

Abstract
Human IL-17 is a cytokine secreted by CD4+-activated memory T cells with the profile of effects of a Th1 cytokine. The effects of IL-17 on many cellular constituents of joints suggest that it may participate in inflammatory joint diseases. Proteins of the complement system are known to be regulated by pro- and anti-inflammatory cytokines. The purpose of this work was to study the effect of IL-17 alone and combined with tumour necrosis factor (TNF) on the expression and synthesis of factor B and C3. Fibroblasts were stimulated with the relevant cytokine or cytokines, pulse labelled with 35S-methionine, and the newly synthesized proteins were immunoprecipitated and subjected to SDS–PAGE. Gene expression was determined by Northern blot analysis. IL-17 10 ng/ml induced increases in gene expression and protein synthesis of C3, 2·25 ± 0·26- and 2·7 ± 0·7-fold, respectively, with concomitant non-significant effects on factor B, 1·5 ± 0·45- and 2·2 ± 1·2-fold, respectively. When both IL-17 and TNF were present simultaneously, the synthesis of factor B increased by 85% more than the expected additive effects of these cytokines separately, while for C3 the effect of both cytokines was 19% lower than the expected additive effect (observed/expected = 0·81). IL-4 reduced the synergistic effect by 50%. We conclude that IL-17 has a regulatory role on C3 expression and synthesis and an amplifying effect on TNF-induced factor B synthesis. Taken together with the evidence that TNF is a major cytokine involved in the inflammation of rheumatoid arthritis, it suggests that IL-17 has a proinflammatory role in the inflammation process of joints. The distinct effects of IL-4, IL-17 and TNF on the synthesis of factor B in fibroblasts suggest that factor B and the alternative pathway of the complement system may play an important role in joint inflammation.
Keywords: IL-17, tumour necrosis factor, complement, fibroblasts
Introduction
Human IL-17 is a recently described cytokine secreted by CD4+ activated memory T cells [1]. This cytokine was found to stimulate the secretion of IL-6, IL-8, prostaglandin E2, and granulocyte colony-stimulating factor (G-CSF) in epithelial, endothelial and fibroblastic cells [2], and to increase surface expression of intercellular adhesion molecule-1 (ICAM-1) in fibroblasts [1], synoviocytes [1,2] and keratinocytes [3]. In addition, IL-17 was shown to increase nitric oxide production in cartilage [4] and in chondrocytes [5]. Stimulation of human chondrocytes with IL-17 is associated with activation of MAP kinases and NF-κ B, and induced gene expression of IL-1β, IL-6 and stromelysin [5]. In human macrophages, IL-17 stimulates IL-1β and tumour necrosis factor-alpha (TNF-α) production as well as IL-6, prostaglandin E2 (PGE2), IL-10, IL-12, IL-R antagonist and stromyelysin secretion [6]. This profile of effects suggests that most, but not all, IL-17 effects are proinflammatory. It was suggested that on the basis of published activities and the cellular source, IL-17 might be classified as a Th1 cytokine [7,8]. The expression of IL-17 in rheumatoid (RA) synovium but not in osteoarthritis (OA) [9] and the effects of IL-17 on many cellular constituents of joints suggest that it may play a major role in rheumatic joint disease. IL-1 and TNF play a pivotal role in local activation leading to joint inflammation and destruction [10]. Among other activities, IL-1 and TNF enhance the synthesis of factor B (Bf), a major activator of the alternative pathway of the complement system, and that of C3 [11]. Anti-inflammatory Th2 cytokines, such as IL-4 and IL-13, decrease TNF-induced Bf synthesis, but not that of C3, in fibroblasts [12,13]. Numerous models demonstrated that IL-4 and IL-13 had favourable effects in reducing joint inflammation and destruction [14–19]. The purpose of the present study was to examine what are the effects of IL-17 alone and in combination with TNF on the synthesis of C3 and Bf in fibroblasts. The nature of the responses may elucidate the role of IL-17 in joint inflammation and validate the assignment of this cytokine as a Th1 cytokine.
MATERIALS and METHODS
Reagents
Dulbecco’s modified Eagle’s medium (DMEM)-high d-glucose, penicillin–streptomycin solution, l-glutamine, and heat-inactivated fetal calf serum (FCS) were purchased from Biological Industries (Kibbutz Beit Haemek, Israel). DMEM without methionine, cysteine and l-glutamine and albumin bovine fraction V were purchased from Sigma Chemical Co. (St Louis, MO). 35S-methionine Redivue pro-mix (sp. act. approx. 1000 Ci/mmol) and α32P-dCTP (sp. act. approx. 3000 Ci/mmol) were acquired from Amersham (Little Chalfont, UK). Goat anti-human C3 and goat anti-human properdin factor B were purchased from Incstar Corp. (Stillwater, MN), and protein A Pansorbin cells from Calbiochem Novabiochem Co. (La Jolla, CA). Recombinant human TNF-α, recombinant human IL-1α and recombinant human IL-17 were purchased from R&D Systems Inc. (Minneapolis, MN).
Cell culture conditions, biosynthetic labelling and immunoprecipitation
Fibroblast cell lines were initiated in our laboratory from skin biopsies of four apparently normal individuals aged 18–60 years as previously described [20]. Monolayers were maintained in DMEM with 10% FCS at 37°C until confluence. Three wells of 2 cm2 each (1·5 × 105 cells) were used per condition. Prior to the addition of cytokines, the medium containing FCS was replaced by DMEM with 1 mg/ml bovine serum albumin (BSA) alone (control) or with various concentrations of cytokines for periods as indicated in each set of experiments.
Biosynthetic labelling for the analysis of the newly synthesized protein was performed essentially as previously described [11]. In brief, following incubation in medium containing cytokines, the cells were incubated in methionine-free medium supplemented with excess of 35S-methionine for 2 h. The medium was then removed, centrifuged and supernatant was kept at −20°C for analysis of proteins secreted from the cells during the labelling period. The cells were then lysed by freeze–thawing in the presence of detergents. Total protein synthesis was estimated by incorporation of 35S-methionine into TCA-insoluble proteins. Specific proteins were sequentially immunoprecipitated from either cell lysates or extracellular medium with monospecific antibody and protein A, and analysed by SDS–PAGE followed by fluorography. Incorporation of 35S-methionine into specific immunoprecipitated protein was determined in gel slices after digestion with 15% hydrogen peroxide for 16 h at 65°C and counted with a β-counter.
Northern blotting
Total RNA was isolated from cells following incubation in medium containing cytokines as indicated, by RNAsol (TEL-TEST Inc., Friends Wood, TX) according to the manufacturer’s instructions. In brief, after fibroblast cells were lysed with RNAzol B (2 ml per 107 cells), the RNA was extracted with chloroform, and the aqueous phase was cold precipitated by isopropanol, dissolved with diethylpyrocarbonate (DEPC)-treated ddH2O and quantified by absorbency at 260 nm. RNA extract (10 μg) was run on agarose-formaldehyde gel and was blotted on nylon membrane (Hybond-N; Amersham) with 10× SSC for 18 h. Equal loading of samples was assured by ethidium-bromide shadowing. The membrane containing the RNA was subjected to UV cross-linking, prehybridized with herring sperm DNA (Promega Corp., Madison, WI) in Rapid-hyb buffer (Amersham) at 65°C and hybridized with random labelled radioactive cDNA probes (redi prime; Amersham). Plasmids pC3.59 with the human C3 cDNA insert (1 kb, BstEII fragment) [21], and BfA28 with the human factor B cDNA insert (1.8 kb, PstI fragment) [22] were used to produce specific probes for the corresponding mRNAs (mRNA masses of C3 and Bf are 5·2 kb and 2·6 kb, respectively, and the cDNA probes do not cross-hybridize). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a constitutively expressed gene that was used as an internal control for mRNA concentration in all Northern blots (1·4 kb, PstI fragment) in pGEM.
Quantification was determined by photodensitometry (computing densitometer, model 300A; Molecular Dynamics, Eugene, OR).
Statistical analysis
The significance of differences between cells treated with IL-17 and TNF together and cells treated with each cytokine alone was determined by Student’s paired t-test.
Results
Effect of hIL-17 on C3 and factor B in skin fibroblasts
hIL-17 at a concentration of 10 ng/ml induced a significant (2·7 ± 0·7, P = 0·001) increase in the synthesis of C3. No additional effect was observed at higher concentrations. However, hIL-17 induced only a minor (2·2 ± 1·2) and non-significant (P = 0·12) increase in Bf synthesis, even at concentrations as high as 100 ng/ml (Fig. 1b). An increase in Bf synthesis could be readily demonstrated in these fibroblast cells following stimulation with 1 ng/ml IL-1α, which has a known stimulatory effect on Bf synthesis in fibroblasts [11] (Fig. 1b). The effect of hIL-17 on the synthesis of C3 and Bf was pretranslational, as demonstrated by a parallel increase in gene expression, 2·25 ± 0·26-fold (P = 0·025) for C3 and 1·5 ± 0·45-fold (P = 0·18) for factor B (Fig. 1c).
Fig. 1.
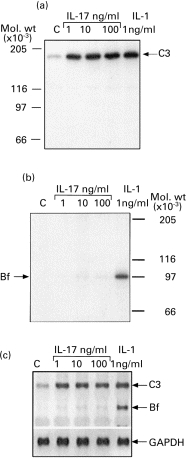
Effect of hIL-17 on C3 and factor B (Bf) synthesis. Skin fibroblasts were incubated in medium containing 1 mg/ml bovine serum albumin (BSA) alone (lane C) or with cytokines for 24 h. In (a) and (b) the cells were subsequently subjected to metabolic labelling in methionine-free medium containing 350 μCi/ml 35S-methionine. Pro C3 protein (a) and Bf protein (b) were immunoprecipitated from cell lysates by specific antibodies and subjected to SDS–PAGE under reducing conditions, followed by impregnating with autofluor and exposure to x-ray film. (c) Monolayers from the same donor and passage were treated equally in parallel, and 10 μg of RNA extracted from those cells were run on agarose-formaldehyde gel and blotted on nylon membrane. mRNA levels of C3 (5·2 kb) and Bf (2·6 kb) were detected following hybridization with 32P-labelled cDNA probes specific to either human C3 or to human Bf. For internal control of mRNA concentration in the various samples, the membrane was hybridized with 32P-labelled glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA, which is a constitutively expressed gene.
Effect of co-incubation in hIL-17 and TNF-α on C3 and Bf synthesis
Co-incubation of fibroblast monolayers in TNF-α and IL-17 induced an increase in the synthesis and secretion of C3 and Bf greater than each cytokine alone. These effects were distinct for each gene, i.e. C3 and Bf. For C3, the observed enhancement was smaller than the sum of enhancements observed for each cytokine alone (Fig. 2a). In contrast, the simultaneous presence of TNF-α and IL-17 had a synergistic effect on the rate of synthesis and secretion of Bf (Fig. 2b). The ratios between the actual value and the expected additive value obtained for Bf and C3 in seven different experiments were summarized and the mean ratio for C3 was 0·81 ± 0·28, suggesting no synergism, whereas the mean ratio for Bf was 1·85 ± 0·56 (P < 0·01). Northern blot scanning analysis confirmed a significant synergistic effect of co-incubation of IL-17 and TNF on factor B gene expression, confirming that the synergistic effect occurred also at the pretranslational level (data not shown).
Fig. 2.
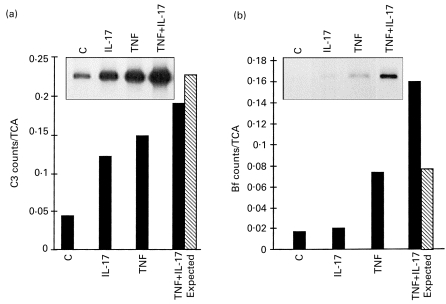
Effect of simultaneous incubation with hIL-17 and tumour necrosis factor-alpha (TNF-α) on C3 and factor B (Bf) synthesis. Fibroblast monolayers were incubated for 24 h in medium/bovine serum albumin (BSA) (lane C) or with 10 ng/ml hIL-17 (IL-17), 1 ng/ml TNF-α (TNF), or both (TNF + IL-17). Cells were subsequently subjected to metabolic labelling with 35S-methionine, and C3 protein (a) and Bf protein (b) were immunoprecipitated from cell lysates by specific antibodies and subjected to SDS–PAGE followed by impregnating with autofluor and exposure to x-ray film (upper inserts). Incorporation of 35S-methionine into specific immunoprecipitated protein was determined in gel slices after digestion and counting with scintillation fluid in a β-counter. Data are expressed as the percent ratio of specific protein to total TCA-precipitable protein in the same condition. The expected additive effects (calculated from counts/TCA from IL-17 alone + TNF-α alone – control, hatched bars) were compared with the actual effects (▪).
The synergistic effect on the synthesis of factor B was specific for the combination of IL-17 and TNF, since stimulation with IL-1β and IL-17 did not induce a synergistic increase in the synthesis of factor B (data not shown).
Dose–response and time course studies
Both dose–response and time-course characteristics of the synergy between hIL-17 and TNF-α on Bf expression and synthesis were studied. Fibroblast cells were incubated with TNF-α and hIL-17 in various concentrations for 24 h before Bf synthesis was analysed. As shown in Fig. 3, TNF-α either alone or combined with IL-17 induced an increase in Bf synthesis in a dose-dependent manner, and the synergism between hIL-17 or TNF was detected in all doses tested.
Fig. 3.
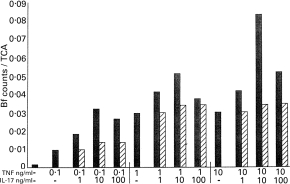
Dose-dependent effects of hIL-17 and tumour necrosis factor-alpha (TNF-α) on factor B (Bf) synthesis in fibroblasts. Fibroblasts were incubated for 24 h in medium/bovine serum albumin (BSA) (control) or with increasing concentrations of TNF-α, hIL-17 or both TNF-α and hIL-17 as indicated. The synthesis of Bf was determined by metabolic labelling, immunoprecipitation and SDS–PAGE as in Fig. 1. Data are expressed as the ratio of Bf specific counts to total TCA-precipitable protein in the same condition. The expected additive effects (calculated from counts/TCA from stimulation with hIL-17 alone + TNF-α alone – control, hatched bars) were compared with the actual effects obtained in each concentration combination (▪). The effect of hIL-17 alone was negligible, as demonstrated in Fig. 1b.
In three separate time–response studies using hIL-17 10 ng/ml, and TNF-α 1 ng/ml, Northern blot analysis of C3 and Bf mRNA expressions indicated that incubation with TNF-α alone or together with IL-17 for a 4-h period induced small increase in Bf gene expression. However, a longer period of incubation was required for the enhancement of C3 mRNA expression (Fig. 4a, a representative experiment). Both Bf and C3 mRNA expression increased in a time-dependent manner. The shortest incubation period tested, namely 4 h, was sufficient to induce a synergistic effect on Bf protein secretion. This effect was maintained throughout all incubation periods examined (Fig. 4b,c). For C3 protein secretion, a 7-h incubation with IL-17 and TNF was required in order to obtain a marked stimulatory effect (Fig. 4d,e). This effect increased in a time-dependent manner, exerting the highest stimulatory effect following 48 h of incubation.
Fig. 4.
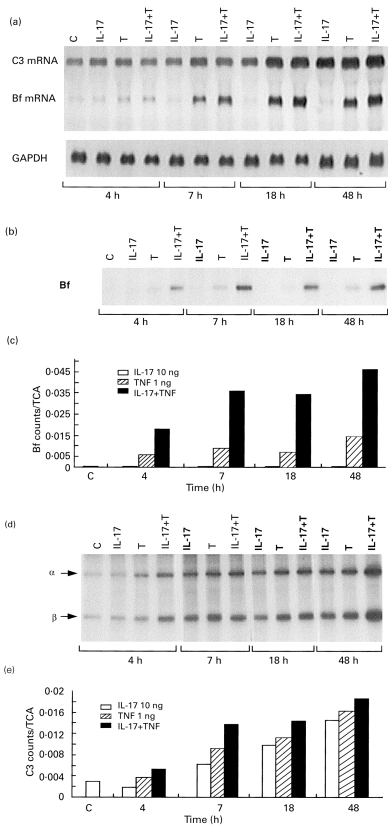
Time to effect of hIL-17 and tumour necrosis factor-alpha (TNF-α) stimulation on C3 and factor-B (Bf) expression and secretion. Fibroblast monolayers were incubated in medium/bovine serum albumin (BSA) or with 10 ng/ml hIL-17, 1 ng/ml TNF-α, or both (TNF-α+hIL-17) for the indicated time intervals. mRNA levels of Bf and C3 were detected by Northern blot analysis of total RNA extracts of these cells as described in the legend to Fig. 1c (a). The secreted Bf protein (b) and C3 protein (α- and β-chains) (d) were determined in the medium by immunoprecipitation and SDS–PAGE, and the data are expressed as the ratio of protein specific counts to total TCA-precipitable protein in the same condition (c,e).
Effect of preincubation with one cytokine on the response of the cells to the other cytokine
We next studied whether the synergistic effect of hIL-17 and TNF-α can be produced by sequential incubation of fibroblasts with the two cytokines. Fibroblasts were first preincubated with either hIL-17 or TNF-α for 12 h. At the end of the first incubation period the media were discarded, the cells were rinsed, and a second cytokine was added for an additional 12 h before C3 and Bf synthesis was analysed. As shown in Fig. 5, the greatest stimulatory effect on C3 synthesis was obtained in cells incubated for 24 h with either hIL-17 or TNF-α or both. Replacing one cytokine with another after 12 h of incubation resulted in a smaller increase of C3 synthesis. This lesser effect was observed regardless of which was the second stimulator, IL-17 or TNF-α. When we looked at the stability of the cytokines’ effect on C3 synthesis after their removal, it appeared that the stimulatory effect of hIL-17 co-incubated with TNF-α on C3 synthesis was better preserved than the effect of each cytokine alone.
Fig. 5.
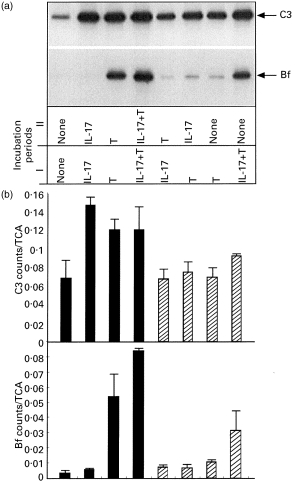
Effect of preincubation with one cytokine on the response to the other cytokine. Fibroblast monolayers were incubated for two consecutive periods of 12 h in varying combinations of control medium, 10 ng/ml hIL-17, 1 ng/ml tumour necrosis factor-alpha (TNF-α), or 1 ng/ml TNF-α+10 ng/ml hIL-17. At the end of period I, the media were discarded and the cells were rinsed before the second medium was added for incubation period II. The cells were then biosynthetically labelled and the synthesis of C3 and factor B (Bf) in the cell lysates was quantified as described in the legend to Fig. 1. (a) A representative experiment. (b) The mean ±s.d. of two separate experiments.
For Bf synthesis, synergism between hIL-17 and TNF-α was demonstrated only when both were present together. Preincubation with one cytokine did not have a significant effect on the subsequent response to the other stimulus. On the other hand, for fibroblasts stimulated with TNF-α and IL-17 for 12 h, the synergistic effect was evident 12 h after the removal of the cytokines.
Effect of hIL-4 on hIL-17 and TNF-α-stimulated factor B and C3 synthesis
We had previously reported that the effect of TNF-α on C3 and Bf synthesis in fibroblasts can be modulated by hIL-4 [12]. C3 synthesis was enhanced by IL-4 in TNF-stimulated fibroblasts, while the synthesis of Bf was decreased. In the current study, we examined whether hIL-4 can affect the synergistic effect of TNF and hIL-17. Fibroblasts were stimulated with TNF-α (1 ng/ml), hIL-17 (10 ng/ml) or hIL-4 (20 ng/ml) alone or in combination for 24 h before C3 and Bf synthesis was determined. As shown in Fig. 6a, hIL-4 alone had no effect on C3 or Bf synthesis, but it augmented TNF-induced increase in C3 synthesis while decreasing TNF-induced synthesis of Bf. hIL-4 also had some augmenting effect on C3 synthesis in hIL-17-stimulated fibroblasts. When monolayers were incubated with TNF and hIL-17, the addition of hIL-4 did not induce further increase in C3 protein synthesis or gene expression.
Fig. 6.
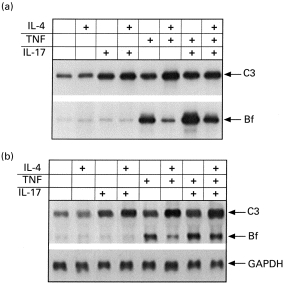
Effect of IL-4 on C3 and factor B (Bf) synthesis in response to costimulation with hIL-17 and tumour necrosis factor-alpha (TNF-α). Fibroblast monolayers were incubated for 24 h with 20 ng/ml IL-4, 10 ng/ml hIL-17, 1 ng/ml TNF-α, or combinations of these cytokines. The cells were subsequently subjected to either metabolic labelling followed by immunoprecipitation of C3 and Bf proteins (a), or to an RNA extraction procedure and Northern blot analysis (b) as described in the legend to Fig. 1.
In contrast, hIL-4 markedly decreased the synergism between hIL-17 and TNF-α on Bf synthesis. These effects were also detected at mRNA levels (Fig. 6b).
Discussion
We report here that hIL-17 induced gene expression and protein synthesis of C3 in fibroblasts. In addition, while hIL-17 had a minor effect on the synthesis of Bf, it had a synergistic effect with TNF on the production of Bf but not of C3. This is the first reported evidence that hIL-17 may have a regulatory role in the synthesis of complement proteins.
The reported effects of hIL-17 were achieved in concentrations similar to other published activities of hIL-17 [1–5], suggesting that these effects may have biological significance. The effect of IL-17 alone or with TNF were demonstrated on skin fibroblasts derived from normal donors as well as on skin and synovial fibroblasts from an RA patient (data not shown).
The distinct effects of hIL-17 on the synthesis of C3 and Bf deserve further clarification. C3 is involved in numerous biological activities, including immune cytolysis, clearance of immune complexes and facilitation of T and B cell proliferation and differentiation [23,24]. The increase in synthesis of C3 in response to hIL-17 suggests a regulatory role of this T cell cytokine.
Bf is a major activator protein of the alternative pathway of the complement system [25], suggesting a proinflammatory role for hIL-17. The fact that a significant effect of hIL-17 on the synthesis of Bf was observed only when the cells were stimulated with TNF-α may suggest that hIL-17 is not an independent proinflammatory cytokine but that it requires co-operation or interaction with other cytokines. Support for this hypothesis was derived from sequential incubation experiments. Most of the synergistic effect was achieved only when hIL-17 was present simultaneously with TNF-α (Fig. 5). Since fibroblasts are a source for various cytokines such as IL-1 and IL-6, which are up-regulated by IL-17, one may suggest that the synergistic effects of IL-17 and TNF are mediated via these cytokines. However, IL-1 did not have a synergistic effect with IL-17 and addition of anti-IL-6 did not inhibit the synergistic effect (data not shown). Furthermore, the synergism between IL-17 and TNF on Bf synthesis and secretion could be detected already following 2 h of stimulation, suggesting that the effect may not require production of new mediators. However, the possibility that IL-1 may play a role in mediating IL-17 effects on C3 synthesis can not be ruled out.
The synergistic effect of IL-17 and TNF is more pronounced at the level of protein synthesis and secretion than at the level of mRNA, i.e. a small increase in the mRNA level resulted in a marked increase in protein synthesis. This may suggest that these cytokines may interact also at levels other than gene transcription [26]. On the other hand, the observation that the effects of IL-17 and TNF are stable for at least 12 h following the removal of cytokines may suggest the involvement of long-life mediators of these effects. The exact nature and level of interaction of these cytokines await further clarification.
The effects of hIL-17 and interferon-gamma (IFN-γ) in respect to Bf synthesis in fibroblasts are similar: a small increase in the synthesis of Bf in the presence of hIL-17 or IFN-γ alone but significant synergism with TNF-α[11]. The effects of IL-17 on the synthesis of Bf are ‘mirror-image’ effects of those of IL-4 and IL-13. Neither IL-4 nor IL-13 alone had an effect on the synthesis of Bf in fibroblasts, but both cytokines significantly decreased the TNF-induced synthesis of Bf [12,13]. This ‘mirror image’ action of hIL-17 on the one hand versus hIL-4 and hIL-13 on the other, and the similarity of their effects to those of IFN-γ, provide support for the classification of IL-17 as a proinflammatory Th1 cytokine [7,8].
Several lines of evidence indicate that TNF-α is the major cytokine involved in the inflammation of RA. First, it was shown that there was no joint inflammation in TNF-α knock-out mice [27]. Second, chronic inflammatory arthritis developed in transgenic mice made to over-express a mutant transmembrane form of the murine TNF-α protein [28]. Finally, in a clinical trial a soluble TNF receptor, TNFR:Fc, was shown to have beneficial effects on chronic RA [29], and blocking of TNF-α in vivo by MoAbs was shown to have a therapeutic effect on clinical severity and joint erosion in a patient with active RA [30] and in a murine model of RA [31].
There are several indications that the distinct effects of Th1 cytokines, IFN-γ and hIL-17, and those of Th2 cytokines, hIL-4 and hIL-13, are specific for Bf and not just a general phenomenon seen in acute-phase proteins. Several examples are the effects of IL-17 on synthesis of IL-6, C3 and C1 inhibitor. hIL-4 has a variable effect on the expression hIL-6, which is the major cytokine involved in acute-phase reaction [14, 32, 33]. The effects of hIL-17 on the synthesis of other proteins of the complement system, which are also considered as acute-phase proteins, are also variable. The synthesis of C3 is increased by hIL-17 but not by hIL-4 or hIL-13 [12, 13], but both hIL-13 and hIL-4 synergise with TNF-α for the synthesis of C3. The synthesis of C1 inhibitor, another acute-phase protein [34], is increased in IFN-γ-stimulated fibroblasts but is not affected by hIL-4 and hIL-13, or by hIL-17.
The alternative pathway of the complement system is one of the main routes to activate the complement system [35,36] and thus may serve as one of the conduction routes for TNF-induced inflammation. The addition of hIL-4 did not abrogate the effect of hIL-17 on TNF-stimulated fibroblasts, but rather exerted its inhibitory activity in an independent manner (Fig. 6). This finding may provide an explanation for the equivocal results of a clinical trial using hIL-4 in RA [37]. This is in contrast with promising results obtained with hIL-10, which was shown to have an effect on cytokine synthesis [38].
The data provided here support the classification of hIL-17 as a Th1 cytokine [7,8]. Furthermore, there is a clear distinction between the responses for Bf synthesis in TNF-stimulated fibroblasts and costimulation with IFN-γ or hIL-17 on the one hand and hIL-4 or hIL-13 on the other. This response can serve as a yardstick to aid classification of a cytokine as being Th1 or Th2.
In conclusion, the data presented here provide evidence for the role of IL-17 in the regulation of Bf and C3 synthesis. IL-17 is a probable amplifier of the proinflammatory effect of TNF and may have a major role in local inflammatory responses such as those present in RA. We also suggest that the nature of the response of the factor B gene can serve as a marker to classify a cytokine that has an effect on fibroblasts as being Th1 or Th2.
Acknowledgments
The authors are indebted to Pnina Koch and Yhudit Katzir for expert technical assistance and to Anji Agajangi for secretarial assistance. This study was supported by grant from German-Israel foundation (GIF, G-374-171.02/94, Y.K. and M.L.).
REFERENCES
- 1.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–6. [PubMed] [Google Scholar]
- 2.Fossiez F, Djossou O, Chomarat P, et al. T cell IL-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teunissen MBM, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JB. Interleukin-17 and interferon-γ synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 4.Attur MG, Patel RN, Abramson SB, Amin AR. IL-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997;40:1050–3. doi: 10.1002/art.1780400609. [DOI] [PubMed] [Google Scholar]
- 5.Shalom-Barak T, Quach J, Lotz M. IL-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-κB. J Biol Chem. 1998;273:27467–73. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 6.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-1β and TNF-α, by human macrophages. J Immunol. 1998;160:3513–21. [PubMed] [Google Scholar]
- 7.Chabaud M, Fossiez F, Taupin J-L, Miossec P. Enhancing effect of IL-17 on IL-1 induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–14. [PubMed] [Google Scholar]
- 8.Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–51. [PubMed] [Google Scholar]
- 9.Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human IL-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 10.Westacott CI, Sharif M. Cytokines in osteoarthritis: mediators or markers of joint destruction? Semin Arthritis Rheum. 1996;25:254–72. doi: 10.1016/s0049-0172(96)80036-9. [DOI] [PubMed] [Google Scholar]
- 11.Katz Y, Strunk RC. IL-1 and TNF: similarities and differences in stimulation of expression of alternative pathway of complement and IFN-b2/IL-6 genes in human fibroblasts. J Immunol. 1989;142:3862–7. [PubMed] [Google Scholar]
- 12.Katz Y, Strunk RC. Enhanced synthesis of factor B by tumor necrosis factor in human skin fibroblasts is decreased by IL-4. J Immunol. 1990;144:4675–80. [PubMed] [Google Scholar]
- 13.Katz Y, Stav D, Barr J, Passwell JH. IL-13 results in differential regulation of the complement proteins C3 and factor B in tumor necrosis factor (TNF)-stimulated fibroblasts. Clin Exp Immunol. 1995;101:150–6. doi: 10.1111/j.1365-2249.1995.tb02291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dechanet J, Taupin JL, Chomarat P, Rissoan MC, Moreau JF, Banchereau J, Miossec P. Interleukin-4 but not interleukin-10 inhibits the production of leukemia inhibitory factor by rheumatoid synovium and synoviocytes. Eur J Immunol. 1994;24:3222–8. doi: 10.1002/eji.1830241247. [DOI] [PubMed] [Google Scholar]
- 15.Sugiyama E, Kuroda A, Taki H, Ikemoto M, Hori T, Yamashita N, Maruyama M, Kobayashi M. IL-10 cooperates with IL-4 to suppress inflammatory cytokine production by freshly prepared adherent rheumatoid synovial cells. J Rheumatol. 1995;22:2020–6. [PubMed] [Google Scholar]
- 16.Isomaki P, Luukkainen R, Toivanen P, Punnonen J. The presence of interleukin-13 in rheumatoid synovium and its antiinflammatory effects on synovial fluid macrophages from patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1693–702. doi: 10.1002/art.1780391012. [DOI] [PubMed] [Google Scholar]
- 17.Bessis N, Boissier MC, Ferrara P, Blankenstein T, Fradelizi D, Fournier C. Attenuation of collagen-induced arthritis in mice by treatment with vector cells engineered to secrete interleukin-13. Eur J Immunol. 1996;26:2399–403. doi: 10.1002/eji.1830261020. [DOI] [PubMed] [Google Scholar]
- 18.Miossec P, Van den Berg W. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum. 1997;40:2105–15. doi: 10.1002/art.1780401203. [DOI] [PubMed] [Google Scholar]
- 19.Bessis N, Chiocchia G, Kollias G, Minty A, Fournier C, Fradelizi D, Boissier MC. Modulation of proinflammatory cytokine production in tumor necrosis factor-alpha (TNF-alpha)-transgenic mice by treatment with cells engineered to secrete interleukin-4, interleukin-10 or interleukin-13. Clin Exp Immunol. 1998;111:391–6. doi: 10.1046/j.1365-2249.1998.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz Y, Strunk RC. Synovial fibroblast-like cells synthesize seven proteins of the complement system. Arthritis Rheum. 1988;31:1365–70. doi: 10.1002/art.1780311104. [DOI] [PubMed] [Google Scholar]
- 21.DeBruijn MHL, Fey GH. Human complement component C3, cDNA coding sequence and derived primary structure. Proc Natl Acad Sci USA. 1985;82:708–12. doi: 10.1073/pnas.82.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woods DE, Markham AF, Ricker AT, Goldberger G, Colten HR. Isolation of cDNA clones for the human complement protein factor B, a class III major histocompatibility complex gene product. Proc Natl Acad Sci USA. 1982;79:5661–5. doi: 10.1073/pnas.79.18.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fishelzon Z. Complement C3: a molecular mosaic of binding sites. Mol Immunol. 1991;28:545–52. doi: 10.1016/0161-5890(91)90169-k. [DOI] [PubMed] [Google Scholar]
- 24.Erdei A, Fust G, Gergely J. The role of C3 in the immune response. Immunol Today. 1991;12:332–7. doi: 10.1016/0167-5699(91)90011-H. [DOI] [PubMed] [Google Scholar]
- 25.Fearon DT, Austen KF. Current concepts in immunology: the alternative pathway of the complement—a system for host resistance to microbial infection. N Engl J Med. 1980;303:259–63. doi: 10.1056/NEJM198007313030505. [DOI] [PubMed] [Google Scholar]
- 26.Katz Y, Cole FS, Strunk RC. Synergism between gamma interferon and lipopolysaccharide for synthesis of factor B, but not C2, in human fibroblasts. J Exp Med. 1988;167:1–14. doi: 10.1084/jem.167.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feltkamp TEW, Aarden LA, Lucas CJ, Verweij CL, De Vries RRP. Genetic risk factors for autoimmune diseases. Immunol Today. 1999;20:10–12. doi: 10.1016/s0167-5699(98)01347-4. [DOI] [PubMed] [Google Scholar]
- 28.Alexopoulou L, Pasparakis M, Kollias G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol. 1997;27:2588–92. doi: 10.1002/eji.1830271018. [DOI] [PubMed] [Google Scholar]
- 29.Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of Etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 30.Kavanaugh AF. Anti-tumor necrosis factor alpha monoclonal antibody therapy for rheumatoid arthritis. Rheum Dis Clin North Am. 1998;24:593–614. doi: 10.1016/s0889-857x(05)70028-4. [DOI] [PubMed] [Google Scholar]
- 31.Camussi G, Lupia E. The future role of anti-tumour necrosis factor (TNF) products in the treatment of rheumatoid arthritis. Drugs. 1998;55:613–20. doi: 10.2165/00003495-199855050-00001. [DOI] [PubMed] [Google Scholar]
- 32.Nakazato Y, Hayashida T, Kanno Y, Okada H, Suzuki H, Saruta T. Interleukin (IL-1)-1 and IL-4 synergistically stimulate NF-IL-6 activity and IL-6 production in human mesangial cells. Kidney Int. 1998;54:71–79. doi: 10.1046/j.1523-1755.1998.00967.x. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki H, Sugiyama E, Tunru IS, Yamashita N, Hamazaki T, Kobayashi M. Suppressive effect of interleukin-4 (IL-4) on IL-6 production by adherent rheumatoid synovial cells. Clin Immunol Immunopathol. 1993;66:67–72. doi: 10.1006/clin.1993.1009. [DOI] [PubMed] [Google Scholar]
- 34.Gabay C, Kushner I. Acute phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 35.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–47. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 36.Carroll MC. The lupus paradox. Nature Genet. 1998;19:3–4. doi: 10.1038/ng0598-3. [DOI] [PubMed] [Google Scholar]
- 37.Van den Bosch F, Russell A, Chair MB, et al. rHIL-4 in subjects with active rheumatoid arthritis: a phase I dose escalating safety study. Arthritis Rheum. 1998;41(Suppl.):S56. [Google Scholar]
- 38.Maini RN, Paulus H, Breedveld FC, et al. rHIL-10 in subjects with active rheumatoid arthritis: a phase I and cytokine response study. Arthritis Rheum. 1997;40(Suppl.):S224. [Google Scholar]