, West Grove, PA) was diluted 1:3000 in 0·2% HSA 0·05% Tween–PBS and incubated for 90 min at room temperature. After washing with 0·05% Tween–PBS, p-nitrophenyl phosphate (Sigma, St Louis, MO) was added and after 30 min incubation absorbance was read at 405 nm. Results were expressed as optical density (OD) units.
Statistical analysis
Comparison of the measured parameters between the different groups was performed by Mann–Whitney U-test or Fisher’s exact test. Spearman’s rank test was used for correlation analyses.
RESULTS
Occurrence of cellular immunity to GAD
Of the 44 patients with APECED 15 (34%), compared with three of the 28 (11%) healthy adult controls, had a positive proliferation response to GAD (10 μ g/ml) when three multiples of median SI (SI ≥ 3) in the control group was used as the cut-off for positivity (P = 0·03, Fisher’s exact test). The distribution of the SIs to GAD differed significantly between patients and controls; median (range) was 1·6 (1–40·0) for the patients and 1·0 (1–13·9) for the controls (Fig. 1; P = 0·004; Mann–Whitney U-test). When the data were analysed as Δ ct/min (ct/min without antigen (medium value) subtracted from the ct/min in the presence of antigen) the results were analogous. Median (range) Δ ct/min for GAD was 397 (0–11 895) for the patients and 0 (0–3599) for the control subjects (P = 0·006; Mann–Whitney U-test). The highest Δ ct/min to GAD were seen in both groups in subjects with the highest SIs to GAD.
Fig. 1.
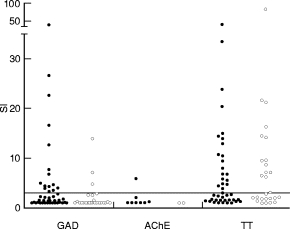
T cell proliferation responses to glutamic acid decarboxylase (GAD), acetylcholinesterase (AChE) and tetanus toxoid (TT) in patients with APECED (•) and in control subjects (○) are expressed as stimulation indices (SI). SI of 3·0 is marked with the horizontal line as a cut-off point for positivity. The distribution of the SIs to GAD differed significantly between patients and controls (P = 0·004; Mann–Whitney U-test). Fifteen of 44 (34%) patients with APECED had a positive SI compared with three of 28 (11%) healthy adults (P = 0·03; Fisher’s exact test).
A subgroup of 29 patients with APECED and 21 control subjects was studied for IFN-γ secretion by GAD-stimulated T cells. Secretion of IFN-γ (≥ 50 pg/ml) by GAD-stimulated PBMC was found in 17 of 29 patients tested (59%) (Fig. 2), and in four of 21 (19%) healthy people studied (P = 0·008, Fisher’s exact test). The levels of GAD-stimulated IFN-γ were high in the patients (median 179 pg/ml, range 0–12·5 ng/ml) and different from the levels seen in controls (median 0, range 0–209 pg/ml) (P = 0·001, Mann–Whitney U-test). In contrast, IFN-γ secretion by TT-stimulated T cells tended to be lower in the patients with APECED than in control subjects (Fig. 2; P = 0·09, Mann–Whitney U-test).
Fig. 2.
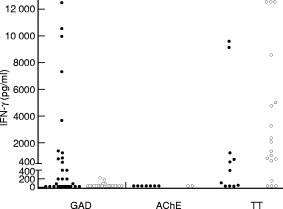
The concentration of IFN-γ secreted by glutamic acid decarboxylase (GAD), acetylcholinesterase (AChE) or tetanus toxoid (TT)-stimulated peripheral blood mononuclear cells (PBMC) in patients with APECED (•) and in control subjects (○). Secretion of IFN-γ (≥ 50 pg/ml) by GAD-stimulated PBMC was found in 17 of 29 patients tested (59%), and in four of 21 (19%) healthy individuals studied (P = 0·008; Fisher’s exact test). The levels of IFN-γ in the patients’ tests were significantly higher than in the controls’ tests (P = 0·001; Mann–Whitney U-test).
Both the proliferation response and IFN-γ secretion by GAD-stimulated T cells occurred in five cases, only proliferation in three cases, only IFN-γ secretion in 12 cases, and neither in nine cases. The levels of IFN-γ by GAD-stimulated PBMC did not differ between the patients with positive (SI ≥ 3) or negative (SI < 3) proliferation response to GAD (P = 0·85; Mann–Whitney U-test). No correlation existed between the proliferation response to GAD and the concentration of IFN-γ secreted by the GAD-stimulated PBMC (r = −0·081, P = 0·68), whereas a positive correlation was seen between the T cell proliferation response to TT and IFN-γ secretion by TT-stimulated T cells (r = 0·741, P = 0·009, Spearman’s correlation).
Medium values and T cell responses to control antigens
Medium values tended to be higher in patients than in the control subjects (median ct/min 591 and 359, respectively; P = 0·06; Mann–Whitney U-test). No difference was seen in T cell proliferation responses to TT expressed as SIs (Fig. 1; median (mean) SIs were 2·9 (7·0) for the patients and 4·7 (9·2) for the control subjects; P = 0·46, Mann–Whitney U-test) or Δ ct/min (P = 0·90, Mann–Whitney U-test) between the groups. The T cell proliferation response to AChE was studied in 10 subjects, five of whom had a positive T cell response to GAD (SI ≥ 3). Only one patient showed a positive proliferative response to AChE (SI = 5·9; Fig. 1). Secretion of IFN-γ (≥ 50 pg/ml) by AChE-stimulated PBMC was not found in any of the nine subjects tested (Fig. 2). None of the 16 individuals tested with the extract of Sf9 cells infected with baculovirus at a concentration of 0·1 μ g/ml and one of the 20 individuals tested at a concentration of 1·0 μ g/ml showed positive response (SI ≥ 3) to this control antigen.
Relationship between cellular and humoral immunity to GAD
Elevated levels of antibodies against GAD were present in 14 of the 44 patients (32%). A negative correlation (r = −0·436, P = 0·03) existed between the levels of antibodies and the SIs to GAD among the patients who responded to GAD (SI ≥ 3 or the level of antibodies ≥ 5; Fig. 3), and the same held true when T cell response to GAD was expressed as Δ ct/min (r = −0·442, P = 0·03). Similarly, the concentration of IFN-γ in the supernatants from the GAD-stimulated PBMC in patients with APECED showed an inverse correlation (r = −0·366) with the levels of antibodies to GAD among the responders (IFN-γ≥ 50 or the level of antibodies ≥ 5), although this was not statistically significant (P = 0·12). The inverse correlation between the levels of antibodies to GAD and the SIs to GAD existed only among the subgroup of APECED patients without IDDM (r = −0·714, P = 0·001), whereas in the subgroup of APECED patients prone to IDDM (eight patients with clinical IDDM and three prediabetic patients) no correlation was seen (r = 0·171, P = 0·69). Antibodies to GAD did not show correlation with T cell responses to TT (r = 0·080, P = 0·6). TT antibody levels did not show any correlation with SIs to TT in the patients with APECED or in control subjects (r = −0·005, P = 0·98 and r = 0·129, P = 0·62, respectively). No difference was seen in TT antibody levels between the groups (median OD was 0·28 for the patients and 0·35 for the control subjects; P = 0·20, Mann–Whitney U-test).
Fig. 3.
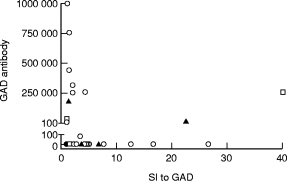
Relation of T cell proliferation response to glutamic acid decarboxylase (GAD; abscissa) and the levels of antibodies to GAD (ordinate) in patients with APECED without insulin-dependent diabetes mellitus (IDDM) (○), with IDDM (▴), and in the patients who developed IDDM within 12 months after testing (□). Elevated levels of antibodies against GAD were seen in 14 of 44 patients (32%). Negative correlation (r = −0·436, P = 0·03) was found between the levels of antibodies and the stimulation indices (SIs) to GAD among patients who responded to GAD (SI ≥ 3 or the level of antibodies ≥ 5).
Association of the HLA DQB1 risk alleles for IDDM with immunity to GAD
Seven of the 41 HLA-typed patients had HLA DQB1*0201/x, 11 patients had HLA DQB1*0302/x, and 23 had DQB1*x/x genotype (x denotes alleles other than DQB1*0201 or 0302). Four of seven patients with DQB1*0201 were also DR7+. We found proliferation response to GAD in five of the seven (71%) patients with DQB1*0201/x, in four of the 11 (36%) patients with DQB1*0302/x, and in only five of the 23 (22%) patients without any risk allele. Positive proliferation response against GAD was significantly more common among the DQB1*0201 allele-positive patients than in those without the risk alleles (P = 0·03; Fisher’s exact test). One of the three healthy individuals with proliferation response to GAD had the DQB1*0302 allele, another had the DQB1*0201 allele, and another had none of the risk alleles.
The secretion of IFN-γ by GAD-stimulated PBMC was not associated with HLA DQB1*0201 or 0302 in the patients or the healthy controls. Secretion of IFN-γ by GAD-stimulated PBMC was detected in two of five patients with DQB1*0201, six of eight with DQB1*0302 and six of 12 without the risk alleles. The proliferation response to GAD (SI = 7·1) and IFN-γ secretion (64 pg/ml) by GAD-stimulated PBMC coincided in only one healthy individual; she had the DQB1*0201 risk allele.
GAD antibody positivity was not associated with the HLA DQB1 risk alleles for IDDM in the patients. GAD antibodies were found in one of seven patients with DQB1*0201, three of 11 with DQB1*0302 and eight of 23 without the risk alleles.
Relation of IDDM, insulin secretion and autoimmunity to GAD in APECED
Eight of the 44 patients (18%) had IDDM diagnosed already years before the T cell testing. The patients with IDDM did not differ from the nondiabetic patients with respect to T cell proliferation, IFN-γ secretion by the PBMC, or antibody-positivity to GAD (Table 1).
Table 1.
Comparison of immunity to glutamic acid decarboxylase (GAD) and the frequencies of HLA DQB1*0201 and 0302 alleles in APECED patients with insulin-dependent diabetes mellitus (IDDM), prediabetic patients and patients without IDDM
| APECED with IDDM | Prediabetic APECED patients | APECED without IDDM | |
|---|---|---|---|
| SI to GAD ≥ 3 | 3/8 (38%) | 1/3 (33%) | 11/33 (33%) |
| GAD-IFN-γ≥ 50 pg/ml | 3/5 (60%) | 1/1 (100%) | 13/23 (57%) |
| GAD antibody-positive | 4/8 (50%) | 2/3 (67%) | 8/33 (24%) |
| HLA DQB1 | |||
| *0201,x | 1/8 (13%) | 2/3 (67%) | 4/30 (13%) |
| *0302,x | 2/8 (25%) | 0/3 (0%) | 9/30 (30%) |
| *x,x | 5/8 (63%) | 1/3 (33%) | 17/30 (57%) |
| Correlation between | r = 0·171, | r = −0·714, | |
| SI to GAD and GAD antibody level | P = 0·83 | P = 0·001 | |
Stimulation index (SI) ≥ 3 is considered a positive proliferation response and GAD antibody level ≥ 5 as a positive humoral response (GAD antibody-positive). No significant differences were found in the frequencies of positive cellular or humoral immunity to GAD or in the frequencies of HLA DQB1 genotypes between patients with IDDM, prediabetic patients and patients without IDDM.
Of the 14 non-diabetic patients who underwent IVGTT, six had a positive proliferation response to GAD. There was no difference in either the 1 + 3 min or 10 min incremental insulin area under the curve (Insarea3’, Insarea10’) during IVGTT between patients with a positive and those with a negative proliferation response. The median (range) of Insarea3’ and Insarea10’ were 85·5 (28–444) and 312 (107–1790) mU/l for patients with SI < 3; and 56·5 (18–465) and 282 (63–2039) mU/l for patients with SI ≥ 3. IFN-γ secretion by GAD-stimulated PBMC was associated with neither decreased Insarea3’ nor Insarea10’ during IVGTT, although the number of patients in this analysis was small (n = 9; data not shown).
Of the three patients who developed diabetes within 12 months after the testing, one had both positive antibody and proliferation response to GAD, another had only positive antibody response, and the only one who was studied for IFN-γ secretion by GAD-stimulated PBMC had no antibody or proliferation response but showed low IFN-γ secretion (86 pg/ml).
IFN-γ secretion by PWM-stimulated PBMC
No difference was seen in IFN-γ levels secreted by PWM-stimulated PBMC between 31 patients and 11 control subjects (medians being 32·3 ng/ml and 32·4 ng/ml, respectively; P = 0·96, Mann–Whitney U-test).
Relation of age and sex of the patients with autoimmunity to GAD
No correlation between age and SI to GAD (r = 0·198, P = 0·20), IFN-γ levels by GAD-stimulated PBMC (r = −0·165, P = 0·39), or GAD antibody levels (r = 0·028, P = 0·86) was seen in this series of APECED patients (Spearman’s correlation). No difference was seen in the SIs to GAD or in the levels of IFN-γ by GAD-stimulated PBMC between the female and male patients (P = 0·97 and P = 0·80, respectively, Mann–Whitney U-test). GAD antibody levels were higher in male patients compared with female patients (P = 0·03, Mann–Whitney U-test).
DISCUSSION
T cell proliferative response to GAD was frequent in our series of patients with APECED (34%) as well as elevated levels of antibodies to GAD (32%). However, coincidence of both responses in the same patient was infrequent, occurring in only four of the 44 patients, and a negative correlation between humoral and cellular immunity to GAD was observed, as previously reported for immunity to GAD67 in subjects at high risk for IDDM [10]. There was also a tendency to an inverse correlation between the concentration of IFN-γ secreted by GAD-stimulated T cells and the level of antibodies to GAD. The dissociation of antigen-specific cellular and humoral immunity is usually explained by reciprocal regulation of these responses by cytokines [18]. Th1-type CD4+ lymphocytes secrete predominantly IFN-γ and IL-2, which activate macrophages and cytotoxic lymphocytes. Th2-type lymphocytes secrete predominantly IL-4 and IL-5, which induce antibody production by B lymphocytes. The cytokines secreted by Th1-type lymphocytes, such as IFN-γ, have been reported to be inhibitory to B cell proliferation and the production of antibodies. Interestingly, the inverse correlation between humoral and proliferative responses to GAD was seen only in the subgroup of APECED patients without IDDM. In this subgroup of patients high antibody levels were seen without proliferative response to GAD, and vice versa, suggesting that the dichotomous responsiveness to GAD does not predict the development of IDDM in these patients.
We observed IFN-γ secretion by GAD-stimulated T cells without detectable proliferation response to GAD in several patients. T cell proliferation reflects the summation of several signals, and activation of lymphokine genes after stimulation with a specific antigen may occur without proliferation [14,19]. GAD-induced secretion of IFN-γ in the absence of T cell proliferation in patients with APECED indicates the presence of specific T lymphocytes recognizing the antigen. In adults immunized against tetanus in childhood a weak responsiveness to a recall antigen is sometimes detected only as activation of cytokine secretion without proliferation response [14]. In the present study GAD-stimulated T cell proliferation was associated with the HLA DQB1*0201 allele, but the secretion of IFN-γ or GAD antibody was in no relation to the HLA DQB1 alleles. In this regard, DQB1*0201 may not be a restrictive element for the recognition of GAD. Others have reported that antibodies to GAD were associated with the HLA DQB1*0201 allele [20] or with DR3 [21], which is in linkage disequilibrium with DQB1*0201. Harrison et al. found an association between both humoral and cellular immune responses to GAD67 and HLA DR3 in their study on first-degree relatives of IDDM patients [10]. It should be emphasized that in our series of APECED patients four of seven patients with HLA DQB1*0201 allele were DR7+, which is not associated with an increased genetic risk for common IDDM.
Our results indicate that most patients with APECED (76%) develop autoimmunity to GAD detected as antibodies, T cell proliferation or GAD-stimulated IFN-γ secretion by T cells. This suggests that immunization to GAD in APECED may be a more common phenomenon than previously thought. The patients with APECED did not show enhanced T cell reactivity in general, since the proliferation responses to TT did not differ between patients and control subjects. In our series, T cell proliferation or IFN-γ response to GAD was found in patients with long-duration IDDM as frequently as in patients without IDDM. All patients with IDDM had had the disease already for several years before T cell testing. It is possible that we could not see the association of GAD reactivity with IDDM because the reactivity had declined after diagnosis. Only three patients developed IDDM within 12 months from T cell testing, and one of them had positive proliferation response to GAD. When patients with decreased insulin response in IVGTT were studied, T cell reactivity to GAD did not show association with this parameter of β-cell dysfunction. Actually, in the present study T cell reactivity to GAD was found in the majority of APECED patients without evidence of IDDM. Others have reported high prevalence of GAD antibodies in APECED whether the patients develop IDDM or not [6,22], and further, it has been reported that the islets of non-diabetic, autoimmune, polyendocrine patients lack immunohistological changes despite the occurrence of GAD antibodies [23]. It thus seems that autoimmunity to GAD does not directly imply clinically detectable β-cell damage in patients with APECED, although based on the present study the occurrence of subclinical, non-destructive insulitis cannot be excluded.
Since chronic candidiasis is a common feature in APECED it could be speculated that there may be a functional defect of cellular immunity in these patients. This functional impairment was not observed in proliferative response to an autoantigen (GAD) or to a foreign antigen (TT) or in IFN-γ secretion by antigen-specific T cells. Further, no defect was seen in the capacity of PWM-stimulated PBMC to secrete IFN-γ in the patients with APECED. Because T cell assays used in the present study reflect mostly the function of T helper cells, these data suggest that the function of these cells is not affected in patients with APECED. It should be emphasized that in the present study we did not measure specific T cell response to candida, and thus no conclusion can be drawn on the function of antigen-specific T cells important in the clearance of candida infection.
The pathogenesis of IDDM in APECED may differ from that in common IDDM because of the single gene defect in APECED and the different HLA background. The gene defect may affect the mechanisms needed for maintaining immune tolerance to self-antigens, thus leading to autoimmune destruction of several cell types, particularly those of endocrine origin. On the other hand, it is possible that the gene defect may lead to destruction of specific cell types and the observed autoimmunity is a consequence of that.
This is the first report of cellular autoimmunity to GAD in APECED. We demonstrate that autoimmunity to GAD is a common feature in APECED and not restricted to the subgroup of patients with IDDM, indicating that not only antigen specificity but other determinants of autoreactivity are important in the development of autoimmune IDDM. However, the pathogenesis of IDDM in APECED may differ from that in common IDDM because of the single gene defect in APECED.
Acknowledgments
We thank Dr Christian Oker-Blom for kindly providing us with the acetylcholinesterase as a control antigen and Dr Thomas Dyrberg for providing us with Baculovirus vector containing GAD gene. The hybridoma developed by Dr David Gottlieb was obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Pharmacology and Molecular Sciences, John Hopkins University School of Medicine, Baltimore, MD 21205, and the Department of Biological Sciences, University of Iowa, Iowa City, IA 52242, under contract N01-HD-2–3144 from NICHD. The skilful technical assistance of Mrs Anneli Suomela is highly appreciated. This study was supported by grants from the Foundation for Paediatric Research in Finland, the 350th Anniversary Foundation of the University of Helsinki, the Foundation for Diabetes Research in Finland, the Juvenile Diabetes Foundation International and the Academy of Finland.
References
- 1.Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–36. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 2.Whitaker J, Landing BH, Esselbom VM, Williams RR. The syndrome of familial juvenile hypoadrenocorticism, hypoparathyroidism and superficial moniliasis. J Clin Endocrin Metab. 1956;16:1374–87. doi: 10.1210/jcem-16-10-1374. [DOI] [PubMed] [Google Scholar]
- 3.Neufeldt M, Maclaren N, Blizzard R. Autoimmune polyglandular syndromes. Pediatr Ann. 1980;9:154–62. [PubMed] [Google Scholar]
- 4.The Finnish‐German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nature Genet. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 5.Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECED gene. Nature Genet. 1997;17:393–8. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 6.Tuomi T, Björses P, Falorni A, Partanen J, Perheentupa J, Lernmark Å, Miettinen A. Antibodies to glutamic acid decarboxylase and insulin-dependent diabetes in patients with autoimmune disease type I. J Clin Endocrin Metab. 1996;81:1488–94. doi: 10.1210/jcem.81.4.8636356. [DOI] [PubMed] [Google Scholar]
- 7.Baekkeskov S, Aanstoot H-J, Christgau S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–6. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 8.Atkinson MA, Kaufman DL, Campbell L, et al. Response of peripheral blood mononuclear cells to glutamate decarboxylase in insulin-dependent mellitus. Lancet. 1992;339:458–9. doi: 10.1016/0140-6736(92)91061-c. [DOI] [PubMed] [Google Scholar]
- 9.Honeyman M, Cram D, Harrison L. Glutamic acid decarboxylase 67-reactive cells: a marker of insulin-dependent diabetes. J Exp Med. 1993;177:535–740. doi: 10.1084/jem.177.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison LC, Honeyman MC, DeAizpura HJ, Schmidli RS, Colman PG, Tait BD, Scram DS. Inverse relation between humoral and cellular immunity to glutamic acid decarboxylase in subjects at risk of insulin-dependent diabetes. Lancet. 1993;341:1365–9. doi: 10.1016/0140-6736(93)90940-i. [DOI] [PubMed] [Google Scholar]
- 11.Worsaae A, Hejnaes K, Moody A, Ludvigsson J, Pociot F, Lorenzen T, Dyrberg T. T cell proliferation responses to glutamic acid decarboxylase-65 in IDDM are negatively associated with HLA DR3/4. Autoimmunity. 1995;22:183–9. doi: 10.3109/08916939508995315. [DOI] [PubMed] [Google Scholar]
- 12.Moody AJ, Hejnaes KR, Marshall MO, Larsen FS, Boel E, Svendsen I, Mortensen E, Dyrberg T. Isolation by anion-exchange of immunologically and enzymatically active human islet glutamic acid decarboxylase 65 overexpressed in Sf9 insect cells. Diabetologia. 1995;38:14–23. doi: 10.1007/BF02369348. [DOI] [PubMed] [Google Scholar]
- 13.Paronen J, Klemetti P, Kantele JM, Savilahti E, Perheentupa J, Åkerblom HK, Vaarala O. Glutamate decarboxylase reactive peripheral blood lymphocytes from patients with insulin-dependent diabetes mellitus express gut-specific homing receptor α4β7-integrin. Diabetes. 1997;46:583–8. doi: 10.2337/diab.46.4.583. [DOI] [PubMed] [Google Scholar]
- 14.Halminen M, Klemetti P, Vaarala O, Hurme M, Ilonen J. Interferon-γ production in antigen specific T cell response: quantitation of specific mRNA and secreted protein. Scand J Immunol. 1997;46:388–92. doi: 10.1046/j.1365-3083.1997.d01-144.x. [DOI] [PubMed] [Google Scholar]
- 15.Kimura A, Sasazuki T. Eleventh International Histocompatibility Workshop reference protocol for the HLA DNA-typing technique. In: Sasazuki T, editor. HLA. Vol. 1. Oxford: Oxford University Press; 1991. pp. 397–42. [Google Scholar]
- 16.Grubin CE, Daniels T, Toivola B, et al. A novel radioligand binding assay to determine diagnostic accuracy of isoform-specific glutamic acid decarboxylase antibodies in childhood IDDM. Diabetologia. 1994;37:344–50. doi: 10.1007/BF00408469. [DOI] [PubMed] [Google Scholar]
- 17.Falorni A, Ortqvist E, Petersson B, Lernmark Å. Radioimmunoassays for glutamic acid decarboxylase (GAD) 65 and GAD autoantibodies using 35S or 3H recombinant human ligands. J Immunol Methods. 1995;186:89–99. doi: 10.1016/0022-1759(95)00139-2. [DOI] [PubMed] [Google Scholar]
- 18.Del Prete GF, De Carli M, Ricci M, Romagnani S. Helper activity for immunoglobulin synthesis of T helper type 1 (Th1) and Th2 human T cell clones: the help of Th1 clones is limited by their cytolytic capacity. J Exp Med. 1991;174:809–913. doi: 10.1084/jem.174.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–9. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 20.Hagopian WA, Sanjeevi CB, Kockum I, et al. Glutamate decarboxylase-, insulin-, and islet cell-antibodies and HLA typing to detect diabetes in a general population-based study of Swedish children. J Clin Invest. 1995;95:1505–11. doi: 10.1172/JCI117822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese S, Bonfanti R, Bazzigaluppi E, Lampasona V, Benazzi E, Bosi E, Chiumello G, Bonifacio E. Association of IA-2 autoantibodies with HLA DR4 phenotypes in IDDM. Diabetologia. 1996;39:1223–6. doi: 10.1007/BF02658510. [DOI] [PubMed] [Google Scholar]
- 22.Björk E, Velloso LA, Kämpe O, Karlsson A. GAD autoantibodies in IDDM, stiff-man syndrome, and autoimmune polyendocrine syndrome type I recognize different epitopes. Diabetes. 1994;43:161–5. doi: 10.2337/diab.43.1.161. [DOI] [PubMed] [Google Scholar]
- 23.Wagner R, McNally J, Bonifacio E, et al. Lack of immunohistochemical changes in the islets of nondiabetic, autoimmune, polyendocrine patients with β-selective GAD-specific islet antibodies. Diabetes. 1994;43:851–6. doi: 10.2337/diab.43.7.851. [DOI] [PubMed] [Google Scholar]