

Abstract
Homeostasis between indigenous intestinal flora and host response may be broken in inflammatory bowel disease. The present study explores whether repeated oral administration of intestinal flora antigens can protect mice against dextran sodium sulphate (DSS)-induced colitis. Sonicates of Gram-positive, Gram-negative, or anaerobic resident bacteria isolated from mouse intestinal flora were fed to BALB/c mice by gastric gavage, with or without cholera toxin. After four weekly doses of 1 mg of these antigen preparations (or of PBS as control), DSS colitis was induced. One week later colitis was evaluated by clinical scores and histology. Mice fed a pool of the three sonicates had decreased inflammation scores (5 (1–14); median (range)) compared with PBS-fed control animals (15 (7–19); P <0·05). Decreased inflammation was observed in mice fed anaerobic bacteria antigens (7 (6–11); P <0·05 versus control), but not in mice fed a pool of Gram-positive and -negative sonicates (16 (12–16)). Inflammation scores of mice fed antigens with cholera toxin were similar to those of PBS-fed control animals. DSS-induced colitis can be suppressed by oral administration of normal intestinal flora antigens containing anaerobes.
Keywords: dextran sulphate, colitis, intestinal flora
INTRODUCTION
The pathogenesis of inflammatory bowel disease (IBD) involves abnormal interactions between the host (genetic susceptibility, immune system dysregulation) and its environment [1]. There is increasing evidence that IBD results from an abnormal immune response to normal intestinal flora, with a break of tolerance towards non-pathogenic bacteria [2]. This hypothesis is supported by the observation that Crohn’s disease (CD) improves following faecal stream diversion and by the fact that infusion of intestinal contents in surgically excluded ileum induces Crohn’s lesions [3,4]. In further support of this hypothesis, administration of antibiotics, IL-10, or antibodies to tumour necrosis factor-alpha (TNF-α) was shown to improve intestinal inflammation [5–7]. However, the search for specific pathogenic microorganisms in IBD has remained so far non-conclusive [8–11]. It seems more likely that a large array of bacteria can be involved in the induction or in the maintenance of the chronic inflammation observed in IBD, the role of specific pathogenic bacteria being one of the many possible triggers of the pathological inflammatory reaction [1,12].
Results obtained in animal models further support that tolerance to autologous flora is broken in IBD. This was shown in a murine model of trinitrobenzene sulphonic acid (TNBS) hapten-induced chronic intestinal inflammation, where tolerance could be restored by treatment with IL-10 or antibodies to IL-12 [13]. Moreover, IL-2 and IL-10 knock-out mice develop colitis spontaneously upon colonization with normal intestinal flora, but inflammation does not occur if the animals are kept in germ-free conditions [14–16]. Similarly, colitis is not detected in HLA-B27 transgenic rats kept under germ-free conditions [17]. In this model, colonization with Bacteroides spp. was shown to induce intestinal inflammation [18]. Severe colitis can be induced in genetically normal mice by oral administration of dextran sodium sulphate (DSS) [19,20]. Exposed mice develop an acute colitis with bloody diarrhoea, shortening of the colon, weight loss, and neutrophilic infiltration of the left colon within 1 week after administration of 5% DSS in drinking water. In this model, concentrations of Bacteroides and Clostridium species are increased in the acute phase of the disease [19]. It has been suggested that after induction of mucosal damage by DSS, intestinal bacteria penetrate the injured mucosa and perpetuate mucosal inflammation [20].
Immune and inflammatory responses in the intestine can be modulated in many ways. To avoid chronic immune activation leading to inflammation, in particular in the gut where bacteria and food antigens are in close proximity to a substantial proportion of the body lymphocytes, the immune system must develop unresponsiveness, tolerance, to such antigenic stimuli [21]. Oral and systemic tolerance are induced by repeated oral administration of antigens, in absence of adjuvant, and can suppress autoimmune diseases [22]. Induction of oral tolerance has been previously reported as an approach to improve colitis in experimental mouse models. Colitis induced by the contact-sensitizing agent TNBS has been prevented by feeding TNBS or TNP-conjugated colonic proteins [23,24]. Cholera toxin, in contrast, has been used as a potent mucosal adjuvant for oral immunization and reported to break peripheral tolerance to co-administered antigens [25,26]. Oral immunization with bacterial antigens and cholera toxin can induce antigen-specific T cells and secretory IgA [27,28] which can prevent contact of bacteria with the intestinal mucosa [29]. Recent evidence suggests that the qualitative composition of the intestinal flora also plays a role in preventing intestinal inflammation. Feeding lactobacilli or nutrients able to modify the intestinal flora composition can prevent the development of inflammatory response in the gut [30].
The present study tests the hypothesis that repetitive feeding with normal intestinal flora antigens can prevent the development of DSS colitis. We observed that mice fed anaerobic bacteria antigens had significantly reduced colitis scores following DSS compared with control mice and that this effect was lost if the antigen preparations were co-administered with cholera toxin (CT).
MATERIALS and METHODS
Preparation of bacterial sonicates
Isolation of facultative and strict anaerobes from the caecal mucosa of BALB/c mice was performed on blood agar, Mac Conkey, vancomycin-nalidixic acid, brilliant green, phenyl-ethyl-alcohol agar, neomycin gelose and Campylobacter media. Bacteria were collected from the cultures and identified. Enterococcus, Staphylococcus, Lactococcus, and Bacillus species were pooled and sonicated under sterile conditions in PBS to yield a Gram-positive bacterial antigen preparation. A Gram-negative preparation was generated by sonication of Escherichia coli and a preparation of anaerobes (A) was obtained by sonication of Bacteroides, Lactobacilli and Clostridia. Further information on the bacteria used in this study is provided in Table 1. Pools of these antigen preparations were generated by mixing equal amounts of the individual preparations (GG =Gram-positive and Gram-negative bacteria, GGA =Gram-positive, Gram-negative and anaerobes).
Table 1.
Bacteria species isolated from BALB/c mice colonic flora
| Gram-positive bacteria | Gram-negative bacteria | Anaerobic bacteria |
|---|---|---|
| Enterococcus durans | Escherichia coli | Bacteriodes ovatus† |
| Staphylococcus intermedius* | Lactobacilli spp.‡ | |
| Lactococcus lactis | Clostridia spp.‡ | |
| Bacillus spp.‡ |
Staphylococcus aureus family.
Bacteriodes fragilis family.
Strain not determined.
Immunization schemes
Eight-week-old BALB/c mice (Velaz, Prague, Czech Republic) were used in this study. Mice were dosed four times by gastric gavage on a weekly schedule with 100 μl of sonicate in PBS, or 100 μl of PBS as a control. Concentrations of sonicates were adjusted so that each mouse received 1 mg of total protein in 100 μl of each oral dose. In addition to the various sonicates and sonicate pools described above, one group of mice was fed GGA supplemented with 10 μg of CT. Two weeks after the last oral dose of sonicate, DSS (5% in distilled sterile water) was administered in drinking water for 7 days. A proportion of the animals in each group were fed with water.
Evaluation of colitis
Immediately after the end of DSS feeding, mice were killed. The presence of rectal prolapse, rectal or colonic bleeding and weight loss was recorded. A symptom score (range 0–4) was constructed attributing one point to each of the following events: rectal prolapse, rectal bleeding, colonic bleeding and death. The length of the colon was measured and sigmoid samples were obtained for histology. Two pathologists, blinded to the treatment schedule of the animals, reviewed the slides. The severity of the colitis was scored, attributing between zero and three points to each of the following parameters: (i) polymorphonuclear infiltrate; (ii) mononuclear infiltrate; (iii) oedema; (iv) erosions and ulcerations; (v) crypt abscess; (vi) crypt destruction; and (vii) distribution of the inflammation (mucosa =1, mucosa and submucosa =2, transmural inflammation =3). Based on the total score (range from 0 to 21), the inflammation was graded as mild (1–5 points), moderate (6–10 points) or severe (>10 points).
RESULTS
Clinical signs of colitis
A significant decrease in median symptom score was observed in the mice fed with the pool of Gram-positive, Gram-negative and anaerobic bacteria antigens, compared with PBS-fed control mice (Fig. 1a). When isolated sonicates were given to mice, colitis was reduced only in mice fed with anaerobic bacteria antigens, but not in animals fed a pool of Gram-positive and Gram-negative bacteria antigens. Animals given pooled antigens of Gram-positive, Gram-negative and anaerobic bacteria with CT had symptom scores that were comparable to PBS-fed controls, suggesting that addition of CT abolished the protective affect of antigen feeding.
Fig. 1.
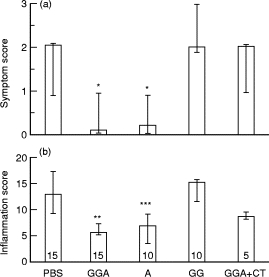
(a) Clinical signs of dextran sodium sulphate (DSS)-induced colitis following oral feeding with bacteria antigen preparations. PBS, Control mice fed PBS instead of bacteria antigens; GGA, mice fed a pool of Gram-positive, Gram-negative, and anaerobic bacteria antigens; A, mice fed anaerobic bacteria antigens; GG, mice fed a pool of Gram-positive and Gram-negative bacteria antigens; CT, cholera toxin. Median symptom scores were calculated attributing one point to each of the following events: rectal prolapse, rectal bleeding, colonic bleeding, mortality, and divided by the number of mice in each treatment group. Results are shown as median symptom scores with 95% confidence interval (CI). *P <0·05 versus PBS, GG or GGA +CT. (b) Inflammation scores in sigmoid biopsies following DSS administration to antigen-fed mice. Inflammation was maximal in control mice (PBS). Administration of GGA and A decreased the severity of DSS-induced inflammation (**P <0·05 versus PBS, GG or GGA +CT; ***P <0·05 versus PBS or GG). Co-administration of CT reversed the protective effect of GGA. Results are shown as medians with 95% CI. Numbers in bars indicate number of animals per group.
Grading of colonic inflammation
In parallel with improved symptom scores, administration of GGA decreased the severity of DSS-induced inflammation scores compared with mice treated with PBS prior to the induction of colitis (Fig. 1b). While severe inflammation, crypt destruction, and ulceration of the mucosa were observed in the PBS-fed group (Fig. 2a,b), minimal inflammation was observed in the GGA-fed group (Fig. 2c,d). Compared with PBS controls, median inflammation scores were also reduced by the administration of A but not by the administration of GG (Fig. 1b). In addition, protection was also associated with decreased splenocyte production of interferon-gamma (IFN-γ) (data not shown). Mice exposed to GGA +CT had an inflammation score that was not statistically different from PBS controls. Mice fed with water instead of DSS had no colonic inflammation, regardless of the antigen they had previously received, indicating that the administration of the bacterial sonicates had no intrinsic harmful effect on the colonic mucosa (data not shown).
Fig. 2.
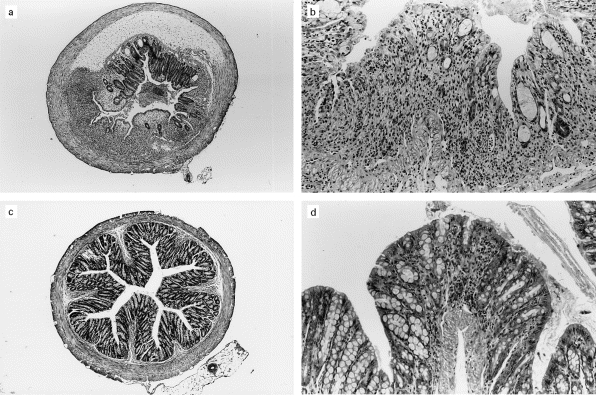
(a,b) Severe inflammation observed in a mouse fed PBS before induction of dextran sodium sulphate (DSS) colitis. (a) Low-power view of a transversal cut section through the sigmoid colon showing oedema of the submucosa and extensive inflammation and ulceration of the mucosal layer. (b) High-power view of the sigmoid mucosa showing severe colitis with subtotal destruction of the crypts and massive inflammatory infiltrate in the lamina propria. (c,d) Minimal inflammation in the colon of a mouse fed Gram-positive, Gram-negative, and anaerobic bacteria antigens (GGA) before DSS colitis induction. (c) Low-power view of a transversal cut section through the sigmoid colon showing no visible alteration of the mucosa. (d) Focal inflammation of the sigmoid mucosa without ulceration or destruction of the crypts at high-power view.
DISCUSSION
In the present study we explore the role of oral administration of normal intestinal flora in the development of DSS-induced colitis. First, we show that oral administration of normal intestinal flora antigens induces resistance to DSS-induced colitis in normal mice. Second, we show that this resistance against of DSS colitis is critically dependent on feeding of anaerobic bacteria antigen preparations. Third, we show that this resistance is abrogated when the antigen preparation is given with CT. This last observation suggests that the decreased inflammation did not result from the development of an immune response to the bacterial antigen preparations, but the mechanisms by which the protective effect occurred remain unclear. One possibility is that oral administration of antigens induced immune tolerance, with decreased immune response to the intestinal flora upon DSS challenge. Administration of CT has been shown to increase gut permeability [31], which in our system could facilitate bacterial antigenic exposure, increasing the severity of DSS colitis. Alternatively, administration of anaerobic bacteria antigens may have altered the intestinal flora homeostasis in a way that rendered mice less susceptible to DSS challenge.
The mechanism by which DSS induces colitis is not well defined, but seems to result from an alteration of colonic epithelial cells, and not from an alteration of T nor B cell responses, because DSS colitis develops in immunodeficient mice [32,33]. This suggests that DSS modifies either the nature or the sampling of luminal antigens [34]. Antibiotics and live bacteria (probiotics) have been used to prevent and ameliorate experimental colitis by changing the equilibrium of commensal bacteria in the intestinal flora [30,35]. In our study, bacteria were washed twice in PBS before sonication, making very unlikely the possibility that the protective effect was due to antibiotics contaminating the final antigen preparation. It is also improbable that live bacteria contained in the sonicates would have modified the intestinal flora of the mice. Sonicates were frozen and thawed before each dosing, a procedure that would have impaired the survival of any bacteria. We cannot rule out however that the administration of bacterial components changed the equilibrium of the intestinal flora, either by providing excess free bacterial antigens to the mucosal immune system, or by competing with live bacteria for intestinal epithelial binding sites.
Tolerance induction has been demonstrated to be an efficacious mechanism of protection in hapten-induced colitis [23,24]. In that model, tolerance induced either to the hapten in solution or to haptenized colonic proteins abrogates hapten-induced colitis, but the antigens used for tolerance induction are unknown to the immune system of the animals before the experiment. In that model, tolerance is thus induced de novo to a foreign antigen. In our model in contrast, the mice should already be tolerant to their endogenous flora, as the antigens used for oral administration were derived from bacteria already present in the lumen. Indeed, in mice physiological tolerance to the intestinal flora establishes during the first weeks of life [36,37]. If immune tolerance participates in the protection observed in this study, then our results suggest that the physiological tolerance to intestinal bacteria can be raised or modified to prevent the development of DSS-related inflammatory response. Oral tolerance can be induced either by clonal deletion or anergy of Th1 cells, or by induction of suppressive Th2 and Th3 clones secreting transforming growth factor-beta (TGF-β), IL-4, and IL-10 [38–40]. In our study, CT abrogated the protective effect of antigenic anaerobic feeding, which would argue against deletion of antigen-specific T cells. Further studies in this model and in T lymphocyte-driven models of colitis will be needed to determine if tolerance to intestinal bacteria antigens takes place in this model and the precise mechanism by which it is induced.
Anaerobic bacteria, especially Bacteriodes spp., have been implicated in the pathogenesis of IBD, and recent evidence suggests that anaerobic bacteria play a role in the initiation of DSS colitis in mice [33]. Our results are compatible with these studies in showing that anaerobic antigens play a crucial role in inducing protection. Whether oral administration of anaerobic antigens led to induction of oral tolerance or to increased resistance of the mucosal barrier remains to be determined. The oral antigen feeding technique described here may represent a novel approach to identify the bacteria and their components involved in the pathogenesis of IBD. Further studies will be required to determine if other intestinal bacteria antigens are able to confer this type of protection.
In conclusion, our data extend the evidence that non-pathogenic luminal bacteria participate in the pathogenesis of chronic intestinal inflammation. Investigations into the mechanisms participating in this protection are in progress. Administration of intestinal flora components may be relevant to the development of novel therapeutic strategies in the management of IBD patients.
Acknowledgments
This work was supported in part by grant nos A7020716 and A7020808 (Grant Agency of Sciences of the Czech Republic), no 310/96/1366, 306/98/0433, and 311/97/0784 (Grant Agency of the Czech Republic), nos 3761-3 and 5051-3 (Ministry of Health of the Czech Republic), and no. VS96149 (Ministry of Education, Youth and Sports). All grants awarded to H.T.-H. Additional support from NIH grant R29-DK53706-01, and from Glaxo Welcome Institute for Digestive Health (P.M.).
REFERENCES
- 1.Sartor RB. Current concepts of the etiology and pathogenesis of ulcerative colitis and Crohn’s disease. Gastroenterol Clinics North Am. 1995;24:475–507. [PubMed] [Google Scholar]
- 2.Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Büschenfelde K-H. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD) Clin Exp Immunol. 1995;102:448–55. doi: 10.1111/j.1365-2249.1995.tb03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee E. Split ileostomy in the treatment of Crohn’s disease of the colon. Ann R Coll Surg Eng. 1975;56:94–102. [PMC free article] [PubMed] [Google Scholar]
- 4.D’Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–7. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 5.Prantera C, Zannoni F, Scribano ML, Berto E, Andreoli A, Kohn A, Luzi C. An antibiotic regimen for the treatment of active Crohn’s disease: a randomized, controlled clinical trial of metronidazole plus ciprofloxacin. Am J Gastroenterol. 1996;91:328–32. [PubMed] [Google Scholar]
- 6.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. NEJM. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 7.van Deventer SJ, Elson CO, Fedorak RN. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology. 1997;113:383–9. doi: 10.1053/gast.1997.v113.pm9247454. [DOI] [PubMed] [Google Scholar]
- 8.Thayer WR, Coutu JA, Chiodini RJ, Van Kruiningen HJ, Merkal RS. Possible role of Mycobacteria in inflammatory bowel disease. Mycobacterial antibodies in Crohn’s disease. Dig Dis Sci. 1984;29:1080–5. doi: 10.1007/BF01317079. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi K, Brown WR, Brennan PJ, Blaser MJ. Serum antibodies to mycobacterial antigens in active Crohn’s disease. Gastroenterology. 1988;94:1404–11. doi: 10.1016/0016-5085(88)90679-8. [DOI] [PubMed] [Google Scholar]
- 10.Brunello F, Pera A, Martini S, et al. Antibodies to Mycobacterium paratuberculosis in patients with Crohn’s disease. Dig Dis Sci. 1991;36:1741–5. doi: 10.1007/BF01296619. [DOI] [PubMed] [Google Scholar]
- 11.Rowbotham DS, Howdle PD, Trejdosiewicz LK. Peripheral cell mediated immune response to mycobacterial antigens in inflammatory bowel disease. Clin Exp Immunol. 1995;102:456–61. doi: 10.1111/j.1365-2249.1995.tb03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Targan SR, Murphy LK. Clarifying the causes of Crohn’s. Nat Med. 1995;1:1241–3. doi: 10.1038/nm1295-1241. [DOI] [PubMed] [Google Scholar]
- 13.Duchmann R, Schmitt E, Knolle P, Meyer zum Büschenfelde K-H, Neurath M. Tolerance towards resident intestinal flora in mice is abrogated in experimental colitis and restored by treatment with interleukin-10 or antibodies to interleukin-12. Eur J Immunol. 1996;26:934–8. doi: 10.1002/eji.1830260432. [DOI] [PubMed] [Google Scholar]
- 14.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–61. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 15.Kühn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10 deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 16.Strober W, Ehrhardt RO. Chronic intestinal inflammation: an unexpected outcome in cytokine or T cell receptor mutant mice. Cell. 1993;78:203–95. doi: 10.1016/0092-8674(93)80062-j. [DOI] [PubMed] [Google Scholar]
- 17.Rath HC, Herfarth HH, Ikeda JS, et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human B2 microglobulin transgenic rats. J Clin Invest. 1996;98:945–53. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sartor RB. The role of endogenous luminal bacteria and bacterial products in the pathogenesis of experimental enterocolitis and systemic inflammation. In: Kagnoff MF, Kiyono H, editors. Essentials of mucosal immunology. New York: Academic Press; 1996. pp. 307–20. [Google Scholar]
- 19.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 20.Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–67. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 21.Cebra JJ, Jiang H-Q, Sterzl J, Tlaskalova-Hogenova H. The role of mucosal microbiota in the development and maintenance of the mucosal immune system. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, editors. Mucosal immunology. New York: Academic Press; 1998. pp. 267–80. [Google Scholar]
- 22.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–40. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 23.Elson CO, Beagley KW, Sharmanov AT, et al. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. J Immunol. 1996;157:2174–85. [PubMed] [Google Scholar]
- 24.Neurath MF, Fuss I, Kelsall BL, Presky DH, Waegell W, Strober W. Experimental granulomatous colitis in mice is abrogated by induction of TGF-beta-mediated oral tolerance. J Exp Med. 1996;183:2605–16. doi: 10.1084/jem.183.6.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierre P, Denis O, Bazin H, Mbongolo Mbella E, Vaerman JP. Modulation of oral tolerance to ovalbumin by cholera toxin and its B subunit. Eur J Immunol. 1992;22:3179–82. doi: 10.1002/eji.1830221223. [DOI] [PubMed] [Google Scholar]
- 26.Elson CO, Ealding W. Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J Immunol. 1984;133:2892–7. [PubMed] [Google Scholar]
- 27.Lycke N. The mechanisms of cholera toxin adjuvanticity. Res Immunol. 1997;148:504–20. doi: 10.1016/s0923-2494(98)80144-2. [DOI] [PubMed] [Google Scholar]
- 28.Hornquist E, Lycke N. Cholera toxin adjuvant greatly promotes antigen priming of T cells. Eur J Immunol. 1993;23:2136–43. doi: 10.1002/eji.1830230914. [DOI] [PubMed] [Google Scholar]
- 29.Michetti P, Porta N, Mahan MJ, Slauch JM, Mekelalanos JJ, Blum AL, Kraehenbuhl JP, Neutra MR. Monoclonal immunoglobulin A prevents adherence and invasion of polarized epithelial cell monolayers by Salmonella typhimurium. Gastroenterology. 1994;107:915–23. doi: 10.1016/0016-5085(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 30.Madsen KL, Foyle JS, Jewell LD, Tavernini MM, Fedorak RN. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology. 1999;116:1107–14. doi: 10.1016/s0016-5085(99)70013-2. [DOI] [PubMed] [Google Scholar]
- 31.Lycke N, Karlsson U, Sjolander A, Magnusson KE. The adjuvant action of cholera toxin is associated with an increased intestinal permeability for luminal antigens. Scand J Immunol. 1991;33:691–8. doi: 10.1111/j.1365-3083.1991.tb02542.x. [DOI] [PubMed] [Google Scholar]
- 32.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sodium sulphate-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 33.Axelsson LG, Landstrom E, Goldschmidt TJ, Gronberg A, Bylund-Fellenius AC. Dextran sulfate sodium (DSS) induced experimental colitis in immunodeficient mice: effects in CD4(+)-cell depleted, athymic and NK-cell depleted SCID mice. Inflamm Res. 1996;45:181–91. doi: 10.1007/BF02285159. [DOI] [PubMed] [Google Scholar]
- 34.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 35.Rath HC, Schultz M, Dieleman LA, Li F, Kolbl H, Falk W, Scholmerich J, Sartor RB. Selective vs. broad spectrum antibiotics in the prevention and treatment of experimental colitis in two rodent models. Gastroenterology. 1998;114:A1067. [Google Scholar]
- 36.Gorbach SL, Nahas L, Lerner PI, Weinstein L. Studies of intestinal microflora. I. Effects of diet, age, and periodic sampling on numbers of fecal microorganisms in man. Gastroenterology. 1967;53:845–55. [PubMed] [Google Scholar]
- 37.Shroff KE, Meslin K, Cebra JJ. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–13. doi: 10.1128/iai.63.10.3904-3913.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and natural recovery from experimental autoimmune encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor beta, interleukin 4, and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355–64. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci USA. 1994;91:6688–92. doi: 10.1073/pnas.91.14.6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–43. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]