

DEFINITION AND CLINICAL DESCRIPTION
SIgAD
SIgAD, using 0.05 g/l of serum IgA as the upper limit for diagnosis in adults and a concomitant lack of secretory IgA, is the most common form of primary immunodeficiency (PID) in the western world and affects approximately 1/600 individuals [1]. However, there is a marked variability in the prevalence in different ethnic groups [2], with a lower frequency in Japanese (1/18 000) and Chinese (1/4000), suggesting a genetic basis for the disorder. The term ‘selective IgAD’ should be reserved for those individuals who do not have identifiable disorders which are known to be associated with low IgA levels (see below). However, in many cases a simultaneous change in the IgG subclass pattern is seen with a lack of specific anti-polysaccharide antibodies of the IgG2 subclass [3] or a total lack of serum IgG2 [4], IgG4 and IgE [5], reflecting a relative or absolute block in switching to genes downstream of the G1.
CVID
CVID affects about 1/25 000 Caucasians, the patients having a marked reduction in serum levels of both IgG (usually < 3 g/l) and IgA (< 0.05 g/l); IgM is also reduced in about half the patients (< 0.3 g/l) [1]. Symptoms of recurring infection can start at any time of life, but there are peaks of onset during 1–5 and 16–20 years of age [6], with equal distribution between the sexes. The condition is clinically more complex than X-linked agammaglobulinaemia (XLA), with patients being prone to chronic inflammatory and autoimmune complications [6,7].
INHERITANCE OF SIgAD AND CVID
Familial inheritance of either SIgAD or CVID occurs in about 20% of cases (Fig. 1). A different population prevalence in various ethnic groups, strong familial clustering of the disorder, a predominant inheritance pattern in multiple-case families compatible with autosomal dominant transmission, and a high relative risk for siblings suggest the involvement of thus far unidentified genetic factors in the pathogenesis of IgAD/CVID [8,9]. In multiple-case families with a dominant transmission of CVID/IgAD, CVID was usually present in the parents accompanied by IgAD in descendants [9]. This is consistent with the hypothesis that CVID may develop later in life as a more severe manifestation of a common, complex genetic defect, most likely involving immunoglobulin class switching. This is supported by a description of a gradual decline of IgG levels that progressed at similar ages in affected siblings [10]. Furthermore, CVID may develop from IgAD [11–13] and occasionally vice versa [14]. In both diseases anti-IgA antibodies have been detected. Since the disease phenotype is persistent and the phenocopy rate is low, chromosome susceptibility loci underlying this complex trait should be detectable by genetic linkage analysis. The recurrence risk of IgAD was found to depend on the gender of parents transmitting the defect: affected mothers are more likely to pass the defect on to their offspring than affected fathers. This was accompanied by a preferential transmission of associated alleles in the MHC, suggesting a role for this region in the parent-of-origin penetrance differences [9]. The role of the MHC is further supported by a higher prevalence of anti-IgA antibodies among females transmitting the disease to their offspring than in female non-transmitters.
Fig. 1.
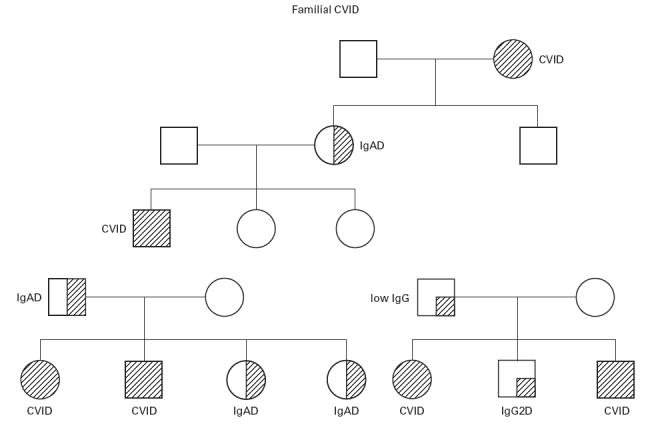
Three pedigrees are shown demonstrating the inheritance patterns for IgAD and CVID. Note that in some families there are relatives with minor IgG abnormalities.
MOLECULAR BASIS
Our recent study involving a large number of multiple-case families supported the presence of a predisposing locus in the class II or class III region [9]. A significant increase in sharing of MHC alleles in affected members of our family dataset was consistent with previous allelic associations observed in case control studies, which appear to be much stronger for IgAD than CVID. It is possible that the gene(s) involved predispose to the production of IgA antibodies.
A number of abnormalities in the cytokine network have been observed in SIgAD and CVID; however, attempts to find a defect in candidate cytokine genes have so far failed, including the available coding region of the cytokine genes in the MHC, such as lymphotoxin α and β (Vorechovsky et al., unpublished). Using the same family material, we were unable to confirm a susceptibility locus on chromosome 18, despite a number of anecdotal reports of patients with gross defects in the 18p region and SIgAD [15]. The finding of immunoglobulin deficiency in only one of two monozygotic twins suggests an environmental, possibly infectious agent as a triggering factor, but longer follow up of these twins is needed [16].
In SIgAD the defect is manifested at the stem cell level, since a bone marrow transplant from an IgA-deficient donor transfers the defect to the recipient [17], whereas bone marrow from a normal individual transplanted into an SIgAD patient will correct the defect [18]. The genes for α1 and α2 can readily be demonstrated in the genome of IgAD patients [19] and these ‘silent’ genes can be re-expressed in the children of SIgAD parents [20], suggesting that the defect is due to a defect in switching or expression of the immunoglobulin genes. This is supported by the production of IgA in vitro by lymphocytes from both SIgAD [21] and CVID [22] patients when cultivated together with anti-CD40 antibodies and IL-10. The physiology of this in vitro system is questionable, but it clearly demonstrates that secretion of IgG1, IgG3, IgG4 [23] and IgA [21,24], with a biased expression of IgA1, can occur if appropriate stimuli are added. Although it is technically difficult to detect the normally small numbers of circulating IgA-bearing B cells, they have been found in SIgAD [25]. Furthermore, T cells from SIgAD patients will support IgA production in vitro by B cells from normal subjects. In a few selected cases the defect is restricted to one of the two IgA subclasses and this is most often, although not invariably [26], due to deletions of the corresponding heavy chain constant region gene [27].
The mechanism of CVID is equally elusive, one problem being that the syndrome probably includes a number of different disorders [28]. At least 30% of patients are lymphopenic, the CD4+ subset being mainly depressed, and this probably accounts for the low levels of IL-2 produced in vitro from stimulated peripheral blood mononuclear cells (PBMC) [29]. The expression of CD40 ligand on activated T cells is usually normal, but is very low in a small group of patients, implying a defect in isotype switching [30]. The B cells from another small subgroup have defective signalling through the CD40 pathway [31]; these patients have raised serum IgM and may be misdiagnosed as XHIM. Levy et al. [32] recently demonstrated somatic hypomutation in B cells from two patients; subsequent work indicates that this occurs in about 20% of patients, but cannot yet be linked to any clinical pattern (Y. Levy, personal communication). Since hypermutation occurs predominantly within the germinal centres (Gcs) of the central lymphoid apparatus, the defect may reflect the fact that splenic Gcs are often poorly developed or disrupted by granulomatous infiltrates in these patients [33]. Another possibility is that inherited subtle defects in DNA repair, which could impair hypermutation, may contribute to an already compromised B cell maturation system. This is supported by the finding of increased chromosomal sensitivity to radiation damage in lymphocytes from some CVID patients [34], and clinical surveys showing an increased susceptibility to some cancers [35]. IgAD, and sometimes a more broader immunoglobulin deficiency, is associated with ataxia telangiectasia and the Nijmegen breakage syndrome, both conditions caused by inherited defects in DNA repair [36].
The majority of patients have a defect in CD4+ T cell priming to antigen, as measured by the numbers of circulating responsive cells following immunization [37]. This could be due to a defect in antigen-presenting cells (APC), and not T cells, since various defects in APC have been reported [38]. There may be a small subgroup of patients with defects in CD3 complex triggering, but this needs to be confirmed [39].
The majority of patients show a pattern of raised production of interferon-gamma by circulating T cells, particularly by the CD8+ subset, increased numbers of DR-expressing CD4+ T cells with up-regulated Fas expression, and an increased rate of apoptosis [40,41]. There is increased chronic ‘activation’ of circulating monocytes producing reactive oxygen [42], and IL-12 after in vitro lipopolysaccharide (LPS) stimulation (Cambronero et al., unpublished). This suggests a ‘pathological’ shift towards a Th1 type of immune response. Tumour necrosis factor (TNF) production from both T cells and monocytes is raised in a subgroup of patients with granulomas [43], probably due to the coincidental inheritance of TNF-α (high) genetic polymorphisms [44]. These abnormalities appear specific to CVID and are not seen in patients with XLA, who have the same therapy and suffer from the same infections.
The recovery of antibody production following HIV infection in CVID patients is important [45]. There have been three published case reports, with a fourth patient currently in our (D.W.'s) clinic with familial CVID, having survived 5 years without immunoglobulin therapy; highly active anti-retroviral therapy (HAART) has now reduced the HIV load to unrecordable levels. Only IgG and IgM antibody production recovered in three of the patients, the IgA remaining unrecordable. These cases demonstrate that CVID is potentially reversible by immunoregulatory factors, and supports the view that SIgAD predisposes to CVID.
ANIMAL MODELS
Although there are a number of reports on IgA-deficient dogs [46–48] and chickens [49], the molecular basis of these deficiencies has not been elucidated. There is no rodent model available yet which resembles the human disease, although knock-out mice with a deleted J chain [50] or Iα region [51] have been described. Only secretory IgA is impaired in the former and serum levels of IgA are up to 30-fold higher than in normal wild-type mice. There seems to be J chain-independent IgA transport in the intestinal, mammary and respiratory epithelial cells of these mice [52], adding to the complexity of the ‘secretory’ IgA machinery. The mice with an Iα region deletion, contrary to what was expected based on mice with deleted I regions or I region promoters for γ1, γ2b and ε, produced normal levels of IgA. This suggests that the I region as such is redundant and can be replaced by other gene sequences [53] and that splicing of germ-line transcripts rather than transcription itself controls DNA rearrangement leading to class switching. Mice with a targeted deletion of the α gene and its associated switch region [54–56] not only lack IgA but also have low serum levels of IgG3 and IgE, but raised levels of IgM and IgG2b, to some extent mimicking the situation in IgAD patients.
Targeted disruption of the IL-5 receptor α gene in mice leads to a reduction in the number of IgA-producing cells at mucosal effector sites such as intestinal lamina propria and nasal mucosa, but normal numbers at inductive sites. Furthermore, serum levels of IgA were normal [57]. Targeted deletions of either the lymphotoxin α [58,59] or lymphotoxin β [59] gene cause reduced serum and secreted IgA with disrupted development of secondary lymphoid organs and a diminished capacity for affinity maturation of the antibodies produced. This has some parallels with a subgroup of CVID patients [32]. Transcription factor knockouts, such as the NF-κB, p65 [60] or p50 Rel-A [61] deficient mice, have defective class switching and could be a model for a subgroup of IgAD/CVID patients [62,63]. Other ‘knockouts’ for various critical immunoregulatory proteins in mice have major general effects on the immune system, but the phenotypes are not consistent with CVID.
DIFFERENTIAL DIAGNOSIS AND DIAGNOSTIC TESTS
The diagnosis is one of exclusion [64]. The family history and the age of onset of symptoms is important, because patients presenting after 15 years are unlikely to have one of the known single gene PIDs such as XLA or X-linked hyper IgM syndrome (X-HIM). There is a typical pattern of immunoglobulin class deficiency, with very low IgA and IgE and variable but usually low IgM. A chest radiograph is necessary to exclude thymoma in patients presenting over 45 years; these patients may only have moderate hypo-immunoglobulinaemia despite having no circulating B cells. This appears to be a distinct entity, with a much worse prognosis than CVID [65]. There is usually no confusion with secondary hypo-immunoglobulinaemia, in which IgA levels are usually only moderately low. Nevertheless, routine screening for nephrotic syndrome, chronic lymphatic leukaemia and myeloma should not be forgotten. Protein losing enteropathy with low immunoglobulins can be confusing, but is usually obvious when the serum IgG fails to rise on immunoglobulin therapy.
IgAD has been associated with a variety of anti-rheumatic and anti-epileptic drugs [66] (Table 1). In about half the cases the deficiency is apparently reversible after cessation of therapy, although full recovery may take months or even years. IgAD was induced by multiple anti-rheumatic drugs in a patient with rheumatoid arthritis [67], suggesting that selected individuals may be genetically predisposed to develop this complication. On the other hand, different drugs with a common molecular mechanism of action (ACE inhibitors) may actually vary in their capacity to induce IgAD in a given patient [68].
Table 1.
Pattern of drug-induced immunoglobulin deficiencies
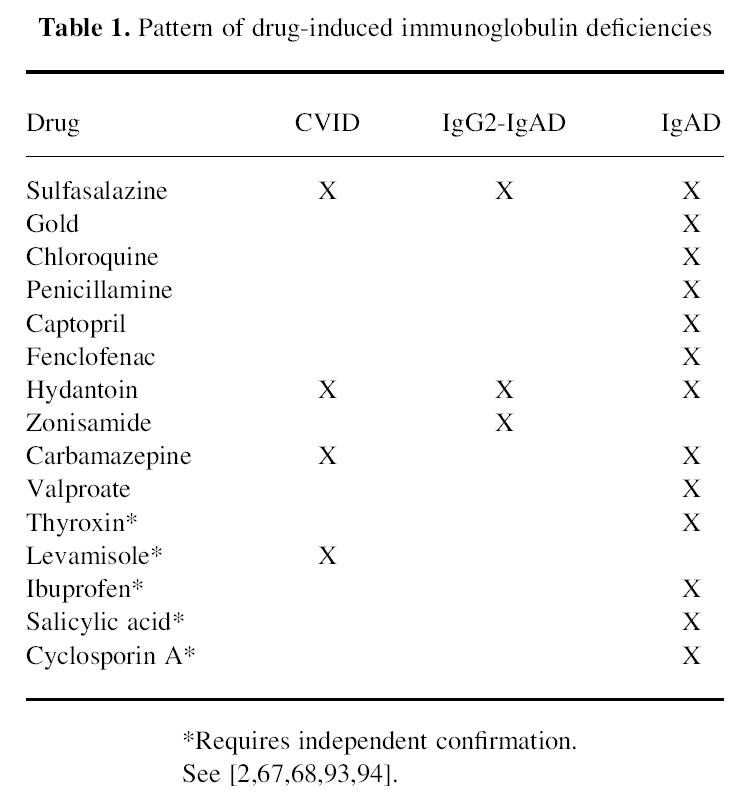
CVID and IgG2-IgA deficiency can also be induced by some of the above drugs [66], and recently zonisamide, a new anti-convulsant [69], was added to the list. The presence of drug-associated pan-hypo-immunoglobulinaemia, IgAD with IgG2 subclass deficiency and selective IgAD suggests that it shares features with the primary forms of SIgAD/CVID and that the pathophysiological process may involve common key steps. There is no common molecular denominator for the drugs used, although a majority appear to act at the level of lysosomes or are lysosomotropic, suggesting that the pathway leading to IgAD involves this organelle and APC. Some of the drugs implicated contain a highly reactive sulphydryl group and it is possible that the immunological dysregulation induced by these agents, including the formation of immune complexes and induction of autoimmune phenomena [70], plays a role in the development of the immunodeficiency in genetically susceptible patients. Sulfasalazine, one of the anti-rheumatic drugs implicated in immunoglobulin deficiency, prevents NF-κB-dependent transcription through inhibition of IκBα degradation [71]. This suggests that this form of IgAD/CVID may be associated with a defect in transcription either of constant region genes or of the switching process.
Various single gene disorders causing hypo-immunoglobulinaemia should be excluded, including ‘leaky’ severe combined immunodeficiency, which can rarely present after childhood [72]. A detailed family history is required. Male patients with low numbers of circulating B cells should be screened for XLA [73], and other autosomal recessive causes of agammaglobulinaemia considered in females [74]. Male patients with X-HIM or X-linked lymphoproliferative syndrome (XLPS) may be confused with CVID, particularly since the former may have normal serum IgM levels [75].
CLINICAL MANAGEMENT
SIgAD
Most individuals with SIgAD are not prone to infection, and are diagnosed during routine tests for other conditions or following family screening of a proband with SIgAD/CVID. There is no consensus on whether they should be routinely screened for anti-IgA antibodies, partly because there is no agreement on what level of antibodies constitutes a risk of anaphylaxis to blood products. A minority are prone to infection, and these should be screened for additional IgG subclass or functional IgG defects (i.e. response to test immunization); however, IgG subclass levels correlate poorly with susceptibility to infection [76]. Most patients can be managed with prophylactic or periodic antibiotics, but a few may benefit from immunoglobulin therapy, regardless of whether an associated IgG functional defect can be demonstrated [77]. Such patients will require immunoglobulin products containing low or minimal IgA if they have high levels of IgA antibodies.
CVID
Respiratory tract
Nearly all patients have recurrent symptoms of bronchitis, and to a lesser extent sinusitis, usually due to non-encapsulated Haemophilus influenzae, although streptococci, Moraxella catarrhalis and mycoplasmas are also important pathogens [78–80]. Until the late 1970s, most patients developed and eventually died from bronchiectasis. Many CVID patients continue to suffer from recurrent bronchitis despite IVIG therapy, and need prophylactic antibiotics to prevent bronchiectasis. Some clinicians favour rotating regimes, but in our experience compliance is poor and breakthrough infection is common. Prophylactic quinolone antibiotics, which have a very low minimum inhibitory concentration (MIC) for H. influenzae, are a better alternative, with amoxycillin for ‘breakthrough’ resistant streptococcal infection (Webster et al., unpublished).
Other infections
About 5% of CVID patients develop mycoplasma infections in the urinary tract and joints, occasionally with systemic spread and deep abscesses [81]. Although most patients respond to doxycyline, they should be referred urgently to a specialist centre where the organism can be characterized and appropriate antibiotics given. There is a promising new pleuromutilin antibiotic under trial for those with resistant organisms (Heilman and Webster, unpublished).
Enteroviral infection of the central nervous system is a rare complication, but can present either acutely or insidiously with signs of encephalitis, seizures, headache, sensory motor disturbances and even personality changes [82]. Cerebrospinal fluid (CSF) should be obtained, but may not grow the virus, particularly if the patient is on IVIG therapy. Polymerase chain reaction (PCR) for enterovirus should be requested routinely [83], and if positive patients should be offered a trial of pleconaril, a new anti-enteroviral drug which appears to have been effective in an open trial (Rotbart and Webster, unpublished).
Granulomas are a special feature of CVID, and do not occur in other primary lymphocyte disorders. In the lungs they can mimic sarcoidosis [84]. Granulomatous infiltration of the spleen occurs in about 20% of patients, and often extends to the liver causing presinusoidal venous congestion with oesophageal varices, sometimes progressing to cirrhosis and liver failure requiring liver transplantation [85]. Steroids can usually control the lung disease but new strategies are needed for liver involvement.
Inflammatory bowel disease is common, with about 30% of patients having some degree of chronic diarrhoea. Although the colon is preferentially involved, the histology showing lymphocytic mucosal infiltration [86], about 10% of patients have a severe gastroenteropathy involving the small and large bowel, with malabsorption, and occasionally fibrotic ileal strictures. The mucosal inflammation often involves the stomach, and a small number of patients develop achlorhydria and pernicious anaemia [87]. This probably explains the apparently raised incidence of carcinoma of the stomach in CVID patients [35], although this is now a very rare complication in the UK and Sweden. Although regular immunoglobulin therapy reduces the susceptibility to giardia and campylobacter enteritis, it does not prevent the unexplained mucosal inflammation; treatment for the latter is currently unsatisfactory and in severe cases involves trying antibiotics, elemental diets and steroids [86].
Autoimmune disease occurs in about 10% of patients, usually immune thrombocytopenia (ITP), haemolytic anaemia or neutropenia. Much rarer complications are red cell aplasia, thyroid disease and neuropathy. Steroids may be useful but in refractory cases high dose IVIG, splenectomy or more aggressive immunosuppressive therapy may be needed [88]. There is a raised incidence of lymphoma [89], which is now more common as patients survive longer. Unexplained fever, weight loss, recent lymphadenopathy and abdominal or chest pain should prompt a search for clonally derived lymphocytes in the blood, followed by lymph node biopsy, and if necessary a diagnostic splenectomy.
Immunoglobulin therapy
Nearly all CVID patients require immunoglobulin replacement therapy, given either 2–4 weekly at a total dose of 400 mg/kg body weight monthly, or as a subcutaneous injection (using an infusion pump) every 1–2 weeks [90,91]. Unfortunately, CVID patients with hepatitis C have a very poor prognosis [92], and most who were infected by contaminated immunoglobulin in the 1980s have died; nowadays only products known to be subjected to formal viral inactivation should be used.
USEFUL CONTACTS
Various European registries, and subregistries, under the auspices of the European Society for Immunodeficiency (ESID), are listed below, together with the contact person for assistance. Also listed are international nursing and patient support groups, which can advise on specific country based groups.
ESID registries
Main CVID/IgAD registry L Hammarstrom, M Abedi
Lennart.Hammarstrom@csb.ki.se
Website: http://www.cbt.ki.se/esidregistry/intro.html
Subregistry for (i) Mycoplasma infection C. Heilmann (carsten_heilmann@online.pol.dk) and
D. Webster (dwebster@rfhsm.ac.uk)
(ii) Enteroviral infection
D. Webster (see above)
International
International Nursing Group for Immunodeficiences (INGID)
Ann Gardulf, Sweden, Fax: + 468 58586850
International Patients Organization for Primary Immunodeficiency (IPOPI): pimmune@dial.pipex.com
REFERENCES
- 1.Primary Immunodeficiency Diseases. Report of a WHO Scientific Group WHO. Clin Exp Immunol. 1997;159:6236–41. [PubMed] [Google Scholar]
- 2.Hammarström L, Smith CIE. Genetic approach to common variable immunodeficiency and IgA deficiency. In: Ochs H, Smith CIE, Puck J, Smith CIE, editors. Primary immunodeficiency diseases, a molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 250–62. [Google Scholar]
- 3.Hammarström L, Persson MAA, Smith CIE. Immunoglobulin subclass distribution of human anti-carbohydrate antibodies: aberrant pattern in IgA deficient donors. Immunology. 1985;54:821–6. [PMC free article] [PubMed] [Google Scholar]
- 4.Oxelius V-A, Laurell AB, Lindqvist B, Henryka G, Axelsson U, Björkander J, Hanson L-Å. IgG subclasses in selective IgA deficiency. Importance of IgG2-IgA deficiency. N Engl J Med. 1981;304:1476–7. doi: 10.1056/NEJM198106113042408. [DOI] [PubMed] [Google Scholar]
- 5.Hammarström L, Grubb R, Jakobsen BK, Oxelius V, Persson U, Smith CIE. Concomitant deficiency of IgG4 and IgE in IgA deficient donors with high titres of anti-IgA. Monogr Allergy. 1986;20:234–5. [Google Scholar]
- 6.Hermaszewski RA, Webster ADB. Primary hypogammaglobulinemia: a survey of clinical manifestations and complications. Quart J Med. 1993;86:31–42. [PubMed] [Google Scholar]
- 7.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. J Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 8.Burrows PD, Cooper MD. IgA deficiency. Adv Immunol. 1997;65:245–76. [PubMed] [Google Scholar]
- 9.Vorechovsky I, Webster ADB, Plebani A, Hammarström L. Genetic linkage of IgA deficiency to the major histocompatibility complex: evidence for allele segregation distortion, parent-of-origin penetrance differences and the role of anti-IgA antibodies in disease predisposition. Am J Hum Genet. 1999;64:1096–109. doi: 10.1086/302326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishizaka A, Nakanishi M, Yamada S, Sakiyama Y, Matsumoto S. Development of hypogammaglobulinemia in a patient with common variable immunodeficiency. Eur J Pediatr. 1989;149:175–6. doi: 10.1007/BF01958274. [DOI] [PubMed] [Google Scholar]
- 11.Johnson ML, Keeton LG, Zhu Z-B, Volanakis JE, Cooper MD, Schroeder JR. Age-related changes in serum immunoglobulins in patients with familial IgA deficiency and common variable immunodeficiency (CVID) Clin Exp Immunol. 1997;108:477–83. doi: 10.1046/j.1365-2249.1997.3801278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espanol T, Catala M, Hernandez M, Caragol I, Bertran JM. Development of a common variable immunodeficiency in IgA deficient patients. Clin Immunol Immunopathol. 1996;80:333–5. doi: 10.1006/clin.1996.0132. [DOI] [PubMed] [Google Scholar]
- 13.Gutierrez MG, Kirkpatrick CH. Progressive immunodeficiency in a patient with IgA deficiency. Ann Allergy Asthma Allergy. 1997;79:297–301. doi: 10.1016/S1081-1206(10)63018-9. [DOI] [PubMed] [Google Scholar]
- 14.Seligmann M, Aucouturier P, Danon F, Preud'homme JL. Changes in serum immunoglobulin patterns in adults with common variable immunodeficiency. Clin Exp Immunol. 1991;84:23–27. doi: 10.1111/j.1365-2249.1991.tb08118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vorechovsky I, Blennow E, Nordenskjold M, Webster ADB, Hammarstrom L. A putative susceptibility locus on chromosome 18 is not a major contributor to human selective IgA deficiency: evidence from meiotic mapping of 83 multiple-case families. J Immunol. 1999;163:2236–42. [PubMed] [Google Scholar]
- 16.Lewkonia RM, Gairdner D, Doe WF. IgA deficiency in one of identical twins. BMJ. 1976;1:311–3. doi: 10.1136/bmj.1.6005.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammarström L, Lönnqvist B, Ringdén O, Smith CIE, Wiebe T. Transfer of IgA deficiency to a bone marrow grafted patient with aplastic anaemia. Lancet. 1985;1:778–81. doi: 10.1016/s0140-6736(85)91446-1. [DOI] [PubMed] [Google Scholar]
- 18.Kurobane I, Riches PG, Sheldon J, Jones S, Hobbs JR. Incidental correction of severe IgA deficiency by displacement bone marrow transplantation. Bone Marrow Transplant. 1991;7:494–5. [PubMed] [Google Scholar]
- 19.Hammarström L, Carlsson B, Smith CIE, Wallin J, Wieslander L. Detection of IgA heavy chain constant region genes in IgA deficient donors: evidence against gene deletions. Clin Exp Immunol. 1985;60:661–4. [PMC free article] [PubMed] [Google Scholar]
- 20.Hammarstrom L, Lange G, Smith CIE. IgA2 allotypes in IgA deficiency. Re-expression of the silent IgA2m2 allotype in the children of IgA deficient patients. J Immunogenet. 1987;14:197–202. doi: 10.1111/j.1744-313x.1987.tb00381.x. [DOI] [PubMed] [Google Scholar]
- 21.Briere F, Bridon J-C, Chevet D, Souillet G, Bienvenu F, Guret C, Martinez-Valdez H, Banchereau J. Interleukin-10 induces B lymphocytes from IgA deficient patients to secrete IgA. J Clin Invest. 1994;94:97–104. doi: 10.1172/JCI117354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zielen S, Bauscher P, Hofmann D, Meur SC. Interleukin 10 and immune restoration in common variable immunodeficiency. Lancet. 1993;342:750–1. [PubMed] [Google Scholar]
- 23.Fujieda S, Zhang K, Saxon A. IL-4 plus CD40 monoclonal antibody induces human B cells g subclass-specific isotype switch: switching to γ1, γ3 and γ4, but not g2. J Immunol. 1995;155:2318–28. [PubMed] [Google Scholar]
- 24.Friman V, Hanson LÅ, Bridon J-M, Tarkowski A, Banchereau J, Briere F. IL-10 driven immunoglobulin production by B lymphocytes from IgA-deficient individuals correlates to infection proneness. Clin Exp Immunol. 1996;104:432–8. doi: 10.1046/j.1365-2249.1996.38746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott LJ, Bryant A, Webster ADB, Farrant J. Failure in secretion by surface-positive B cells in common variable immunodeficiency (CVID) Clin Exp Immunol. 1994;95:10–13. doi: 10.1111/j.1365-2249.1994.tb06007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammarström L, Holm G, Palmblad J, Persson MAA, Smith CIE. Lack of IgG in a healthy adult: a rare case of dysgammaglobulinemia with undetectable serum IgG, IgA2 and IgE. Clin Immunol Immunopathol. 1984;30:1–10. doi: 10.1016/0090-1229(84)90001-1. [DOI] [PubMed] [Google Scholar]
- 27.Engström P-E, Norhagen-Engström G, Bottaro A, et al. Subclass distribution of antigen-specific IgA antibodies in normal donors and individuals with homozygous IgH Cα1 or Cα2 gene deletions. J Immunol. 1990;145:109–16. [PubMed] [Google Scholar]
- 28.Spickett GP, Farrant J, North ME, Zhang J, Morgan L, Webster ADB. Common variable immunodeficiency: how many diseases? Immunol Today. 1997;19:325–8. doi: 10.1016/s0167-5699(97)01086-4. [DOI] [PubMed] [Google Scholar]
- 29.North ME, Ivory K, Funauchi M, Webster ADB, Lane AC, Farrant J. Intracellular cytokine production by human CD4+ and CD8+ T cells from normal and immunodeficient donors using directly conjugated anti-cytokine antibodies and three-colour flow cytometry. Clin Exp Immunol. 1996;105:517–22. doi: 10.1046/j.1365-2249.1996.d01-795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrington M, Grosmaire LS, Nonoyama S, et al. CD40 ligand expression is defective in a subset of patients with common variable immunodeficiency. Proc Natl Acad Sci USA. 1994;91:1099–103. doi: 10.1073/pnas.91.3.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durandy A, Hivroz C, Mazerolles F, et al. Abnormal CD-40 mediated pathway in B lymphocytes from patients with hyper IgM syndrome and normal CD40-ligand expression. J Immunol. 1997;158:2576–84. [PubMed] [Google Scholar]
- 32.Levy Y, Gupta N, Le Deist F, Fischer A, Weill J-C, Reynaud C-A. Defect in IgV gene somatic hypermutation in common variable immunodeficiency syndrome. Proc Natl Acad Sci USA. 1998;95:13135–40. doi: 10.1073/pnas.95.22.13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webster ADB, Amlot P. Granulomas in primary immunodeficiency. In: James DG, Zumla A, editors. The granulomatous disorders. Cambridge: Cambridge University Press; 1999. pp. 387–98. [Google Scholar]
- 34.Vorechovsky I, Scott D, Haeney MR, Webster ADB. Chromosomal radiosensitivity in common variable immunodeficiency. Mutation Res. 1993;290:255–64. doi: 10.1016/0027-5107(93)90166-d. [DOI] [PubMed] [Google Scholar]
- 35.Kinlen LJ, Webster ADB, Bird AG, Haile R, Peto J, Soothill JF, Thompson RA. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet. 1985;2:263–6. doi: 10.1016/s0140-6736(85)91037-2. [DOI] [PubMed] [Google Scholar]
- 36.Wegner RD, Chrzanowska KH, Sperling K, Stumm M. Ataxia telangiectasia variants (Nijmegen Breakage Syndrome) In: Ochs HD, Smith CIE, Puck JM, editors. Primary immunodeficiency diseases. A molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 324–34. [Google Scholar]
- 37.Kondratenko I, Amlot PL, Webster ADB, Farrant J. Lack of specific antibody response in common variable immunodeficiency (CVID) associated with failure in production of antigen-specific memory T cells. Clin Exp Immunol. 1997;108:9–13. doi: 10.1046/j.1365-2249.1997.d01-993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spickett GP, Webster ADB, Farrant J. Cellular abnormalities in Common variable immunodeficiency. In: Rosen FS, Seligmann M, editors. Immunodeficiencies. Philadelphia: Harwood Academic Publishers; 1993. pp. 111–26. [Google Scholar]
- 39.Fischer MB, Wolf HM, Hauber I, Eggenbauer H, Thon V, Sasgary M, Eibl MM. Activation via the antigen receptor is impaired in T cells, but not in B cells from patients with common variable immunodeficiency. Eur J Immunol. 1996;26:231–7. doi: 10.1002/eji.1830260136. [DOI] [PubMed] [Google Scholar]
- 40.North ME, Webster ADB, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): abnormalities in intracellular production of interferon-gamma CD28+ (‘cytotoxic’) CD28( (‘suppresor’) CD8+ subsets. Clin Exp Immunol. 1998;111:70–75. doi: 10.1046/j.1365-2249.1998.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iglesias J, Matamoros N, Raga S, Ferrer JM, Mila J. CD95 expression and function on lymphocyte subpopulations in common variable immunodeficiency (CVID); related to increased apoptosis. Clin Exp Immunol. 1999;117:138–46. doi: 10.1046/j.1365-2249.1999.00946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aukrust P, Muller F, Frøland SS. Enhanced generation of reactive oxygen species in monocytes from patients with common variable immunodeficiency. Clin Exp Immunol. 1994;97:232–8. doi: 10.1111/j.1365-2249.1994.tb06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aukrust P, Kristoffersen AK, Müller F, Haug CJ, Espevik T, Frøland SS. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency—possible immunologic and clinical consequences. Blood. 1996;87:674–81. [PubMed] [Google Scholar]
- 44.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-α-polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997;159:6236–41. [PubMed] [Google Scholar]
- 45.Webster ADB, Lever A, Spickett G, Beattie R, North M, Thorpe R. Recovery of antibody production after HIV infection in common variable hypogammaglobulinaemia. Clin Exp Immunol. 1989;77:309–13. [PMC free article] [PubMed] [Google Scholar]
- 46.Felsburg PJ, Glickman LT, Jezyk PF. Selective IgA deficiency in the dog. Clin Immunol Immunopathol. 1985;36:297–305. doi: 10.1016/0090-1229(85)90050-9. [DOI] [PubMed] [Google Scholar]
- 47.Moroff SD, Hurvitz AI, Peterson ME, Saunders L, Noone KE. IgA deficiency in shar-pei dogs. Vet Immunol Immunopathol. 1986;13:181–8. doi: 10.1016/0165-2427(86)90071-1. [DOI] [PubMed] [Google Scholar]
- 48.Glickman LT, Shofer FS, Payton AJ, Laster LL, Felsburg PJ. Survey of serum IgA, IgG, and IgM concentrations in a large beagle population in which IgA deficiency had been identified. Am J Vet Res. 1988;49:1240–5. [PubMed] [Google Scholar]
- 49.Luster MI, Leslie GA, Cole RK. Selective IgA deficiency in chickens with spontaneous autoimmune thyroiditis. Nature. 1976;263:331. doi: 10.1038/263331a0. [DOI] [PubMed] [Google Scholar]
- 50.Hendrickson BA, Conner DA, Ladd DJ, et al. Altered hepatic transport of immunoglobulin A in mice lacking the J chain. J Exp Med. 1995;182:1905–12. doi: 10.1084/jem.182.6.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harriman GR, Bradley A, Das S, Rogers-Fani P, Davis AC. IgA class switch in Iα exon-deficient mice. Role of germline transcription in class switch recombination. J Clin Invest. 1996;97:477–85. doi: 10.1172/JCI118438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendrickson BA, Rindisbacher L, Corthesy B, Kendall D, Waltz DA, Neutra MR, Seidman JG. Lack of association of secretory component with IgA in J chain-deficient mice. J Immunol. 1996;157:750–4. [PubMed] [Google Scholar]
- 53.Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825–8. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- 54.Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, Zhang Y, Mbawuike IN. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J Immunol. 1999;162:2521–9. [PubMed] [Google Scholar]
- 55.Mbawuike IN, Pacheco S, Acuna CL, Switzer KC, Zhang Y, Harriman GR. Mucosal immunity to influenzae without IgA: an IgA knockout mouse model. J Immunol. 1999;162:2530–7. [PubMed] [Google Scholar]
- 56.Blanchard TG, Czinn SJ, Redline RW, Sigmund N, Harriman G, Nedrud JG. Antibody-independent protective mucosal immunity to gastric Helicobacter infection in mice. Cell Immunol. 1999;191:74–80. doi: 10.1006/cimm.1998.1421. [DOI] [PubMed] [Google Scholar]
- 57.Hiroi T, Yanagita M, Iijima H, Iwatani K, Yoshida T, Takatsu K, Kiyono H. Deficiency of IL-5 receptor α-chain selectively influences the development of the common mucosal immune system independent IgA-producing B-1 cell in mucosa associated tissues. J Immunol. 1999;162:821–8. [PubMed] [Google Scholar]
- 58.Banks TA, Rouse BT, Kerley MK, et al. Lymphotoxin-α-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol. 1995;155:1685–93. [PubMed] [Google Scholar]
- 59.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins α and β revealed in lymphotoxin b deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 60.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-kB Rel-A deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–61. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. Cells from p50/NF-kB knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156:183–91. [PubMed] [Google Scholar]
- 62.Islam KB, Baskin B, Christensson B, Hammarström L, Smith CIE. In vivo expression of human immunoglobulin germ-line mRNA in normal and immunodeficient individuals. Clin Exp Immunol. 1994;95:3–9. doi: 10.1111/j.1365-2249.1994.tb06006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Z, Yunis D, Irigoyen M, Kitchens B, Bottaro A, Alt FW, Alper CA. Discordance between IgA switching at the DNA level and IgA expression at the mRNA level in IgA deficient patients. Clin Immunol. 1999;91:263–70. doi: 10.1006/clim.1999.4702. [DOI] [PubMed] [Google Scholar]
- 64.Chapel HM, Webster ADB. Assessment of the immune system. Primary immunodeficiency diseases. In: Ochs HD, Smith CIE, Puck JM, editors. A molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 419–31. [Google Scholar]
- 65.Asherson GL, Webster ADB. Diagnosis and treatment of immunodeficiency diseases. London: Blackwell Scientific Publications; 1980. pp. 78–98. [Google Scholar]
- 66.Truedsson L, Baskin B, Pan Q, Rabbani H, Vorechovsky I, Smith CIE, Hammarström L. Genetics of IgA deficiency. APMIS. 1995;103:833–42. doi: 10.1111/j.1699-0463.1995.tb01442.x. [DOI] [PubMed] [Google Scholar]
- 67.Farr M, Kitas GD, Tuhn EJ, Bacon PA. Immunodeficiencies associated with sulphasalazine therapy in inflammatory arthritis. Br J Rheumatol. 1991;30:413–7. doi: 10.1093/rheumatology/30.6.413. [DOI] [PubMed] [Google Scholar]
- 68.Hammarström L, Smith CIE, Berg U. Captopril-induced IgA deficiency. Lancet. 1991;336:436. doi: 10.1016/0140-6736(91)91220-o. [DOI] [PubMed] [Google Scholar]
- 69.Maeoka Y, Hara T, Dejima S, Takeshita K. IgA and IgG2 deficiency associated with zonisamide therapy: a case report. Epilepsia. 1997;38:611–3. doi: 10.1111/j.1528-1157.1997.tb01147.x. [DOI] [PubMed] [Google Scholar]
- 70.Smith CIE, Hammarström L. Immunologic abnormalities induced by D-penicillamine. In: Dukor P, Kallos P, Schlumberger HD, West GB, editors. Pseudoallergic reactions. Involvement of drugs and chemicals. Vol. 4. Basel: S Karger AG; 1985. pp. 138–80. [Google Scholar]
- 71.Wahl C, Liptay S, Adler G, Schmid RM. Sulfasalazine: a potent inhibitor of nuclear factor kappa B. J Clin Invest. 1998;101:1163–74. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shovlin CL, Simmonds HA, Fairbanks LD, et al. Adult onset immunodeficiency due to inherited adenosine deaminase (ADA) deficiency. J Immunol. 1994;153:2331–9. [PubMed] [Google Scholar]
- 73.Gaspar W, Conley ME. CEI review. 2000 in press. [Google Scholar]
- 74.Conley ME. Autosomal recessive agammaglobulinaemia. In: Smith CIE, Ochs HD, Puck JM, editors. Primary immunodeficiency diseases. A molecular and genetic approach. Oxford: Oxford University Press; 1999. pp. 285–91. [Google Scholar]
- 75.Notarangelo LD, Hayward AR. X-linked immunodeficiency with hyper-IgM: biological and clinical aspects. Clin Exp Immunol. 2000 doi: 10.1046/j.1365-2249.2000.01142.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aittoniemi J, Koskinen S, Laippala P, Laine S, Miettinen A. The significance of IgG subclasses and mannan-binding lectin (MBL) for susceptibility to infection in apparently healthy adults with IgA deficiency. Clin Exp Immunol. 1999;116:505–8. doi: 10.1046/j.1365-2249.1999.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gustafson R, Gardulf A, Granert C, Hansen S, Hammarstrom L. New strategy for prophylactic therapy to patients with ‘selective’ IgA deficiency. Lancet. 1997;350:865. doi: 10.1016/S0140-6736(05)62034-X. [DOI] [PubMed] [Google Scholar]
- 78.Bjorkander J, Blake B, Hanson LA. Primary hypogammaglobulinaemia: impaired lung function and body growth with delayed diagnosis and inadequate treatment. Eur J Resp Dis. 1984;65:529–36. [PubMed] [Google Scholar]
- 79.Samuelson A, Borelli S, Gustafson R, Hammarstrom L, Smith CIE, Jonasson J, Lindberg AA. Characterisation of Haemophilus influenzae isolates from the respiratory tract of patients with primary antibody deficiencies: evidence for persistent colonisation. Scand J Infect Dis. 1995;27:303–13. doi: 10.3109/00365549509032722. [DOI] [PubMed] [Google Scholar]
- 80.Roifman CM, Rao CP, Lederman HM, Lavi S, Quinn P, Gelfand EW. Increased susceptibility to mycoplasma infection in patients with hypogammaglobulinaemia. Am J Med. 1986;80:590–4. doi: 10.1016/0002-9343(86)90812-0. [DOI] [PubMed] [Google Scholar]
- 81.Franz A, Webster ADB, Furr PM, Taylor-Robinson D. Mycoplasmal arthritis in patients with primary immunoglobulin deficiency: clinical features and outcome in 18 patients. Br J Rheumatol. 1997;36:661–8. doi: 10.1093/rheumatology/36.6.661. [DOI] [PubMed] [Google Scholar]
- 82.Rudge P, Webster ADB, Revez T, Warner T, Espanol T, Cunningham-Rundles C, Hyman N. Encephalomyelitis in primary hypogammaglobulinaemia. Brain. 1996;119:1–15. doi: 10.1093/brain/119.1.1. [DOI] [PubMed] [Google Scholar]
- 83.Webster ADB, Rotbart HA, Warner T, Rudge P, Hyman N. Diagnosis of enterovirus brain disease in hypogammaglobulinaemic patients by polymerase chain reaction. Clin Infect Dis. 1993;17:657–61. doi: 10.1093/clinids/17.4.657. [DOI] [PubMed] [Google Scholar]
- 84.Leen CLS, Bath JCJL, Brettle RP, Yap PL. Sarcoidosis and primary hypogammaglobulinaemia: a report of two cases and a review of the literature. Sarcoidosis. 1985;2:91–95. [PubMed] [Google Scholar]
- 85.Smith MS, Webster ADB, Dhillon AP, Dusheiko G, Boulton R, Rolles K, Burroughs AK. Orthotopic liver transplantation for HCV cirrhosis in two patients with common variable hypogammaglobulinaemia. Gastroenterology. 1995;108:879–89. doi: 10.1016/0016-5085(95)90464-6. [DOI] [PubMed] [Google Scholar]
- 86.Teahon K, Webster AD, Price AB, Weston J, Bjarnason I. Studies on the enteropathy associated with primary hypogammaglobulinaemia. Gut. 1994;35:1244–9. doi: 10.1136/gut.35.9.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Webster ADB. Immunodeficiency and gastrointestinal disease. In: Triger DR, editor. Clinical immunology of the liver and gastrointestinal tract. Bristol: John Wright; 1986. pp. 127–49. [Google Scholar]
- 88.Webster ADB, Platts-Mills TAE, Janossy G, Morgan M, Asherson GL. Autoimmune blood dyscrasias in five patients with hypogammaglobulinaemia: response of neutropenia to vincristine. J Clin Immunol. 1981;1:113–8. doi: 10.1007/BF00915388. [DOI] [PubMed] [Google Scholar]
- 89.Sander CA, Medeiros LJ, Weiss LM, Yano T, Sneller MC, Jaffe ES. Lymphoproliferative lesions in patients with common variable immunodeficiency. Am J Surg Pathol. 1992;16:1170–82. doi: 10.1097/00000478-199212000-00004. [DOI] [PubMed] [Google Scholar]
- 90.Chapel H, Brennan V, Delson E. Immunoglobulin replacement therapy by self-infusion at home. Clin Exp Immunol. 1988;73:160–2. [PMC free article] [PubMed] [Google Scholar]
- 91.Gardulf A, Andersen V, Bjorkander J, et al. Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: safety and cost. Lancet. 1995;345:365–9. doi: 10.1016/s0140-6736(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 92.Bjoro K, Skaug T, Haaland T, Froland SS. Long-term outcome of chronic hepatitis C virus infection in primary immunodeficiency. Quart J Med. 1999;92:433–41. doi: 10.1093/qjmed/92.8.433. [DOI] [PubMed] [Google Scholar]
- 93.Kondo N, Takao A, Orii T. Case report: immunoglobulin A deficiency in patients with rheumatoid arthritis treated with aspirin. Biother. 1993;7:59–62. doi: 10.1007/BF01878155. [DOI] [PubMed] [Google Scholar]
- 94.Murphy EA, Morris AJ, Walker E, Lee FD, Sturrock RD. Cyclosporine A induced colitis and acquired selective IgA deficiency in a patient with juvenile chronic arthritis. J Rheumatol. 1993;20:1397–8. [PubMed] [Google Scholar]