

Abstract
Epidemiological and experimental studies suggest that diesel exhaust particles (DEP) may play an active role in the increased respiratory mortality and morbidity. We have shown that DEP augmented the production of inflammatory cytokines by human airway epithelial cells in vitro. ICAM-1 has been shown to play an important role in the local accumulation of inflammatory cells. We studied the effect of DEP on ICAM-1 gene expression and surface expression in human bronchial epithelial cell line BEAS-2B. DEP (5–50 μg/ml) showed a stimulatory effect on ICAM-1 mRNA levels as evaluated by reverse transcription-polymerase chain reaction (RT-PCR). Flow cytometric analysis demonstrated an increased ICAM-1 expression on the epithelial cell surfaces. The soluble form of ICAM-1 molecules was also increased by the stimulation of DEP. In vitro neutrophil attachment onto DEP-stimulated epithelial cells was augmented, which was partially blocked by anti-ICAM-1 neutralizing antibody. Finally, these events were significantly inhibited by pretreatment with anti-oxidants pyrrolidine dithiocarbamate and N-acetyl cysteine, and p38 mitogen activated protein kinase (MAPK) inhibitor SB203580. These findings suggested that DEP induced up-regulation of ICAM-1 gene, and this process might be largely dependent on oxidant-mediated NF-κB activation and p38-MAPK pathways.
Keywords: diesel exhaust particle, ICAM-1, airway epithelium
INTRODUCTION
Suspended particulate matters (PM), especially PM2.5 (particles collected with an efficiency of 50% for those with a diameter of 2·5 μ m) have attracted special attention, since they precipitate throughout the whole lung, including the periphery, to elicit disease [1–5]. Diesel exhaust particles (DEP), the major component of PM2.5 in urban areas, have been demonstrated to have a stimulatory effect on cytokine production in vitro and in vivo[6–9]. Acute exposure of DE produced airway inflammatory responses with cell infiltration and increased mediator production in human healthy volunteers [10]. Recently, we and others [7–9] demonstrated that DEP directly stimulated human airway epithelial cells to release cytokines and mediators such as IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF) and soluble form of ICAM-1. Increased production of such inflammatory mediators was via, at least in part, the activation of transcription factors such as NF-κB and/or AP-1 [11,12]. ICAM-1, one of adhesion molecules belonging to the immunoglobulin superfamily, has been considered to play a critical role in the attachment and migration of immune and inflammatory cells to the airway mucosa [13,14]. In the present study, we investigated the effects of DEP on ICAM-1 expression in human airway epithelial cells in vitro.
MATERIALS AND METHODS
Preparation of DEP
The engine used for preparation of DEP was 4JB1 type (Isuzu Automobile Co., Tokyo, Japan), light-duty (2740 cm3), four-cylinder diesel engine. The engine was connected to an EDYC dynamometer (Meiden-Sya, Tokyo, Japan), and was operated using a standard diesel fuel with the speeds of 2000 rev/min under the load of 6 torque (kg/m). The exhaust was introduced into a stainless steel dilution tunnel (300 mm diameter × 8400 mm). The DEPs were collected on glassfibre filter (203 × 254 mm) in a constant-volume sampler system equipped to the end of the dilution tunnel [6]. The temperature at the sampling point was below 50°C. The samples were harvested by gently scooping the accumulated DEP powders. There was no contamination of glass fibres when assessed by scanning electron microscopy (data not shown).
Culture of a bronchial epithelial cell line BEAS-2B
A human bronchial epithelial cell line BEAS-2B [6,15] (kind gift of Drs J. F. Lechner and C. C. Harris, National Cancer Institute, Bethesda, MD) was plated onto collagen-coated 24-well flat-bottomed tissue culture plates (Koken, Tokyo, Japan) at a density of 5 × 104 cells/well in hormonally defined Ham's F12 medium (HD-F12) as reported [6,12]. HD-F12 contained 1% penicillin–streptomycin, 5 μg/ml insulin (Gibco, Grand Island, NY), 5 μg/ml transferrin (Gibco), 25 ng/ml epidermal growth factor (Collaborative Research Corp., Lexington, MA), 15 μg/ml endothelial cell growth supplement (Collaborative Research Corp.), 2 × 10−10 m triiodothyronin (Gibco), and 10–7 m hydrocortisone (Gibco). The cells were incubated in a humidified atmosphere at 37°C and 5% CO2. The medium was changed at day 1 and subsequently every 2 days.
Reverse transcription-polymerase chain reaction for ICAM-1 mRNA in airway epithelial cells
To assess the ICAM-1 mRNA levels in human airway epithelial cells, a semiquantitative assay utilizing reverse transcription-polymerase chain reaction (RT-PCR) was performed as reported previously [16,17]. Total RNA was isolated from epithelial cell samples by the guanidinium thiocyanate-phenol-chloroform extraction method as described by Chomczynski & Sacchi [18]. Briefly, after the counting and assessment of viability, the cells (5·0 × 105 viable cells) were lysed in solution D (4 m guanidinium thiocyanate, 25 mm sodium citrate, pH 7; 0·5% sarcosyl, 0·1 m 2-mercaptoethanol). The mixture was extracted with chloroform-phenol (1:1, v/v) and chloroform-isoamyl alcohol (24:1, v/v). The total RNA was precipitated in isopropanol and the pellet was washed twice with 70% ethanol, dried, and dissolved in diethylpyrocarbonate-treated water. Extracted RNA was reverse transcribed to cDNA by using Takara RNA-PCR kit according to the manufacturer's recommendation. Briefly, total RNA (8 μg), random hexadeoxynucleotides, and avian myeloblastosis virus reverse transcriptase in total 20 μl mixture were used for cDNA synthesis. The specific primer pairs used for PCR amplification are listed below:
ICAM-1
(5′ primer) 5′-TATGGCAACGACTCCTTCT-3′
(3′ primer) 5′-CATTCAGCGTCACCTTGG-3′
β-actin
(5′ primer) 5′-ATCTGGCACCACACCTTCTACAATGAGCTGCG-3′
(3′ primer) 5′-CGTCATACTCCTGCTTGCTGATCCACATCTGC-3′ (Clontech, Palo Alto, CA)
The reaction mixture contained 10 mm Tris–HCl (pH 8·3 at 25°C), 50 mm KCl, 1·5 mm MgCl2, 1 mg/ml gelatin, 0·4 μm of each primer, 0·25 m diethyl-p-nitrophenyl monothiophosphate (dNTP), cDNA (2 μl of PCR products), and 1 U of Taq polymerase (Perkin Elmer Cetus, Norwalk, CT) in 25 μl. Amplification was performed for allotted cycles of denaturation (94°C, 2 min), annealing (60°C, 30 s) and extension (72°C, 1·5 min) using a thermal cycler (Progene; Techne, Cambridge, MA). The PCR cycle was determined by preliminary experiments showing a linear relationship between PCR cycles and intensity of signals on ethidium bromide-stained agarose gels. For semiquantitative evaluation of ICAM-1 and β-actin mRNAs, 30 and 25 cycles were chosen, respectively. The PCR product was separated using agarose gel (1·0%) electrophoresis, and the intensity of ethidium bromide fluorescence was evaluated by NIH Image version 1.61.
Flow cytometry for the detection of ICAM-1 on the epithelial cells
The surface expression of ICAM-1 on BEAS-2B cells was evaluated by a flow cytometry analysis using a FACScan system (Becton Dickinson Immunocytometry Systems, San Jose, CA) as reported previously [19]. Briefly, the cells were treated with DEP for 18 h, and washed twice with PBS. After trypsinization, suspended BEAS-2B cells (105 cells) were incubated in 200 μl of PBS containing 10 μl of rat anti-human ICAM-1 antibody (M7063; Dako, Carpinteria, CA) or control IgG1 for 15 min. After washing, the cells were incubated in 200 μl of PBS containing the FITC-conjugated anti-rat IgG1 MoAb (10084D; PharMingen, San Diego, CA) as the second antibody for 15 min. After washing, the cells were then suspended in 500 μl of PBS and analysed.
Assay for soluble ICAM-1
Specific immunoreactivity for soluble ICAM-1 (sICAM-1) in culture supernatants was measured by ELISA kits (R&D Systems, Inc., Minneapolis, MN). Each sample was assayed in duplicates as recommended by the manufacturer.
Effect of DEP on neutrophil attachment onto BEAS-2B
The BEAS-2B cells were cultured onto collagen-coated 96-chamber flat-bottomed culture plates until confluence. Neutrophils were isolated from peripheral blood of healthy volunteers by the methods of density gradient Percoll centrifugation at the purity of not less than 97% as described elsewhere [20]. The epithelial cells were treated with DEP (5–50 μg/ml) for 18 h. After the epithelial cells were rinsed twice, purified neutrophils (100 μl of 2·0–105/ml) were applied onto culture plates. The plate was incubated for 30 min in a humidified atmosphere at 37°C and 5% CO2, then rinsed twice, and 0·01% Triton X was added for cell lysis. Activity of myeloperoxidase (MPO) was measured by spectrophotometric assay based on MPO-catalysed oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) by H2O2 reported previously [20]. Preliminary experiments showed that this MPO method showed a good positive correlation with the results of direct counting of attached neutrophils on epithelial cells (r = 0·976, P < 0·001, Pearson correlation test) [20].
Effects of the drugs acting on signal transduction pathways
To study the mechanisms of ICAM-1 gene expression in DEP-stimulated BEAS-2B cells, effects of several agents were studied. These included a protein kinase C inhibitor staurosporin (Sigma Chemical Co., St Louis, MO), anti-oxidant drugs with an inhibitory effect on NF-κB activation, pyrrolidine dithiocarbamate (PDTC) [21] and N-acetyl cysteine (NAC) [22] (Sigma), a p38-MAPK inhibitor SB203580 [23] (a kind gift by Smithkline Beecham), and a MEK inhibitor PD98059 [24] (Sigma). Various concentrations of each drug were added to the cells 1 h before stimulation with DEP, and the effects of the drugs on ICAM-1 mRNA expression and protein production were studied.
Statistical analysis
The results were analysed by Student's t-test for comparison between the two groups, and by non-parametric equivalents of analysis of variance (anova) for multiple comparison as reported [6].
RESULTS
Effects of DEP on ICAM-1 mRNA levels in BEAS-2B cells
To evaluate the effects of DEP on ICAM-1 mRNA levels, a semiquantitative RT-PCR study was performed. As shown in Fig. 1a,b, DEP (50 μg/ml) showed a stimulatory effect on ICAM-1 mRNA levels in BEAS-2B cells as corrected by β-actin transcripts. The message peaked at 12 h after stimulation with DEP, then returned to basal levels. DEP (5–50 μg/ml) showed a dose-dependent stimulatory effect on ICAM-1 mRNA levels when evaluated 12 h after DEP treatment (Fig. 1c,d). DEP at 100 μg/ml failed to increase the ICAM-1 mRNA levels, although trypan blue dye exclusion technique did not show significant cytotoxicity. Preincubation of BEAS-2B cells with PDTC and NAC significantly attenuated DEP (50 μg/ml)-induced increase in ICAM-1 mRNA levels (Fig. 2a,b). SB203580, but not staurosporin or PD98059, showed an inhibitory effect on DEP-induced ICAM-1 expression (Fig. 3a,b). The baseline expression of ICAM-1 was not significantly affected by these agents (data not shown).
Fig. 1.
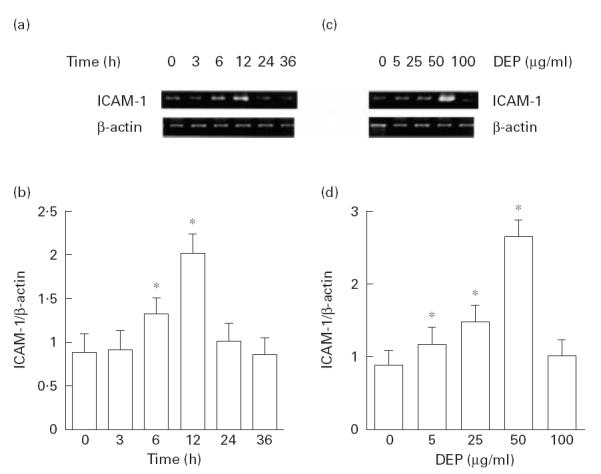
Diesel exhaust particles (DEP) induced an increased expression of ICAM-1 mRNA as assessed by semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR). Addition of DEP (50 μg/ml) to BEAS-2B showed increased levels of ICAM-1 mRNA as corrected by β-actin transcripts that peaked after 12 h. (a) A representative result. (b) Results of three independent experiments. *P < 0·05 compared with naive cells, anova. DEP showed a dose-dependent stimulatory effect on ICAM-1 mRNA levels when studied after 12 h. (c) A representative result. (d) Results from three independent experiments. *P < 0·05, anova.
Fig. 2.
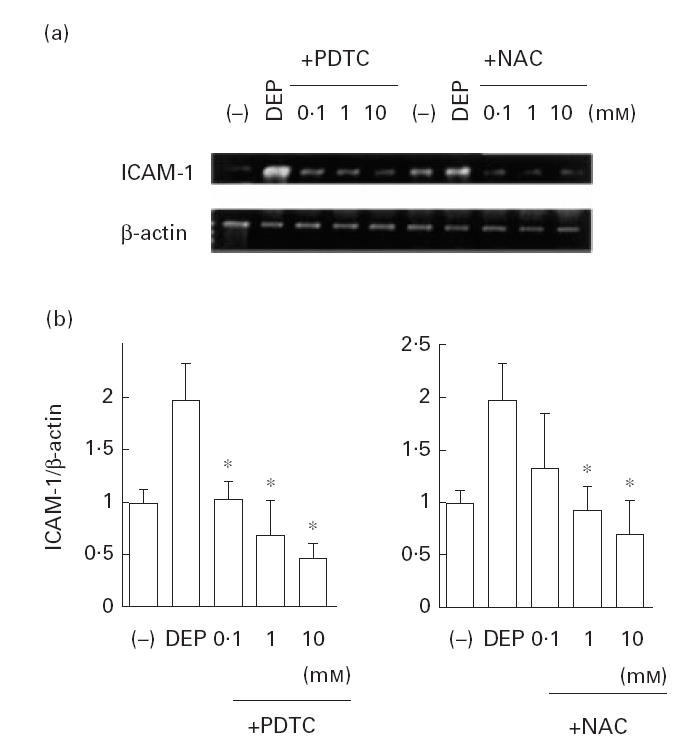
Effect of the anti-oxidant drugs on diesel exhaust particle (DEP)-induced ICAM-1 gene expression. Both pyrrolidine dithiocarbamate (PDTC) and N-acetyl cysteine (NAC) showed a significant suppressive effect on DEP (50 μg/ml)-induced ICAM-1 mRNA levels corrected by β-actin transcripts. (a) Representative results. (b) Data from three independent experiments. *P < 0·05 compared with DEP-treated cells, anova.
Fig. 3.
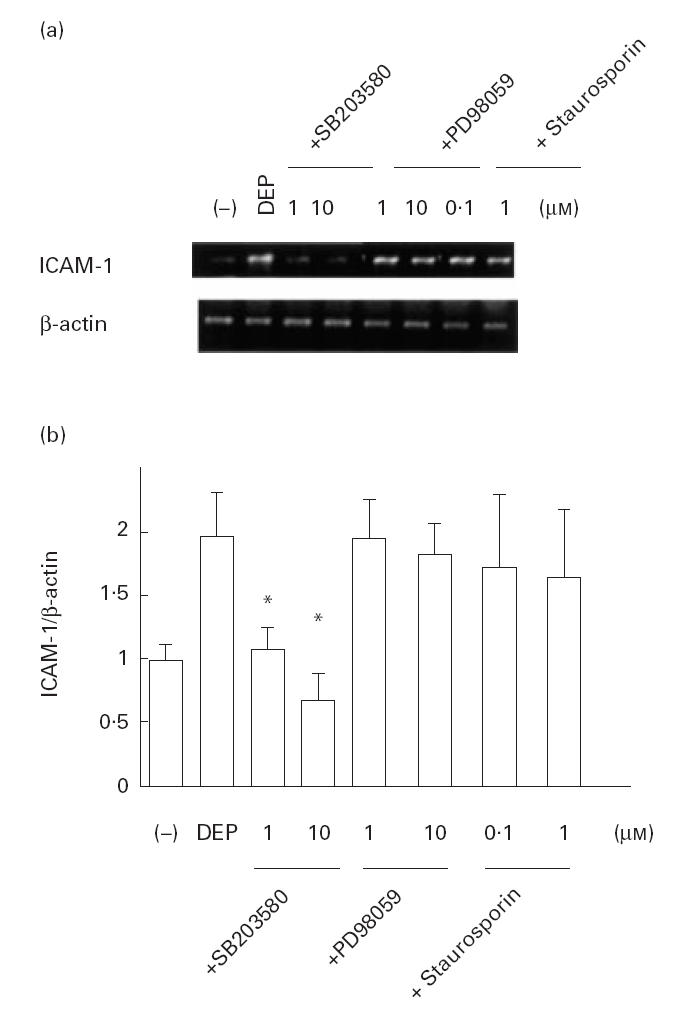
Effect of the drugs acting on signal transduction on diesel exhaust particle (DEP)-induced ICAM-1 gene expression. SB203580 showed a significant suppressive effect on DEP (50 μg/ml)-induced ICAM-1 mRNA levels corrected by β-actin transcripts. In contrast, PD98059 or staurosporin had no effect. (a) Representative results. (b) Data from three independent experiments. *P < 0·05 compared with DEP-treated cells, anova.
Flow cytometry for the detection of ICAM-1 on the epithelial cells
The surface expression of ICAM-1 on BEAS-2B cells was evaluated by flow cytometry analysis 18 h after the cells were treated with DEP. Untreated cells constitutively expressed ICAM-1, and treatment with DEP induced increased expression of this adhesion molecule on the cell surfaces (Fig. 4a) (% positive cells: 29·0 ± 4·54% in untreated cells, 65·4 ± 10·2% in DEP (50 μg/ml)-treated cells). As shown in Fig. 4b, the mean fluorescence intensity (MFI) was significantly augmented by the addition of DEP at 10–50 μg/ml in a dose-dependent fashion. PDTC, NAC and SB203580 showed an inhibitory effect on DEP-induced increase in MFI, but staurosporin or PD98059 showed no significant effect (Fig. 4).
Fig. 4.
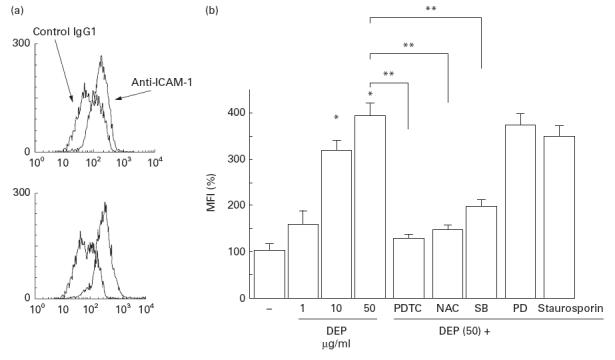
Diesel exhaust particles (DEP) induced an increased expression of surface ICAM-1 molecules as assessed by flow cytometric analysis. (a) A representative flow cytometric histogram for untreated (upper panel) and DEP (50 μg/ml)-treated (lower panel) BEAS-2B cells. (b) Specific fluorescence was expressed as the mean fluorescence intensity (MFI) as shown in the ordinate (MFI of controls expressed as 100%). DEP showed a dose-dependent stimulatory effect on ICAM-1 expression, which was significantly suppressed by the 1-h pretreatment with pyrrolidine dithiocarbamate (PDTC; 10 mm), N-acetyl cysteine (NAC; 10 mm) and SB203580 (SB) (10 μm), whereas PD98059 (PD) (10 μm) and staurosporin (1 μm) had no effect. *P < 0·05 compared with naive cells; **P < 0·05 compared with DEP (50 μg/ml)-treated cells, anova.
Effects of DEP on soluble form of ICAM-1 release by human bronchial epithelial cells
DEP showed a significant stimulatory effect on the release of sICAM-1 by BEAS-2B cells (Fig. 5), as reported previously [8]. NAC, PDTC and SB203580 showed an inhibitory effect on the release of sICAM-1 from the epithelial cells (Fig. 5).
Fig. 5.
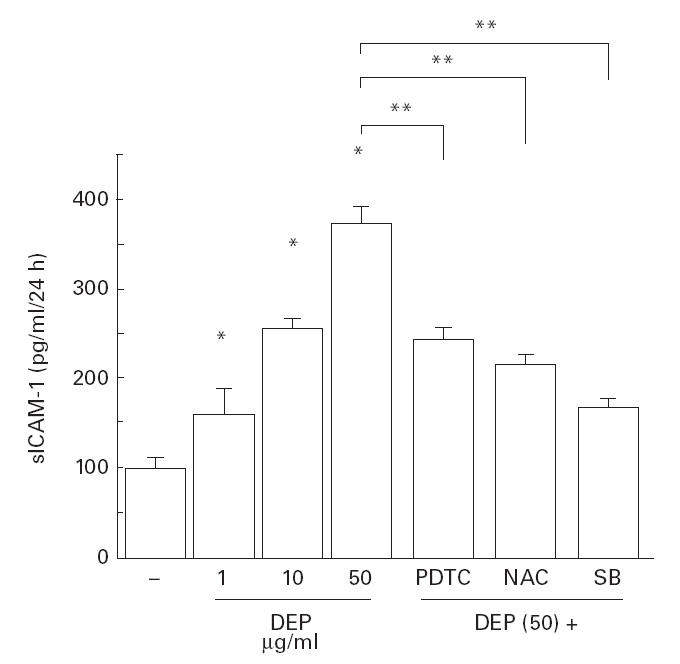
Diesel exhaust particles (DEP) increased the release of soluble ICAM-1 into media. DEP showed a dose-dependent stimulatory effect on sICAM-1 release when evaluated after 24 h, which was significantly suppressed by pretreatment with pyrrolidine dithiocarbamate (PDTC; 10 mm), N-acetyl cysteine (NAC; 10 mm) and SB203580 (10 μm). *P < 0·05 compared with naive cells; **P < 0·05 compared with DEP (50 μg/ml)-treated cells, anova.
Effect of DEP on neutrophil attachment onto BEAS-2B
To assess the biological activity of DEP-induced ICAM-1 expression, an in vitro assay for the neutrophil attachment to BEAS-2B cells was performed. As shown in Fig. 6, the degree of neutrophil attachment to the epithelial cells was assessed by MPO activity. DEP induced a significant increase in MPO activity when evaluated 18 h after DEP treatment, suggesting that DEP increased neutrophil attachment onto the epithelial cell layer by inducing adhesion molecules such as ICAM-1 on the epithelial cells. To evaluate this possibility, anti-ICAM-1 neutralizing antibody (Immunotech, Marseille, France) was added to the cells and neutrophil attachment was evaluated. The results showed that this antibody partially, but significantly, blocked DEP-induced increase of neutrophil attachment to epithelial cells (Fig. 6).
Fig. 6.
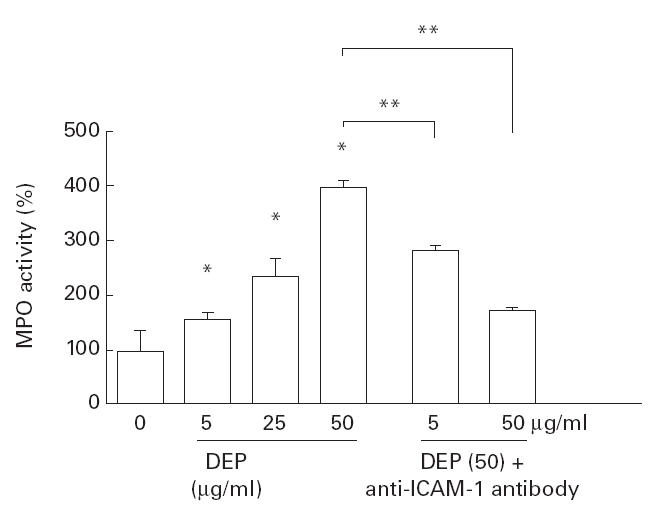
Neutrophil attachment onto diesel exhaust particle (DEP)-treated BEAS-2B cells in vitro. Neutrophil attachment evaluated as myeloperoxidase (MPO) activity (naive cells = 100%) was increased by the 18-h preincubation of BEAS-2B cells with various doses of DEP. When the neutralizing antibody (5 and 50 μg/ml) was used, significant inhibition of DEP (50 μg/ml)-induced attachment was seen, suggesting that ICAM-1 expression was involved in this process. *P < 0·05 compared with naive cells; **P < 0·05 compared with DEP (50 μg/ml)-treated cells, anova.
DISCUSSION
In the present study, we found that DEP up-regulated ICAM-1 mRNA levels and surface protein expression in human bronchial epithelial cells. Neutrophil attachment onto DEP-treated epithelial cells was increased, and studies with anti-ICAM-1 antibodies suggested that this process was at least in part via an increased expression of ICAM-1 molecules on airway epithelial cells.
ICAM-1 (CD54), one of the adhesion molecules belonging to the immunoglobulin superfamily, has been postulated to play an important role in the accumulation of inflammatory cells, including neutrophils, eosinophils and T lymphocytes. Tosi et al. [14] demonstrated that neutrophil adhesion onto the epithelial cell layer was regulated by ICAM-1. They showed that the treatment with anti-ICAM-1 antibody abrogated neutrophil attachment to epithelial cells. Taguchi et al. [25] showed that T cell adhesion and subsequent transmigration were regulated by lymphocyte function-associated antigen-1 (LFA-1) on T cells and ICAM-1 on epithelial cells. Anti-ICAM-1 antibody significantly blocked inflammatory responses found in animal models of asthma [26]. Finally, increased levels of serum and/or bronchoalveolar lavage fluid sICAM-1 have been reported in a variety of inflammatory lung disorders such as sarcoidosis and bronchial asthma [27,28]. Taken together, ICAM-1 expression might be crucial in the regulation of local inflammatory responses in the lung. Although the blocking effect of antibody against ICAM-1 was not complete in our experiment, our findings strongly suggest that DEP-induced up-regulation of ICAM-1 in epithelium had a potency leading to an increased attachment of neutrophils in vitro.
There is accumulating evidence that suggests an important role of ICAM-1 in the pathogenesis of cardiovascular diseases [29]. ICAM-1, CD54, is also involved in mucosal immunity as one of the important costimulatory molecules for the activation of T cells. Therefore, DEP-induced up-regulation of this adhesion molecule might be related to the adjuvant activity of DEP previously shown in animals [30].
Previous reports [6,12,31] demonstrated that DEP stimulated IL-8 gene expression in human bronchial epithelial cells. This effect was largely due to increased transcriptional rates of the mRNA as evaluated by nuclear run on assay [12]. Studies with electrophoretic mobility shift assay suggested that DEP induced the activation of the transcription factor NF-κB [12], which is considered to play an important role in the gene regulation of IL-8. Genomic human ICAM-1 gene has a specific binding motifs to NF-κB at their promotor regions [32]. Therefore, it was likely that activation of NF-κB by DEP shown in IL-8 gene expression might also be involved in ICAM-1 gene up-regulation found in this study. In accordance with this hypothesis, anti-oxidants PDTC and NAC significantly suppressed DEP-induced ICAM-1 expression at mRNA and protein levels. These compounds are believed to inhibit activation of NF-κB by interfering with the actions of reactive oxygen radicals in the cells [21,22]. However, further experiments such as reporter gene assay are necessary to verify this hypothesis. SB203580, a specific inhibitor of p38MAPK pathway, also showed an inhibitory effect, whereas PD98059, an inhibitor of MEK, showed no effect. Recent reports [33,34] suggested that p38MAPK pathways are involved in signal transduction for cytokine expression. Our present data suggest that DEP-induced ICAM-1 expression was dependent, at least in part, on p38MAPK and NF-κB activation pathways. Reports by Nel and co-workers [11,35] showed that DEP induced activation of Jun-N-terminal kinase (JNK) in macrophage cell lines in vitro. Several reports have demonstrated that the other air pollutants induced increased ICAM-1 expression [36,37] and NF-κB activation [37]; these effects of pollutants on respiratory cells may be via common pathways, but the exact mechanisms remain unelucidated.
In conclusion, DEP up-regulated ICAM-1 expression in human bronchial epithelial cells, which may be one important mechanism to induce inflammatory reactions in the airways. Further studies would be necessary to link MAP kinase activation and activation of transcription factors for the elucidation of the intracellular pathways in cell activation by DEP.
Acknowledgments
This work was supported in part by the Pollution-Related Health Damage Compensation and Prevention Association of Japan, and The Manabe Medical Foundation.
REFERENCES
- 1.Draft of Technical Report II-A. Particulate air quality: the 1994 air quality management plan. Diamond Bar, CA: South Coast Air Quality Management District; March 1994. [Google Scholar]
- 2.Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society. Health effects of outdoor air pollution. Am J Respir Crit Care Med. 1996;153:3–50. doi: 10.1164/ajrccm.153.1.8542133. [DOI] [PubMed] [Google Scholar]
- 3.Churg A, Brauer M. Human lung parenchyma retains PM2.5. Am J Respir Crit Care Med. 1997;155:2109–11. doi: 10.1164/ajrccm.155.6.9196123. [DOI] [PubMed] [Google Scholar]
- 4.Dockery DW, Pope AC, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Speizer FE. An association between air pollution and mortality in six US Cities. N Engl J Med. 1993;329:1753–9. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 5.Pope AC, Thun MJ, Namboodiri MN, Dockery DW, Evans JS, Speizer FE, Heath CW. Particulate air pollution as predictor of mortality in a prospective study of US adults. Am J Respir Crit Care Med. 1995;151:669–74. doi: 10.1164/ajrccm/151.3_Pt_1.669. [DOI] [PubMed] [Google Scholar]
- 6.Ohtoshi T, Takizawa H, Okazaki H, Kawasaki S, Takeuchi N, Ohta K, Ito K. Diesel exhaust particles (DEP) stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. J Allergy Clin Immunol. 1998;101:778–85. doi: 10.1016/S0091-6749(98)70307-0. [DOI] [PubMed] [Google Scholar]
- 7.Bayram H, Devalia JL, Sapsford RJ, Ohtoshi T, Miyabara Y, Sagai M, Davis RJ. The effect of diesel exhaust particles on cell function and release of inflammatory mediators from human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol. 1998;18:441–8. doi: 10.1165/ajrcmb.18.3.2882. [DOI] [PubMed] [Google Scholar]
- 8.Steerenberg PA, Zonnenberg JAJ, Dormans JAMA, Joon PNP, Wouters IM, Van Bree L, Scheepers PTJ, Van Loveren H. Diesel exhaust particles induced release of interleukin 6 and 8 by (primed) human bronchial epithelial cells (BEAS 2B) in vitro. Exp Lung Res. 1998;24:85–100. doi: 10.3109/01902149809046056. [DOI] [PubMed] [Google Scholar]
- 9.Diaz-Sanchez D, Tsien A, Casillas A, Doston AR, Saxon A. Enhanced nasal cytokine production in human beings after in vivo challenge with diesel exhaust particles. J Allergy Clin Immunol. 1996;98:114–23. doi: 10.1016/s0091-6749(96)70233-6. [DOI] [PubMed] [Google Scholar]
- 10.Salvi S, Blomberg A, Rudelt B, Kelly F, Sandstrom T, Holgate ST, Frew A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–9. doi: 10.1164/ajrccm.159.3.9709083. [DOI] [PubMed] [Google Scholar]
- 11.Nel A, Diaz-Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. 1998;102:539–54. doi: 10.1016/s0091-6749(98)70269-6. [DOI] [PubMed] [Google Scholar]
- 12.Takizawa H, Ohtoshi T, Kawasaki S, et al. Diesel exhaust particles (DEP) induce nuclear factor-kappa B (NFκB) activation in human bronchial epithelial cells in vitro: importance in cytokine transcription. J Immunol. 1999;162:4705–11. [PubMed] [Google Scholar]
- 13.Bloemen PG, van den Tweel MC, Henricks PA, Engels F, Wagenaar SS, Rutten AA, Nijkamp FP. Expression and modulation of adhesion molecules on human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1993;9:586–93. doi: 10.1165/ajrcmb/9.6.586. [DOI] [PubMed] [Google Scholar]
- 14.Tosi MF, Stark JM, Smith CW, Hamedani A, Gruenert DC, Infeld MD. Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil–epithelial cell adhesion. Am J Respir Cell Mol Biol. 1992;7:214–21. doi: 10.1165/ajrcmb/7.2.214. [DOI] [PubMed] [Google Scholar]
- 15.Reddel RR, Ke Y, Gerwin BI, et al. Transformation of human bronchial epithelial cells by infection with SV 40 or adenovirus-12 SV 40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV 40 early region genes. Cancer Res. 1988;48:1904–9. [PubMed] [Google Scholar]
- 16.Chelly J, Montarras D, Pinset C, Berwald-Netter Y, Kaplan J, Kahn A. Quantitative estimation of minor mRNAs by cDNA-polymerase chain reaction. Application to dystrophin mRNA in cultured myogenic and brain cells. Eur J Biochem. 1990;187:691–8. doi: 10.1111/j.1432-1033.1990.tb15355.x. [DOI] [PubMed] [Google Scholar]
- 17.Takizawa H, Desaki M, Ohtoshi T, et al. Erythromycin modulates IL-8 expression in human bronchial epithelial cells: studies with normal and inflamed airway epithelium. Am J Respir Crit Care Med. 1997;156:266–71. doi: 10.1164/ajrccm.156.1.9612065. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;161:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima J, Ono M, Takeda M, Kawauchi M, Furuse A, Takizawa H. Role of costimulatory molecules on airway epithelial cells acting as alloantigen-presenting cells. Transplantation Proc. 1997;29:2297–300. doi: 10.1016/s0041-1345(97)00334-5. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki S, Takizawa H, Ohtoshi T, et al. Roxithromycin inhibits cytokine production and neutrophil attachment with human bronchial epithelial cells in vitro. Antimicrob Agents Chemother. 1998;42:1499–502. doi: 10.1128/aac.42.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schreck R, Meier B, Maennel DN, Droege W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J Exp Med. 1992;175:1181–94. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackwell TS, Holden EP, Blackwell TR, Christman BW, Christman JW. Activation of NF-κB in rat lungs by treatment with endotoxin: modulation by treatment with N-acethylcysteine. J Immunol. 1996;157:1630–7. [PubMed] [Google Scholar]
- 23.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1996;372:739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 24.Allesi DR, Cuenda A, Cohen P, Dundley DT, Saltiel AR. PD98059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 25.Taguchi M, Sampath D, Koga T, Castro M, Look DC, Nakajima S, Holtzman MJ. Patterns for RANTES secretion and intercellular adhesion molecule 1 expression mediate transepithelial T cell traffic based on analyses in vitro and in vivo. J Exp Med. 1998;187:1927–40. doi: 10.1084/jem.187.12.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wegner CD, Gundel RH, Reilly P, Haynes N, Letts LG, Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science. 1990;247:456–9. doi: 10.1126/science.1967851. [DOI] [PubMed] [Google Scholar]
- 27.Shijubo N, Imai K, Shigehara K, Hinoda Y, Abe S. Circulating soluble intercellular adhesion molecule-1 (sICAM-1) in patients with sarcoidosis. Clin Exp Immunol. 1996;106:549–54. doi: 10.1046/j.1365-2249.1996.d01-872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oymar K, Bjerknes R. Differential patterns of circulating adhesion molecules in children with bronchial asthma and acute bronchiolitis. Ped Allergy Immunol. 1998;9:73–79. doi: 10.1111/j.1399-3038.1998.tb00307.x. [DOI] [PubMed] [Google Scholar]
- 29.Rohde LE, Hennekens CH, Ridker PM. Cross-sectional study of soluble intercellular adhesion molecule-1 and cardiovascular risk factors in apparently healthy men. Arteriosclerosis, Thrombosis Vascular Biol. 1999;19:1595–9. doi: 10.1161/01.atv.19.7.1595. [DOI] [PubMed] [Google Scholar]
- 30.Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Miyamoto T, Ikemori R, Muranaka M. Diesel-exhaust particulates inoculated by the intranasal route have an adjuvant activity for IgE production in mice. J Allergy Clin Immunol. 1987;79:6139–45. doi: 10.1016/s0091-6749(87)80161-6. [DOI] [PubMed] [Google Scholar]
- 31.Boland S, Baeza-Squiban A, Fournier T, et al. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am J Physiol. 1999;276(4, Part 1):L604–13. doi: 10.1152/ajplung.1999.276.4.L604. [DOI] [PubMed] [Google Scholar]
- 32.Barnes PJ. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 33.Rawadi G, Ramez B, Lemercier B, Roman-Roman S. Activation of mitogen-activated protein kinase pathways by Mycoplasma fermentans membrane lipoproteins in murine macrophages: involvement in cytokine synthesis. J Immunol. 1998;160:1330–9. [PubMed] [Google Scholar]
- 34.Gon Y, Hashimoto S, Matsumoto K, Nakayama T, Takeshita I, Horie T. Cooling and rewarming-induced IL-8 expression in human bronchial epithelial cells through p38 MAP kinase-dependent pathway. Biochem Biophys Res Commun. 1998;249:156–60. doi: 10.1006/bbrc.1998.9115. [DOI] [PubMed] [Google Scholar]
- 35.Ng D, Hiura N, Faris M, Saxon A, Nel A. Macrophage activation by polycyclic aromatic hydrocarbons: evidence for the involvement of stress-activated protein kinases, activator protein-1, and antioxidant response elements. J Immunol. 1998;161:942–51. [PubMed] [Google Scholar]
- 36.Treadwell MD, Mossman BT, Barchowsky A. Increased neutrophil adherence to endothelial cells exposed to asbestos. Toxicol Appl Pharmacol. 1996;139:62–70. doi: 10.1006/taap.1996.0143. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy T, Ghio AJ, Reed W, et al. Copper-dependent inflammation and nuclear factor-kappaB activation by particulate air pollution. Am J Respir Cell Mol Biol. 1998;19:366–78. doi: 10.1165/ajrcmb.19.3.3042. [DOI] [PubMed] [Google Scholar]