

Abstract
The theoretical risk of triggering vasculitis resulting from administration of G-CSF and GM-CSF to patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV), such as Wegener's granulomatosis (WG), who develop agranulocytosis due to cytotoxic therapy, is unknown. Since there is strong evidence that activation of polymorphonuclear neutrophils (PMN) induced by binding of ANCA to PR3 or myeloperoxidase (MPO) expressed on their plasma membrane is involved in the pathogenesis of systemic vasculitides (SV), we studied the surface expression of PR3 and MPO on PMN from healthy donors in response to G-CSF and GM-CSF in vitro by flow cytometric analysis. Increasing doses of G-CSF did not alter PR3 expression on either untreated or tumour necrosis factor-alpha (TNF-α)-primed donor PMN significantly. In contrast, GM-CSF significantly increased PR3 membrane expression on both intact PMN and neutrophils primed with TNF-α. MPO expression was not significantly altered by either G-CSF or GM-CSF. In summary, these data demonstrate that GM-CSF, but not G-CSF, induces plasma membrane expression of PR3 on PMN in vitro. Since in AAV accessibility of the antigen (PR3 or MPO) to the antibody (ANCA) on the plasma membrane of PMN is thought to be essential for neutrophil activation by ANCA, the results of the present study suggest that administration of GM-CSF to patients with WG with neutropenia implies a definite theoretical risk of deterioration of vasculitis via this mechanism.
Keywords: proteinase 3, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, polymorphonuclear neutrophils
INTRODUCTION
Administration of the recombinant protein form of human G-CSF (rhG-CSF) and human GM-CSF (rhGM-CSF) to patients with both congenital and acquired neutropenias is considered a safe and effective way to hasten recovery from neutropenia and curtail infectious complications [1]. rhG-CSF and rhGM-CSF not only stimulate the proliferation and differentiation of haematopoietic precursors, but also modulate functional features like microbicidal killing, chemotaxis or expression of adhesion molecules as well as survival of mature polymorphonuclear neutrophils (PMN) [1–5]. However, individual patients treated with rhG-CSF or rhGM-CSF for severe chronic neutropenia or Felty's syndrome developed apparently treatment-induced cutaneous vasculitis or neutrophilic dermatoses like Sweet's syndrome [6–11]. Since rhGM-CSF and rhG-CSF also modulate various functions of mature neutrophils [1–5], which are considered the primary effector cells in vasculitis, a causal association of growth factor administration and development of vasculitis has been suggested and a cautious use of haematopoietic growth factors in autoimmune diseases has recently been proposed [11].
Since severe infections are common in patients with systemic vasculitides (SV) and are often related to severe neutropenia resulting from cytotoxic therapy [12–16], administration of G-CSF to SV patients with agranulocytosis might be a useful therapeutic strategy. We recently reported the results of a retrospective analysis of patients with Wegener's granulomatosis (WG) treated with low doses of rhG-CSF for cyclophosphamide (CYC)-induced agranulocytosis, showing that recombinant human G-CSF can shorten the duration of CYC-induced severe neutropenia in a clinically significant manner (reduction of infectious complications) without inducing a disease flare [17]. Recent experimental protocols using high-dose CYC as a potential therapy for patients with severe autoimmune disease, including vasculitis, involve the administration of rhG-CSF for stem cell mobilization, which also may cause a flare up of the underlying rheumatic disease [18]. Despite the growing importance of haematopoietic growth factors for the treatment of patients' vasculitides, the specific risk of administration of these powerful agents in view of the pathogenesis and pathophysiology of vasculitides like WG has not been defined.
Anti-neutrophil cytoplasmic antibodies (ANCA), which are directed against granule proteins translocated on the surface of PMN, are thought to play an important pathogenic role in primary small vessel vasculitides via activation of PMN [18–23]. Cytoplasmic (classic) C-ANCA and perinuclear P-ANCA, directed against PR3 or myeloperoxidase (MPO), respectively, are strongly associated with SV like WG (mostly C-ANCA directed against PR3), microscopic polyangiitis (mostly P-ANCA directed against MPO) or Churg–Strauss syndrome (CSS) [19,20]. We and others demonstrated the presence of PR3 and MPO on the plasma membrane of PMN from patients with active WG [21,23,24]. Since certain cytokines like tumour necrosis factor-alpha (TNF-α) and IL-8 can induce a translocation of PR3 from the intragranular loci to the cell surface of PMN [23], we wondered whether G-CSF and/or GM-CSF could also induce the membrane surface expression of PR3 and MPO on PMN. In this case, GM-CSF/G-CSF administration to patients with ANCA-associated vasculitides would indeed bear a definite risk of exacerbating vasculitis by binding of G-CSF/GM-CSF-induced PR3 to C-ANCA, as leucocyte membrane expression of PR3 correlates with disease activity in patients with WG [24].
Thus, we studied the in vitro effect of rhG-CSF and rhGM-CSF on the surface expression of PR3 and MPO on neutrophils.
MATERIALS AND METHODS
Volunteers
Blood samples were obtained from nine healthy donors (four males, five females, mean age 36·0 ± 5·1 years (range 22–58 years)). Donors were working in departments of the Forschungsinstitut Borstel other than the laboratory where the in vitro stimulation experiments where performed. None of the donors had contact with PR3, to avoid contamination. There was no evidence of an inflammatory or malignant disorder in any of the subjects. None of the donors took any medication.
Cytokines and monoclonal antibodies
For in vitro studies rhTNF-α, rhG-CSF, and rhGM-CSF purchased from TEBU GmbH (Frankfurt, Germany) were used. MoAbs against PR3 (designated WGM1 and WGM2) and MPO were raised in our laboratory as previously described [25].
Preparation of human PMN
Heparinized blood samples (20 U heparin/ml blood) were diluted 1:2 with Hanks' balanced salt solution (HBSS) and the peripheral blood mononuclear cells (PBMC) were separated in a first step from erythrocytes and polymorphonuclear granulocytes (PMN) by Ficoll–Hypaque density gradient centrifugation. Then the PMN-containing erythrocyte sediments were mixed with two volumes of polyvinylalcohol. The PMN-containing supernatant was collected after sedimentation for 20 min. Contaminating erythrocytes were removed by hypotonic lysis. Cell purity was >98% as determined by Pappenheim staining and microscopical examination at ×630 magnification using oil immersion. PMN viability was >95% as determined by trypan blue exclusion. All media and buffers used for the neutrophil separation were free from endotoxin in order to avoid any contamination which might affect PR3 expression.
In vitro stimulation experiments
PMN freshly isolated as described above were resuspended in PBS with Ca2+ and Mg2+ at a final concentration of 2 × 106 cells/ml. The cell suspension was then incubated with supraphysiological doses of rhG-CSF (0·03, 0·1, 0·3, 1 and 4 ng/ml), rhGM-CSF (0·03, 0·1, 0·3, 1 and 4 ng/ml), TNF-α (1, 3 and 10 ng/ml) or buffer in a shaking water bath at 37°C for 10 min. The concentrations of the haematopoietic growth factors used in all experiments were chosen according to the range of serum levels of rhG-CSF/rhGM-CSF observed during treatment with these cytokines [26]. To assess the effect of these cytokines on preactivated PMN, the cell suspension was preincubated with TNF-α (1 ng/ml) as the priming agent for 10 min. Subsequently, G-CSF (1 and 4 ng/ml), GM-CSF (1 and 4 ng/ml), or buffer alone were added and the incubation was continued for a further 10 min at 37°C. After rapid cooling to 4°C with excess of ice-cold PBS the cells were centrifuged at 300 g for 10 min and the cells were used for subsequent immunofluorescence staining and flow cytometric analysis.
Flow cytometric analysis
Quantitative flow cytometric analysis of surface antigens was performed as previously described [27]. Cytokine-treated or control PMN (1 × 106/ml) were incubated with MoAbs against PR3 and MPO, respectively, or with isotype control antibodies for 20 min. After washing, the cells were incubated with the second antibody (dichlorotriazinyl aminofluorescein (DTAF)-conjugated F(ab)2 goat anti-mouse IgGFc or DTAF-conjugated F(ab)2 goat anti-mouse IgMFc; Dianova, Hamburg, Germany). The labelled cells were washed and then fixed with PBS containing 1·5% paraformaldehyde. In order to minimize any activation of cells all procedures were performed on ice. The flow cytometric analysis was performed within the subsequent 24 h using a Cytofluorograf System 50 (Ortho Diagnostic Systems, Raritan, NJ). The expression of PR3 is given as the percentage of PMN with fluorescence intensities above the threshold fluorescence determined as the upper level of fluorescence intensity of PMN treated with isotype control antibodies. In addition, median fluorescence intensities are shown.
Statistical analysis
Data are expressed as mean ± s.e.m. The Kolmogorov–Smirnov Z-test was used to analyse the distribution of the samples. The t-test for paired samples was used to compare values between two groups. P < 0·05 was considered significant. Statistical analysis was performed using SPSS for Windows, version 7.5 (SPSS Inc., Chicago, IL).
RESULTS
Flow cytometric analysis revealed native membrane expression of PR3 on 18·0 ± 3·5% of untreated human neutrophils (Fig. 1). Addition of supraphysiological (therapeutic) concentrations of rhG-CSF (0·03–4 ng/ml) to untreated PMN in vitro did not alter membrane expression of PR3 significantly (P > 0·2) (Fig. 1a, Table 1). In contrast, a higher percentage of PR3-expressing PMN was found in response to GM-CSF, with a statistically significant difference at a GM-CSF dose of 1 ng/ml (18·0 ± 3·5% versus 22·2 ± 4·9%; P = 0·049) (Fig. 1a, Table 1). Dose–response analyses demonstrated no significant stimulating effect of either G-CSF or GM-CSF at doses lower than 1 ng/ml (Fig. 2). Incubation of PMN in the presence of increasing doses of TNF-α (1–10 ng/ml) caused a dose-dependent increase of total PR3 surface expression (Table 2). After treatment with TNF-α at a dose of 10 ng/ml the percentage of PMN expressing PR3 (52·3 ± 2·2%) was significantly higher than after stimulation with G-CSF at doses of 1 ng/ml (18·8 ± 4·2%; P = 0·005) and 4 ng/ml (17·18 ± 2·4%; P = 0·016), and GM-CSF at a dose of 1 ng/ml (22·2 ± 4·9%; P = 0·001) and 4 mg/ml (22·6 ± 2·6%; P = 0·001), each without prior priming with TNF-α (Fig. 1a).
Fig. 1.
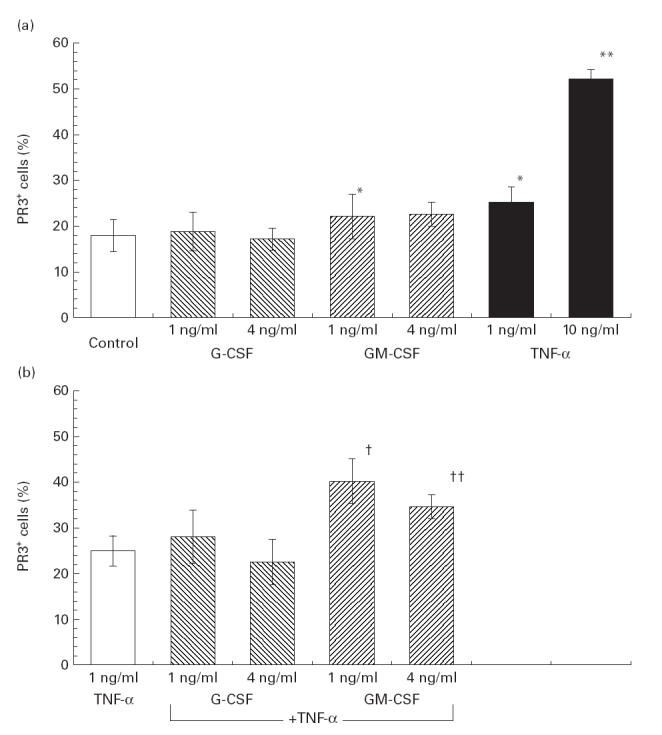
Flow cytometric analysis of the membrane expression of PR3 on polymorphonuclear neutrophils (PMN) derived from healthy donors. Cells were incubated in the presence of increasing doses of tumour necrosis factor-alpha (TNF-α), G-CSF and GM-CSF as described in Materials and Methods. (a) Unstimulated intact PMN. (b) Primed PMN, re-incubated with TNF-α (1 ng/ml) for 10 min. Antigen expression is given as the percentage of total PMN with fluorescence intensities above the threshold fluorescence determined as the upper level of fluorescence intensity of PMN treated with the isotype control antibodies. Results represent means ±s.e.m. of nine individual experiments, by using cells from nine different donors. Statistically significant difference of *P < 0·05 or **P < 0·005 versus control PMN (unstimulated). Statistically significant difference of †P < 0·05 or ††P < 0·001 versus PMN primed with TNF-α (1 ng/ml for 10 min) alone.
Table 1.
Flow cytofluorometric analysis of the membrane expression of PR3 on polymorphonuclear neutrophils (PMN) from healthy donors after incubation with G-CSF and GM-CSF in vitro
| Percentage of PMN expressing PR3 | |||
|---|---|---|---|
| Condition (concentration of cytokine) | G-CSF | GM-CSF | P |
| Basal (1 ng/ml) | 18·0 ± 3·5 | 22·2 ± 4·9 | 0·003* |
| Basal (4 ng/ml) | 17·18 ± 1·9 | 22·7 ± 2·6 | 0·04* |
| Primed (1 ng/ml) | 28·1 ± 5·2 | 40·2 ± 7·5 | 0·004* |
| Primed (4 ng/ml) | 22·6 ± 2·8 | 34·7 ± 3·8 | 0·001* |
PMN were incubated with TNF-α in increasing concentrations ranging from 1 to 10 ng/ml for 10 min. Subsequently, PMN (1 × 106/ml) were incubated with MoAbs to PR3 (WGM2) for 20 min. After washing, the second antibody (fluorescein (DTAF)-conjugated F(ab)2 goat anti-mouse (IgG) was added. Subsequently the labelled cells were fixed with PBS containing 1·5% paraformaldehyde. The surface expression of PR3 is given as the percentage of PMN showing PR3 expression on flow cytometric analysis. Data are means ±s.e.m. from nine experiments using cells from nine different donors.
Statistically significant difference between stimulated (TNF-α) and unstimulated PMN (basal).
Fig. 2.
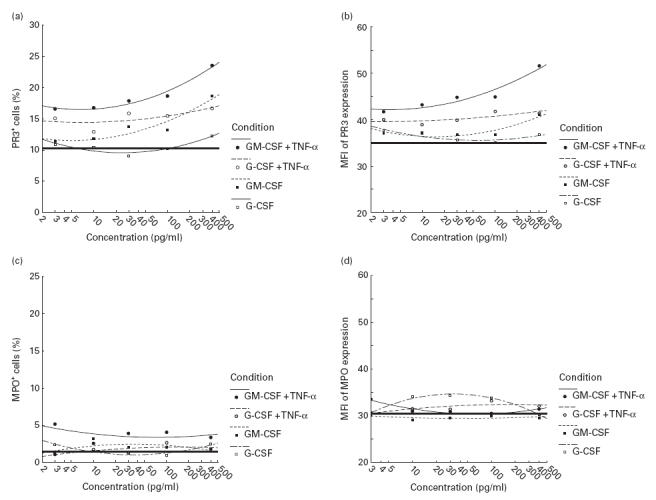
Flow cytometric analysis of the membrane expression of PR3 and myeloperoxidase (MPO) on polymorphonuclear neutrophils (PMN) derived from three healthy donors. Both intact and tumour necrosis factor-alpha (TNF-α)-primed (1 ng/ml for 10 min) PMN were incubated in the presence of increasing doses (0·03–4 ng/ml, respectively) of G-CSF and GM-CSF as described in Materials and Methods. Antigen expression is given as the percentage of total PMN (a,c) with fluorescence intensities above the threshold fluorescence determined as the upper level of fluorescence intensity of PMN treated with the isotype control antibodies, and the median fluorescence intensity (MFI) (b,d). The solid horizontal line represents the basal level of PR3 or MPO expression without any stimulation. Results represent means ±s.e.m. of three individual experiments, by using cells from three different donors.
Table 2.
Flow cytofluorometric analysis of the membrane expression of PR3 on polymorphonuclear neutrophils (PMN) from healthy donors after incubation with tumour necrosis factor-alpha (TNF-α) in vitro
| Condition (concentration of TNF-α) | Percentage of PMN expressing PR3 | P |
|---|---|---|
| Basal | 18·0 ± 3·5 | – |
| TNF-α (1 ng/ml) | 25·3 ± 3·3 | 0·044* |
| TNF-α (3 ng/ml) | 36·0 ± 0·8 | 0·014* |
| TNF-α (10 ng/ml) | 52·3 ± 2·2 | 0·002* |
PMN were incubated with TNF- a in increasing concentrations ranging from 1 to 10 ng/ml for 10 min. Subsequently, PMN (1 × 106/ml) were incubated with MoAbs to PR3 (WGM2) for 20 min. After washing, second antibody (fluorescein (DTAF)-conjugated F(ab)2 goat anti-mouse (IgG) was added. Subsequently the labelled cells were fixed with PBS containing 1·5% paraformaldehyde. The surface expression of PR3 given as the percentage of PMN showing PR3 expression on flow cytometric analysis. Data are means ± s.e.m. from nine experiments using cells from nine different donors.
Statistically significant difference between stimulated (TNF-α) and unstimulated PMN (basal).
To assess the effect of rhG-CSF and rhGM-CSF on activated neutrophils PMN were pretreated with TNF-α (1 ng/ml) as the priming agent. After pretreatment with TNF-α, incubation of the primed PMN with rhG-CSF (1 ng/ml) did not alter PR3 expression compared with PMN treated with the same concentration of TNF-α only (28·1 ± 5·2% versus 25·3 ± 3·3%; P > 0·2). Higher doses of G-CSF (4 ng/ml) showed no additional effect (22·6 ± 2·8% versus 25·3 ± 1·4%; P = 0·1) (Fig. 1b). In contrast, rhGM-CSF significantly increased PR3 membrane expression on activated PMN, resulting in 40·3 ± 7·5% (1 ng/ml GM-CSF; P = 0·04) and 34·7 ± 3·8% (4 ng/ml GM-CSF; P = 0·001) PR3+ cells compared with 25·3 ± 3·3 PR3+ cells after stimulation with TNF-α alone (Fig. 1b). Direct comparison between the stimulatory effect of both haematopoietic cytokines on PR3 expression on primed PMN demonstrated a significantly higher percentage of PR3+ cells in response to GM-CSF compared with G-CSF at both 1 and 4 ng/ml of each cytokine, respectively (Table 1). In contrast to the observations made for TNF-α, higher doses of rhG-CSF (up to 4 ng/ml), and rhGM-CSF (up to 4 ng/ml) showed no additional stimulating effect on membrane expression of PR3 on either native or primed PMN (Fig. 1). Dose–response analyses revealed also that with G-CSF and GM-CSF doses <1 ng/ml significant stimulation of PR3 expression on primed PMN could not be achieved (Fig. 2). Analysis of the median fluorescence intensity of PR3 expression (Fig. 2b) demonstrated that the higher total PR3 expression was mainly a result of an increased subset of PR-expressing PMN (% PR3+ PMN) than a result of enhanced surface expression on individual cells.
Surface expression of MPO was found in <4% of untreated PMN. Addition of G-CSF and GM-CSF at five different doses (0·03–4 ng/ml) did not result in significant stimulation of MPO expression (percentage of MPO-expressing cells and median fluorescence intensity), either in native PMN or in TNF-α-primed neutrophils (Fig. 2c,d).
DISCUSSION
ANCA-induced activation of primed neutrophils is thought to be the initiating event in the development of vasculitis in WG [19,21–25,28–30, ]. We previously reported that certain cytokines like TNF-α can induce a translocation of PR3 to the plasma membrane of PMN [23]. Other cytokines, like IL-8 and transforming growth factor-beta (TGF-β), which alone do not increase PR3 expression, can stimulate PR3 expression significantly if PMN are primed [23,31]. The results of the present study demonstrate a divergent effect of the two haematopoietic growth factors G-CSF and GM-CSF on PR3 expression in vitro. While even high concentrations of rhG-CSF given as single agent or after priming with TNF-α did not alter the expression of PR3 on the plasma membrane of PMN in vitro, rhGM-CSF, but not rhG-CSF, induced significant expression of PR3 on plasma membranes of both intact and preactivated PMN primed with TNF-α. In agreement with our observation, it has been reported that the subset of PMN expressing PMN on their surface membrane largely varies among healthy probands, but stays relatively stable over time in each individual [21]. The results of the present study indicate that GM-CSF can shift this bimodal expression pattern of PR3 towards a higher percentage of PMN expressing PR3 which may become possible targets of ANCA-induced neutrophil activation. The reasons for the differential effect of rhGM-CSF and rhG-CSF on PR3 surface expression on neutrophils are unclear. Despite recent progress regarding structure and function of the human PR3 genome and protein [32–34], the factors modulating production, packaging and surface expression on PMN are poorly understood. Interestingly, the same divergent effect of G-CSF and GM-CSF for PR3 expression on neutrophils observed in the present study has been reported for the production of MPO, another target antigen of ANCA [35]. Treweeke et al. not only demonstrated a five-fold increase in fMLP-induced MPO production in PMN in response to GM-CSF, which was not seen in the presence of G-CSF, but also found several other functions of mature PMN including chemiluminescence and O2− production which were stimulated by GM-CSF, but not by G-CSF [35]. However, expression of MPO on plasma membranes of PMN was found not to be influenced by either G-CSF or GM-CSF in the present study. The different modes of stimulation (fMLP versus TNF-α) and that the fact that increased MPO production not necessarily implies increased surface expression may explain the different findings of our study and that of Treweeke. While GM-CSF and G-CSF share many common properties regarding the production and function of PMN, several differential effects of these haematopoietic growth factors on mature PMN have been shown in vitro and in vivo, which have been extensively reviewed recently [2]. Different signal transduction mechanisms of GM-CSF (via tyrosine kinase) and G-CSF (involvement of G-proteins) have been demonstrated in PMN and may possibly account for the different functional pattern of the two factors [36].
Our results are of clinical relevance in view of recent observations describing an increased incidence of cutaneous vasculitides in patients treated with rhG-CSF or rhGM-CSF for severe neutropenia due to different disorders, including autoimmune neutropenia due to Felty's syndrome [6–11]. Thus, the question has been raised whether administration of rhG-CSF or rhGM-CSF to patients with SV for neutropenia due to cytotoxic medication like CYC might possibly trigger the inflammatory process and aggravate the vasculitic disease [11]. For rhGM-CSF, our data indicate a substantially higher theoretical risk of exacerbating the vasculitis when given to patients with active C-ANCA-associated vasculitides. In the present study we used (TNF-α) primed PMN, which are also found in active C-ANCA-associated vasculitides, where activated PMN as well as increased levels of proinflammatory cytokines like TNF-α or TGF-β are found [31,37]. In view of the general limitation that information gathered from in vitro studies is not always correlated with results obtained during administration of the drug in vivo, the results of the present study indicate that rhGM-CSF given to WG patients with drug-induced neutropenia is likely to increase the relative and absolute amount of PMN expressing PR3 on their plasma membrane. Since in C-ANCA-associated vasculitides accessibility of the antigen (PR3) to the antibody (ANCA) on the plasma membrane of PMN is thought to be essential for neutrophil activation by ANCA, the results of the present study suggest that deterioration of vasculitis appears possible, at least via this mechanism in patients with WG receiving rhGM-CSF. In contrast, our data make a clinically relevant C-ANCA-mediated neutrophil activation induced by rhG-CSF less likely. However, the short incubation times chosen for the present in vitro experiments may not necessarily reflect the in vivo situation where the concentrations of haematopoietic growth factors are elevated for a longer time during therapeutic administration. The data of the present study support the results of a recent clinical study showing that administration of rhG-CSF to WG patients with CYC-induced agranulocytosis was not associated with an increase of clinical and laboratory chemical signs of disease activity during up to 6 months of follow up [17].
However, it has to be pointed out that our data do not exclude the possibility that rhG-CSF given to patients with C-ANCA-associated vasculitides in vivo might aggravate vasculitic disease by raising the neutrophil count far above normal or by modulating the function of mature PMN via other mechanisms. Clinical observations as well as data from comprehensive in vitro and in vivo studies investigating the influence of rhG-CSF and rhGM-CSF on mature neutrophils suggest than the first rather than the latter might be the case [6,38–43, ]. Mechanisms whereby rhG-CSF and GM-CSF theoretically could modulate neutrophil function in WG apart from direct stimulation of PR3 expression include direct stimulation of neutrophil function as well as indirect mechanisms such as release of proinflammatory mediators.
First, expression of Fcγ receptor IIIb (CD16), which is involved in initiation of ANCA-induced neutrophil activation [41], has been shown to be markedly decreased after administration of rhG-CSF to neutropenic patients and healthy volunteers [2], suggesting inhibition rather than stimulation of ANCA-induced neutrophil activation via this mechanism. However, stimulation of ANCA-induced neutrophil activation by other mechanisms remains possible and merits further exploration. Second, a reduced expression of leucocyte adhesion molecule-1 (LAM-1) [37] and a reduction of directed migration and chemotaxis of PMN from healthy donors given rhG-CSF in vivo [2,39] argue against a significant promoting effect of rhG-CSF on leucocyte adherence and migration during the initial phase in the development of necrotizing vasculitis. Third, G-CSF induces a shift towards an anti-inflammatory cytokine response (reduced secretion of TNF-α and interferon-gamma (IFN-γ), increased expression of IL-8, IL-10 and soluble TNF-α receptors), making an indirect impact of G-CSF on proinflammatory properties of PMN from patients receiving G-CSF less likely [39,40]. Finally, G-CSF or GM-CSF alone is unable to initiate a respiratory burst in native PMN [2]. Recent in vivo studies however demonstrated that following administration of G-CSF in vivo superoxide anion production in response to several physiological stimuli can be different and depends on the stimulus used, resulting in enhanced (fMLP), normal (opsonized zymosan) or reduced (phorbol myristate acetate, platelet-activating factor/fMLP) respiratory burst activity [38]. Numerous in vitro studies describe many other pro- and anti-inflammatory capabilities of G-CSF [1–5], but data derived from several recent studies in healthy volunteers investigating the effect of G-CSF on functional parameters of mature PMN in vivo indicate that G-CSF administration imparts only modest effects on neutrophil function in vivo [38–40].
In conclusion, these data demonstrate that rhGM-CSF, but not rhG-CSF, induces plasma membrane expression of PR3 on activated PMN in vitro. Thus, patients with ANCA-associated vasculitis receiving GM-CSF instead of rhG-CSF for drug-induced agranulocytosis may be at higher risk of exacerbating vasculitic disease by increased binding of ANCA to PR3 and subsequent neutrophil activation. In view of the data shown here and our clinical experience reported earlier [17], we recommend the use of short courses of low-dose rhG-CSF in severely neutropenic patients with C-ANCA-associated vasculitides (if clinically indicated) or for progenitor cell rescue in experimental protocols of stem cell transplantation, until greater experience with the use of haematopoietic growth factors in vasculitic disorders is available.
REFERENCES
- 1.Welte K, Gabrilove J, Bronchud MH, Platzer E, Morstyn G. Filgrastim (r-metHuG-CSF): the first 10 years. Blood. 1996;88:1907–29. [PubMed] [Google Scholar]
- 2.Spiekermann K, Rosler J, Emmendorfer A, Elsner J, Welte K. Functional features of neutrophils induced by G-CSF and GM-CSF treatment: differential effects and clinical implications. Leukemia. 1997;11:466–78. doi: 10.1038/sj.leu.2400607. [DOI] [PubMed] [Google Scholar]
- 3.Frampton JE, Lee CR, Faulds D. Filgrastim. A review of its pharmacological properties and therapeutic efficacy in neutropenia. Drugs. 1994;48:731–60. doi: 10.2165/00003495-199448050-00007. [DOI] [PubMed] [Google Scholar]
- 4.Anderlini P, Przepiorka D, Champlin R, Körbling M. Biological and clinical effects of granulocyte colony-stimulating factor in normal individuals. Blood. 1996;88:2819–25. [PubMed] [Google Scholar]
- 5.Lieschke GJ, Burgess AW. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor (first of two parts) N Engl J Med. 1991;327:28–35. doi: 10.1056/NEJM199207023270106. [DOI] [PubMed] [Google Scholar]
- 6.Jain KK. Cutaneous vasculitis associated with granulocyte colony-stimulating factor. J Am Acad Dermatol. 1994;31:213–5. doi: 10.1016/s0190-9622(94)70149-0. [DOI] [PubMed] [Google Scholar]
- 7.Johnson MM, Grimwood CR. Leukocyte colony stimulating factors. A review of associated dermatoses and vasculitides. Arch Dermatol. 1994;130:77–81. doi: 10.1001/archderm.130.1.77. [DOI] [PubMed] [Google Scholar]
- 8.Hellmich B, Schnabel A, Gross WL. Treatment of severe neutropenia due to Felty's syndrome and systemic lupus erythematosus with granulocyte colony-stimulating factor. Semin Arthritis Rheum. 1999;29:82–99. doi: 10.1016/s0049-0172(99)80040-7. [DOI] [PubMed] [Google Scholar]
- 9.Viddarson B, Geirsson AJ, Önundarson PT. Reactivation of rheumatoid arthritis and development of leukocytoclastic vasculitis in a patient receiving granulocyte colony-stimulating factor for Felty's syndrome. Am J Med. 1995;98:589–91. doi: 10.1016/s0002-9343(99)80019-9. [DOI] [PubMed] [Google Scholar]
- 10.Dreicer R, Schiller JH, Carbone PP, Mueller W, Odenwald E, Souza L, Riehm H. Granulocyte-macrophage colony stimulating factor and vasculitis (letter) Ann Intern Med. 1989;111:91–92. doi: 10.7326/0003-4819-111-1-91_2. [DOI] [PubMed] [Google Scholar]
- 11.Merkel PA. Drugs associated with vasculitis. Curr Opin Rheumatol. 1998;10:45–50. doi: 10.1097/00002281-199801000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, Rottem M, Fauci AS. Wegener's granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488–99. doi: 10.7326/0003-4819-116-6-488. [DOI] [PubMed] [Google Scholar]
- 13.Schmitt WH, Gross WL. Vasculitis in the seriously ill patient: diagnostic approaches and therapeutic options in ANCA-associated vasculitis. Kidney Int. 1998;53(S64):39–44. [PubMed] [Google Scholar]
- 14.Hoffmann GS. Treatment of Wegener's granulomatosis: time to change the standard of care? Arthritis Rheum. 1997;40:2099–104. doi: 10.1002/art.1780401202. [DOI] [PubMed] [Google Scholar]
- 15.Guillevin L, Cordier J-F, Lohte F, et al. A prospective, multicenter randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener's granulomatosis. Arthritis Rheum. 1997;40:2187–98. doi: 10.1002/art.1780401213. [DOI] [PubMed] [Google Scholar]
- 16.Bradley JD, Brandt KD, Katz BP. Infectious complications of cyclophosphamide treatment for vasculitis. Arthritis Rheum. 1989;32:45–53. doi: 10.1002/anr.1780320108. [DOI] [PubMed] [Google Scholar]
- 17.Hellmich B, Schnabel A, Gross WL. Granulocyte colony-stimulating factor treatment for cyclophosphamide-induced severe neutropenia in Wegener's granulomatosis. Arthritis Rheum. 1999;42:1752–6. doi: 10.1002/1529-0131(199908)42:8<1752::AID-ANR26>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 18.Snowden JA, Brooks PM. Hematopoetic stem cell transplantation in rheumatic diseases. Curr Opin Rheumatol. 1999;11:167–72. doi: 10.1097/00002281-199905000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clin Exp Immunol. 1993;91:1–2. doi: 10.1111/j.1365-2249.1993.tb03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Woude FJ, Rasmussen N, Lobatto S, et al. Autoantibodies against neutrophils and monocytes: tools for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet. 1985;i:425–9. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 21.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Letters. 1995;374:29–33. doi: 10.1016/0014-5793(95)01073-n. [DOI] [PubMed] [Google Scholar]
- 22.Keogan MT, Esnault VLM, Green AJ, Lockwood CM, Brown DL. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. 1992;90:228. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csernok E, Ernst M, Schmitt WH, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller Kobold AC, Kallenberg CG, Tervaert JW. Leukocyte membrane expression of proteinase 3 correlates with disease activity in patients with Wegener's granulomatosis. Br J Rheumatol. 1998;37:901–7. doi: 10.1093/rheumatology/37.8.901. [DOI] [PubMed] [Google Scholar]
- 25.Csernok E, Lüdemann J, Gross WL, Bainton DF. Ultrastructural localization of proteinase 3, the target of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am J Pathol. 1990;137:1113. [PMC free article] [PubMed] [Google Scholar]
- 26.Takatani H, Soda H, Fukuda M, et al. Levels of recombinant human granulocyte colony stimulating factor in serum are inversely correlated with circulating neutrophil counts. Antimicrob Agents Chemother. 1996;40:988–91. doi: 10.1128/aac.40.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst M, Kern P, Flad H, Ulmer AJ. Effects of systemic in vivo interleukin-2 (IL-2) reconstitution in patients with acquired immune deficiency syndrome (AIDS) and AIDS-related complex (ARC) on phenotypes and functions of peripheral blood mononuclear cells (PBMC) J Clin Imunol. 1986;6:170–81. doi: 10.1007/BF00918750. [DOI] [PubMed] [Google Scholar]
- 28.Kallenberg CGM, Heeringa P, Tervaert JWC. Are antineutrophil cytoplasmic antibodies pathogenic in Wegener's granulomatosis? Lessons from in vitro and in vivo experimental findings. In: Shoenfeld Y, editor. The decade of autoimmunity. Amsterdam: Elsevier; 1999. pp. 227–34. [Google Scholar]
- 29.Jenette CJ, Falk RJ. Pathogenesis of the vascular and glomerular damage in ANCA-positive vasculitis. Nephrol Dial Transplant. 1998;13(S1):16–20. doi: 10.1093/ndt/13.suppl_1.16. [DOI] [PubMed] [Google Scholar]
- 30.Gross WL, Trabandt A, Csernok E. Pathogenesis of Wegener's granulomatosis. Ann Intern Med (Paris) 1998;149:280–6. [PubMed] [Google Scholar]
- 31.Csernok E, Szymkowiak CH, Mistry N, Daha MR, Gross WL, Kekow J. Transforming growth factor-beta (TGF-beta) expression and interaction with proteinase 3 (PR3) in anti-neutrophil cytoplasmic antibody (ANCA)-associated vascultitis. Clin Exp Immunol. 1996;105:104–11. doi: 10.1046/j.1365-2249.1996.d01-715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szymkowiak CH, Johnston TW, Csernok E, Gross WL. Expression of the human autoantigen of Wegener's granulomatosis (PR3) in a baculovirus expression system. Biochem Biophys Res Commun. 1996;219:283–9. doi: 10.1006/bbrc.1996.0224. [DOI] [PubMed] [Google Scholar]
- 33.Specks U, Fass DN, Fautsch MP, Hummel AM, Viss MA. Recombinant human proteinase 3, the Wegener's autoantigen, expressed in HMC-1 cells is enzymatically active and recognized by c-ANCA. FEBS Letters. 1996;390:265–70. doi: 10.1016/0014-5793(96)00669-2. [DOI] [PubMed] [Google Scholar]
- 34.Jenne DE. Structure of the azurocidin, proteinase 3, and neutrophil elastase genes. Implications for inflammation and vasculitis. Am J Respir Crit Care Med. 1994;150:S147–54. doi: 10.1164/ajrccm/150.6_Pt_2.S147. [DOI] [PubMed] [Google Scholar]
- 35.Treweeke AT, Aziz KA, Zuzel M. The role of G-CSF in mature neutrophil function is not related to GM-CSF-type cell priming. J Leuk Biol. 1994;55:612–6. doi: 10.1002/jlb.55.5.612. [DOI] [PubMed] [Google Scholar]
- 36.Balazovich KJ, Almeida HI, Boxer LA. Recombinant human G-CSF and GM-CSF prime human neutrophils for superoxide production through different signal transduction mechanisms. J Lab Clin Med. 1991;118:576–84. [PubMed] [Google Scholar]
- 37.Kekow J, Szymkowiak CH, Gross WL. Involvement of cytokines in granuloma formation within primary systemic vasculitis. In: Romagnani S, editor. Cytokines: basic principles and clinical applications. New York: Raven Press; 1992. pp. 341–8. [Google Scholar]
- 38.Leavey PJ, Sellins KS, Thurman G, et al. In vivo treatment with granulocyte colony stimulating factor results in divergent effects on neutrophil functions measured in vitro. Blood. 1998;92:4366–74. [PubMed] [Google Scholar]
- 39.Pollmächer T, Korth C, Mullington J, et al. Effects of granulocyte colony-stimulating factor on plasma cytokine and cytokine receptor levels and on the in vivo host response to endotoxin in healthy men. Blood. 1996;86:900–5. [PubMed] [Google Scholar]
- 40.Hartung T, Döcke WD, Gantner F, et al. Effect of granulocyte colony stimulating factor treatment on ex vivo blood cytokine response in human volunteers. Blood. 1995;85:2482–9. [PubMed] [Google Scholar]
- 41.Kocher M, Edberg JC, Fleit HB, Kimberly RP. Antineutrophil cytoplasmic antibodies preferentially engage Fc gamma RIIIb on human neutrophils. J Immunol. 1998;161:6909–14. [PubMed] [Google Scholar]
- 42.Ohsaka A, Saionjii K, Sato N, et al. Granulocyte colony stimulating factor downregulates the surface expression of human leukocyte adhesion molecule 1 on human neutrophils in vitro and in vivo. Br J Haematol. 1993;84:574–80. doi: 10.1111/j.1365-2141.1993.tb03130.x. [DOI] [PubMed] [Google Scholar]
- 43.Yong KL, Linch DC. Differential effects of granulocyte- and granulocyte-macrophage colony-stimulating factors (G- and GM-CSF) on neutrophil adhesion in vitro and in vivo. Eur J Haematol. 1991;49:251–9. doi: 10.1111/j.1600-0609.1992.tb00057.x. [DOI] [PubMed] [Google Scholar]