

Abstract
Extracellular calreticulin (CRT) as well as anti-CRT antibodies have been reported in patients with various autoimmune disorders and CRT has been implicated in ‘epitope spreading’ to other autoantigens such as the Ro/SS-A complex. In addition, antibodies against parasite forms of the endoplasmic reticulum chaperone, CRT, have been found in patients suffering from onchocerciasis and schistosomiasis. In this study, we screened sera for anti-CRT antibodies from patients with active and inactive systemic lupus ertythematosus (SLE) and primary or secondary Sjögren's syndrome. Approximately 40% of all SLE patients were positive for anti-CRT antibodies. The antigenic regions of CRT were determined using full length CRT and fragments of CRT prepared in yeast and Escherichia coli, respectively. Synthetic 15mer peptides corresponding to the major autoantigenic region of CRT (amino acids 1–289), each one overlapping by 12 amino acids, were used to map the B cell epitopes on the CRT protein recognized by autoimmune sera. Major antigenic epitopes were found to be associated with the N-terminal half of the protein in 69% of the SLE sera from active disease patients, while the C-domain was not antigenic. Major epitopes were found to be reactive with antibodies in sera from SLE patients with both active and inactive disease, spanning different regions of the N and P-domains. Sera from both healthy and disease controls and primary Sjögren's syndrome patients were non-reactive to these sequences. Limited proteolysis of CRT with two major leucocyte serine proteases, elastase and cathepsin G, demonstrated that an N-terminal region of CRT is resistant to digestion. Interestingly, some of the epitopes with the highest reactivity belong to the fragments of the protein which bind to C1q and inhibit complement activation. Whether C1q association with CRT is a pathological or protective interaction between these two proteins is currently under investigation.
Keywords: systemic lupus erythematosus, calreticulin, epitope, autoimmune disease
INTRODUCTION
Autoantibodies against calreticulin (CRT) are present in the sera of a number of autoimmune conditions including systemic lupus erythematosus (SLE), Sjögren's syndrome (SS) [1–4], coeliac disease [5] and congenital heart block [6]. Anti-CRT antibodies are also found in the human hosts of a number of parasitic diseases, such as malaria [7], onchocerciasis [8], schistosomiasis [9] and tick infections [10]. The amino acid sequence identity between the parasite forms of CRT and human CRT is approximately 60–70%, which might point to a cross-reactive immune response. The possible pathological roles of CRT in autoimmunity have recently been reviewed [11], in which CRT appears to associate physically with a number of other autoantigens, including peptides of the Ro/SS-A complex and human cytoplasmic RNAs (hY RNAs). In particular, CRT can facilitate the binding of the 60-kD polypeptide component of the Ro/SS-A RNP (Ro60) to hY RNA. In addition, CRT and the 52-kD Ro/SS-A polypeptide (Ro52) appear to be capable of interacting through direct protein–protein binding in vivo[12]. Calreticulin is implicated in the phenomenon of ‘epitope spreading’, in which initiation of immunity to either Ro52 or Ro60 can lead to reciprocal spreading of autoimmunity to Ro60 or Ro52, respectively, and induce the production of anti-CRT autoantibodies in some strains of mice [13].
Protein sequences of many animal and plant forms of CRT are known, but the structure and fine specificity of the regions of these sequences which act as B cell epitopes are not well defined. Calreticulin has arbitrarily been divided into three domains, N- (amino acids 1–180); P- (amino acids 181–290) and C- (amino acids 291–400). In previous studies a number of antigenic regions of CRT have been proposed. Lieu et al. [14] found that a single peptide corresponding to residues 7–24 of the human CRT sequence contained an epitope reactive to a number of SLE sera and suggested the N-terminal region of CRT contains a major autoantigenic region. Lieu also identified other antigenic epitopes spanning the N-domain and P-domain of CRT [15].
Systemic autoimmune diseases evoke an immune response to a diverse, yet selective group of intracellular antigens of which CRT is a member. The initiating stimulus for diseases such as SLE has been a focus of attention by a number of researchers. The study of cells undergoing apoptosis has revealed that endoplasmic reticulum (ER) blebs contain high concentrations of CRT and other target antigens. Calreticulin is known to form complexes with other proteins leading to epitope spreading and immune intolerance, possibly leading to the initiation of a primary immune response against CRT and its associated molecules. It is therefore important to determine the specific regions of CRT that induce a primary immune response in autoimmune patients and investigate the stability of the molecule's structure in an inflammatory environment. In this study, by using 15mer overlapping synthetic peptides, we have determined the primary structure of the antigenic epitopes present in the N- and P-domains of CRT which are recognized by antibodies in SLE, and in primary and secondary SS patient sera. We have also assessed if more frequent reactions with certain epitopes are associated with specific clinical manifestations and disease subsets. Finally, CRT may be released into the extracellular environment from cells undergoing necrosis and apoptosis [16], therefore the ability of CRT to form aggregates and resist proteolytic digestion by inflammatory proteases was also examined.
PATIENTS AND METHODS
Patient sera
Sixteen sera from patients with active SLE, 18 with inactive SLE, four with primary Sjögren's syndrome (pSS), and nine with secondary Sjögren's syndrome (sSS) were examined. All patients with SLE fulfilled the American College of Rheumatology revised criteria for the classification of SLE [17]. Active SLE was defined as a SLE Disease Activity Index (SLEDAI) score >3 according to Petri et al. [18]. All patients fulfilled four or more of the classification criteria for SS proposed by the European Community Study Group in 1993 [19], and those with sSS were initially diagnosed with SLE. Ten sera with primary anti-phospholipid syndrome (APS) that fulfilled the criteria of Harris [20], but that were negative for antinuclear antibodies, were used as a disease control group. In addition, 20 normal sera were collected from healthy blood bank donors, 14 females and four males between the ages of 20 and 35 years.
CRT protein and peptide expression, purification and synthesis
Construction of CRT expression vector for the Pichia system
The cDNA of full length rabbit CRT, without a signal sequence, was obtained by restriction endonuclease digestion of pGB-CRT [21] with EcoRI and gel purification of the resulting DNA fragment. The Pichia expression plasmid, pPIC-9, was digested with EcoRI. The CRT DNA fragment was then ligated into the prepared pPIC-9 plasmid to generate CRT-pPIC. The presence of CRT cDNA and its orientation in the plasmid was determined by endonuclease digestion. All sequences were confirmed by DNA sequencing. CRT-pPIC was transformed into the KM71 strain of Pichia. Spheroplasts were prepared according the protocol outlined by the manufacturer (Invitrogen, San Diego, CA). Secreted protein was purified by DEAE–Sepharose column chromatography and ion exchange chromatography using a Resource Q FPLC column (Pharmacia, Milton Keynes, UK). Pure CRT was pooled and concentrated.
Expression of N- and P-domain
A maltose-binding protein (MBP) fusion system was used consisting of the pMAL-c2 plasmid transformed in E. coli to express and isolate recombinant forms of the N-terminal domain (N-domain; amino acids 1–181) and proline-rich domain (P-domain; amino acids 182–292), as previously described [22].
Purification of native CRT
Human native CRT was isolated and purified from U937 tissue culture cells grown in vitro in RPMI 1640 medium (Gibco, Paisley, UK) supplemented with 2 mm l-glutamine (Sigma, Poole, UK), 10% (v/v) fetal calf serum (FCS; TCS Botolph Claydon, Oxford, UK), penicillin (100 μg/ml) and kanamycin (100 μg/ml). Briefly, U937 cells were centrifuged and resuspended in PBS (without Ca2+ or Mg2+) protease inhibitor cocktail tablets (Boehringer Mannheim, Lewes, UK) for 1 h on ice. After washing, the cells were suspended in relaxation buffer (100 mm KCl, 3 mm NaCl, 1 mm ATP (Na)2, 3·5 mm MgC12, 10 mm PIPES) and placed in a Bioneb cell disrupter (ThermoMetric Ltd, Northwich, UK), which was used according to the manufacturer's instructions. Aliquots of cells (2 × 109/ml; 5 ml in total) were passed through the nebulizer three times to obtain an efficient subcellular suspension containing a mixture of microsomal, plasma membrane, ER, Golgi and nuclei subfractions. This suspension was centrifuged at 1400 g for 10 min and the resulting pellet containing unbroken cells, nuclei and cell debris was discarded. The supernatant was further centrifuged at 15 000 g for 30 min. The resulting supernatant enriched in microsomal fractions was carefully collected and centrifuged at 70 000 g for 45 min to obtain a microsomal pellet. Approximately 700 μl of the microsomal fraction (containing 70 mg protein) were mixed with 5 ml lysis buffer (0·1 m NaCl, 0·02 m Tris, 0·005 m CaCl2, 0·005 m MgC12 and 1% v/v Nonident P-40, supplemented with protease cocktail inhibitors (Boehringer), and mixed on a rotary mixer at 4°C for 1 h in Eppendorf tubes. The lysate was then subjected to centrifugation at 10 000 g for 30 min at 4°C. The lysate was then dialysed overnight at 4°C in 10 mm sodium phosphate buffer pH 7·4, containing 0·2 mm EDTA. Then the sample was passed through a Sephadex G25 column equilibrated in sodium phosphate buffer and the protein fractions collected. The pooled protein fractions were then fractionated on a Mono Q FPLC column (Pharmacia) equilibrated with 10 mm sodium phosphate buffer pH 7·4, containing 0·2 mm EDTA. The protein fractions were eluted with a linear gradient (40 ml) of 0–1 m NaCl in sodium phosphate buffer. CRT-enriched fractions normally eluted at approximately 470 mm NaCl. The pool containing CRT was concentrated by ultracentrifugation and the concentrated sample applied to a Superose-12 (Pharmacia) FPLC gel-filtration column (300 × 10 mm diameter). Gel filtration was carried out in 50 mm sodium phosphate buffer pH 7·4 containing 2 mm EDTA, 150 mm NaCl.
Peptide synthesis
A series of 90 peptides, 15 amino acid residues long and overlapping by 12 residues, spanning the whole N- and P-domains of CRT, was synthesized using Fmoc-based solid-phase peptide synthesis as described [23].
ELISA
ELISAs for antibodies against CRT
A solid-phase indirect ELISA was used to detect binding of autoantibodies against full-length native and recombinant human CRT. Briefly, Nunc Polysorb microtitre plate wells were coated overnight with 100 μl/well of CRT, fusion proteins MBP-N, P or C domain CRT (1–5 μg/ml) or irrelevant control antigens, bovine serum albumin (BSA) and mannan binding lectin in sodium carbonate buffer (0·015 m Na2CO3/0·035 m NaHCO3) pH 9·6. Synthetic CRT 15mer peptides (50 μg in 50 μl water) were coated by air drying onto microtest flexible 96-well plates overnight at 25°C. Unoccupied absorption sites were blocked by 2 h incubation at 37°C with a 5% (w/v) powdered milk/glycine blocking solution and then the plates were washed three times with PBS containing 0·05% v/v Tween 20 (PBS–T). Serum samples were diluted 1:50 in PBS–T pH 7·4 and 50-μl volumes were then added to the microtitre wells in triplicate and incubated at 37°C for 2 h. The wells were washed and incubated with 100 μl of peroxidase-conjugated anti-human IgG (1:5000; Sigma) for 1 h at 37°C. After washing three times, 50 μl of 3,3′,5,5′,-tetra-methyl-benzidine (TMB; BioRad, Hemel Hempstead, UK) were added to each well, plates were incubated for 20 min at 21°C, and the enzyme reaction was terminated by addition of 150 μl of 2 n H2SO4. The optical density (OD) of the wells was read at 450 nm using a TITERTEK ELISA plate reader.
Competitive ELISA assays were performed as described above with one exception. During the blocking buffer incubation step, human sera from SLE and SS patients positive for anti-CRT were preincubated with 1 μg/ml native CRT (final concentration in diluted serum) before their addition to the solid-phase CRT or control antigen plates.
Peptide competitive assay
In order to investigate if the synthetic epitope analogues could inhibit binding to full-length CRT, sera from active SLE patients (7193, 10357 and 10681) selected for high anti-CRT activity against single, triple or multiple epitope sites were diluted at a ratio of 1:250 (giving 50% of the maximal OD (ranging from 0·6 to 0·1)), and were incubated for 3 h at 37°C with a mixture of the autoreactive peptides (concentrations ranging from 0·01 to 5 μg/ml). The mixtures were centrifuged for 5 min at 10 000 g and the supernatants transferred into microtitre plates precoated with BSA or CRT. ELISA run in triplicate measured the level of binding of anti-CRT. Percentage of inhibition (I) was calculated from the formula I (%) = 100 − 100 × o/O, where o corresponds to the OD of the peptide incubated sample and O to the OD of the positive control (without peptide).
Limited proteolytic digestion of CRT
Fixed amounts of CRT (15 μg) in 45 μl of Tris–HCl containing 2 mm Ca2+ were digested with either 0·75 μg and 0·05 μg of elastase (activity 625 U/mg) or cathepsin G (activity 120 U/mg) for 2 h at 37°C at pH 6·4 or pH 7·4. The CRT:enzyme ratios were 20:1 and 320:1 w/w. The reaction was stopped by the addition of an equal volume of SDS-loading PAGE buffer. SDS–PAGE was carried out using 15% (w/v) acrylamide gels under reducing conditions and stained by coomassie blue. To confirm that the protein expressed was the N-terminal region of CRT, an Edman degradation was carried out using an Applied Biosystems 470A protein sequencer and on-line Applied Biosystems 120A analysis of the amino acid derivatives.
Statistical analysis
During initial screening procedures, the OD of each clinical sample was expressed as: positive OD ≥ (OD sample−OD background)/(mean OD + 2 s.d. of normal sera). In accordance with this formula, an OD450 nm of 0·21 was the cut-off point for each assay. Statistical differences between patients and normal individuals were calculated using the non-parametric t-test. More stringent critera were applied to ELISAs using synthetic peptides, where the cut-off point of positivity was calculated as the mean OD of 10 autoimmune disease control sera +6 s.d. The sensitivity of the ELISA with the synthetic peptides was calculated as follows: sensitivity (%) = (number of positive samples × 100)/total samples.
RESULTS
Antigenicity of the domains of CRT
An initial ELISA screen of sera from 57 patients with autoimmune disorders and 20 healthy controls was carried out for autoantibodies against native human CRT (Fig. 1). Forty-four percent of patients with active SLE, 33% of patients with inactive SLE and 33% with sSS were found positive for anti-CRT antibodies against native CRT. None of the normal sera or sera from patients with sSS and primary APS (ANA−) had elevated anti-CRT antibody titres. The same patient sera were re-screened using a full-length yeast recombinant form of rabbit CRT. Similar results were obtained (Table 1), but overall, lower numbers of patients in all clinical groups had antibodies against the recombinant form of the protein.
Fig. 1.
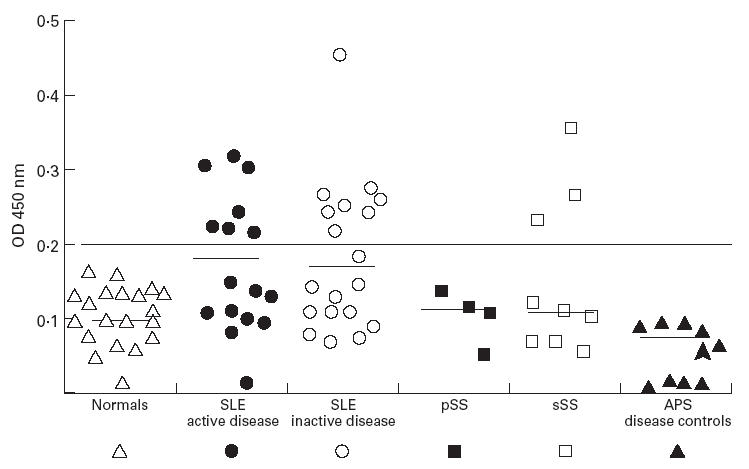
Prevalence of antibodies to human full-length calreticulin (CRT) in sera from patients with active and inactive systemic lupus erythematosus (SLE), primary Sjögren's syndrome (pSS), secondary Sjögren's syndrome (sSS) and anti-phospholipid syndrome without anti-nuclear antibodies (disease controls) and normal controls. The cut-off point was calculated using the mean +2 s.d. of 20 normal human sera. Solid bar represents the mean optical density (OD) for each patient group.
Table 1.
Prevalence of raised titres of autoantibodies (> mean +2 s.d. of healthy controls) to full-length and N- and P-domains of calreticulin (CRT) in sera from patients with active and inactive forms of systemic lupus erythematosus (SLE), patients with primary Sjögren's syndrome (pSS), secondary Sjögren's syndrome (sSS) and disease control patients (positive for anti-phospholipid antibodies but negative for anti-La/SSB antibodies)
| Protein on plate—250 ng/well | ||||
|---|---|---|---|---|
| Patient group | Recombinant CRT | Native CRT | N-domain CRT | P-domain CRT |
| SLE, active | 6/16 (38%) | 7/16 (44%) | 11/16 (69%) | 2/16 (12·5%) |
| SLE, inactive | 4/18 (22·3%) | 6/18 (33%) | 2/18 (11%) | 1/18 (5·5%) |
| pSS | 0/4 (0·0%) | 0/4 (0·0%) | 0/4 (0·0%) | 0/4 |
| sSS | 1/9 (11%) | 3/9 (33%) | 3/9 (33%) | 0/9 |
| Disease controls | Not done | 0/10 (0·0%) | 3/10 (33%) | 0/10 |
| Mean OD ± 2 s.d. | ||||
| for 10 healthy controls | 0·367 | 0·20 | 0·177 | 0·022 |
To ensure that the sera identified as having anti-CRT antibodies were not false positives due to the presence of rheumatoid factor, or some other non-specific reactant, each positive serum was preincubated with 100 μl of a 1-μg/ml human CRT. As expected, CRT in the fluid phase competed for specific anti-CRT binding with the solid-phase bound material, resulting in a lowering of the OD obtained (Fig. 2).
Fig. 2.
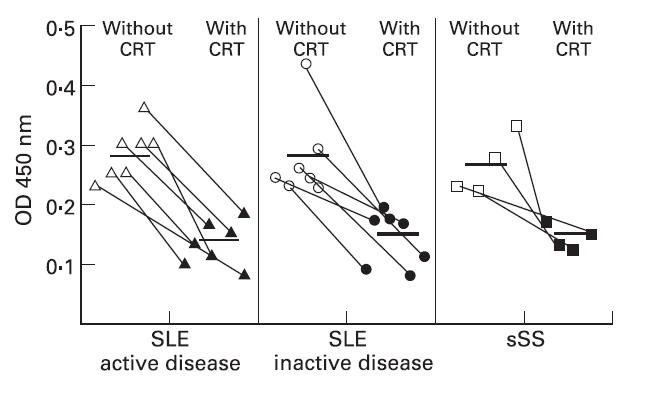
Competitive inhibition of anti-calreticulin (CRT) antibodies in sera from systemic lupus erythematosus (SLE) and secondary Sjögren's syndrome (sSS) patients from binding to immobilized CRT in the presence of 100 ng of free fluid-phase CRT. Solid bar represents the mean optical density (OD) for each patient group.
As outlined in Table 1, approximately one third of sera from both active and inactive SLE patients contained antibodies against native calreticulin. However, over two thirds of sera from active SLE patients contained antibodies against the N-domain fragment of CRT. This result was unexpected, as it indicated that more sera from active SLE patients are positive against the domain fragments of CRT than the whole intact molecule. Gel filtration analysis of the N-domain of CRT indicates that the fragment is unfolded, and therefore the apparent increase in immunogenicity may to due to the exposure of ‘cryptic’ epitopes not normally revealed in the folded whole molecule. The P-domain was only weakly antigenic, with only 5% of all patient sera appearing to possess anti-P-domain antibodies. None of the sera tested had antibodies against the C-domain (data not shown) and they were not considered in further studies presented.
Identification of antigenic determinants
Ninety 15mer peptides, each overlapping by 12 contiguous residues and covering the full sequence of the N- and P-domain of CRT (amino acids 1–290) were prepared and bound to ELISA plates. All solid-phase peptides were tested against 1:50 diluted sera from all autoimmune patients earlier found to have anti-CRT antibodies. Ten primary APS disease and healthy control groups were also examined. Multiple antigenic recognition sites were recognized by antibodies from patients with active or inactive SLE and sSS (Fig. 3). Antibodies from patient sera with either active or inactive SLE bound several of the 15 epitopes located in the first 280 amino acids of CRT, while sera from other patients bound fewer peptides. Antibodies to peptide 48 – KDIRKCKDDEFTHLYT (142–156) were found in 6/16 (sensitivity = 37·5%) of SLE sera with anti-CRT activity. Antibodies to peptide 65 – DPDASKPEDWDERAK (193–207) were detected in 11/16 (sensitivity = 69%) of the SLE sera. Peptide 85 – EYKGEWKPRQIDNPD (253–267) was also a target of antigenicity in 56 of the anti-CRT SLE+ sera. Peptide 90 – YKGTWIHPEIDNPEY (268–282) was recognized by 10/16 (sensitivity = 63%) SLE sera and by all three patients with sSS, positive for anti-CRT antibodies. None of the disease or healthy control sera (n = 20) reacted with any of these or the other 86 15mer peptides tested.
Fig. 3.
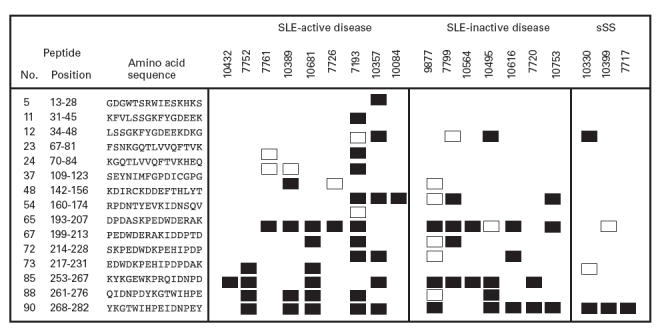
Prevalence of antibodies as detected by ELISA to multiple antigenic calreticulin (CRT) synthetic peptides in sera from patients with active and inactive systemic lupus erythematosus (SLE), as well as secondary Sjögren's syndrome (sSS). Ten healthy and 10 autoimmune disease control sera were used as negative controls. The mean optical density (OD at 450 nm) of samples which were ≥mean +6 s.d. (□) and mean +≥ 8 s.d. (▪), higher than disease control Ods, is presented. Peptides with which no reactions were found are omitted from the figure.
One of the SLE sera (7193) reacted with 11 of the 15 antigenic peptides identified from the 90 CRT peptides screened, 8/16 SLE sera reacted with between four and eight peptides, 3/16 SLE sera reacted with three peptides. The remaining patient sera reacted against only one or two peptides. None of the reactive peptides acted as a marker of active and inactive SLE in patients. The specificity of the epitopes recognized by antibodies from specific individuals was investigated by inhibition experiments. Three patients A, B and C whose sera reacted with one, three and five synthetic peptides, respectively, were incubated with mixtures of the appropriate peptides (concentrations ranging from 0·125 to 25 μg/ml) and the degree of inhibition of binding to CRT was assessed. As shown in Fig. 4, a dose-dependent inhibition of anti-CRT antibody binding to native CRT was achieved using the reactive epitope analogues.
Fig. 4.
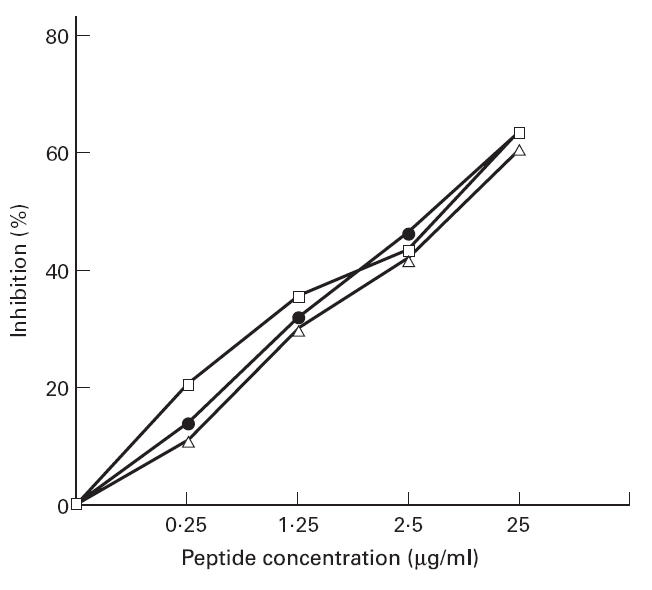
Inhibition of anti-calreticulin (CRT) antibody binding to immobilized native CRT using as inhibitors different concentrations of synthetic peptides of CRT. Synthetic peptides were selected which were autoreactive for individual patients, sera from three patients were diluted 1:250 and mixed with the appropriate peptides (final concentrations 25 μg/ml): patient A (□, peptide 48); patient B (•, peptides 65, 67, 73, 85, 88 and 90); and patient C (Δ, peptides 23, 24 and 26).
The four most antigenic epitope regions recognized in this study by antibodies from five or more SLE patients in relation to the whole protein sequence are presented (bold) in Table 2, together with antigenic regions previously identified by other workers (italics). These were compared with predicted antigenic amino acid residues (underlined) based on hydrophilic accessible sites by the method of Parker et al. [24].
Table 2.
Amino acid sequence of human calreticulin (CRT) showing the previously inferred autoantigenic sites and sites reactive in this study (bold) to anti-CRT antibodies from systemic lupus erythematosus (SLE) and secondary Sjögren's (sSS) sera
| N-domain | |||||
| 1 | EPAVYFKEQF | LDGDGWTSRW | IESK HKSDFG | KFVLSSGKFY | 40 |
| 41 | GDEEKDKGLQ | TSQDARFYAL | SASFEPFSNK | GQTLVVQFTV | 80 |
| 81 | KHEQNIDCGG | GYVKLFPNSL | DQTDMHGDSE | YNIMFGPDIC | 120 |
| 121 | GPGTKKVHVI | FNYKGKNVLI | NKDIRCKDDE | FTHLYTLIVR | 160 |
| 161 | PDNTYEVKID | NSQVESGSLE | 180 | ||
| P-domain | |||||
| 181 | DDW DFLPPKK | IKDPDASKPE | DWDERAKIDD | PTDSKPEDWD | 220 |
| 221 | KPEHIPDPDA | KKPEDWDEEM | DGEWEPPVIQ | NPEYKGEWKP | 260 |
| 261 | RQIDNPDYKG | TWIHPEIDNP | EYSPDPSIYA | 290 | |
| C-domain | |||||
| 291 | YDNFGVLGLD | LWQVKSGTIF | DNFLITNDEA | YAEEFGNETW | 330 |
| 331 | GVTKAAEKQM | KDKQDEEQRL | KEEEEDKKRK | EEEEAEDKED | 370 |
| 371 | DEDKDEDEED | EEDKEEDEEE | DVPGQAKDEL | 400 | |
Enzymatic resistant fragments of CRT
In an attempt to understand why antibodies are made against the N domain of CRT a number of physical properties of CRT were examined. Calreticulin, once released from cells at a site of inflammation, would be susceptible to proteolytic attack by leucocyte secretory enzymes such as cathepsin G and elastase. Full-length CRT was therefore treated with different amounts of these inflammatory enzymes for 2 h and then the fragments identified on a SDS–PAGE gel. Treatment with either enzyme, at the highest protein:enzyme ratio tested (20:1) generated 20–30-kD fragments of CRT that were resistant to proteolytic digestion (Fig. 5). N-terminal sequencing confirmed that these fragments were portions of the N-terminal half of CRT. The N-terminal sequence of other smaller fragments analysed began with the sequence KIDD (amino acids 207–210), suggesting that the 25-kD fragment might be approximately 206 amino acids in length and represent the sequence of the protein covered by the first 65 15mer overlapping peptides. This part of the protein encompasses nine out of the 15 reactive epitopes (60%) to anti-CRT antibodies.
Fig. 5.
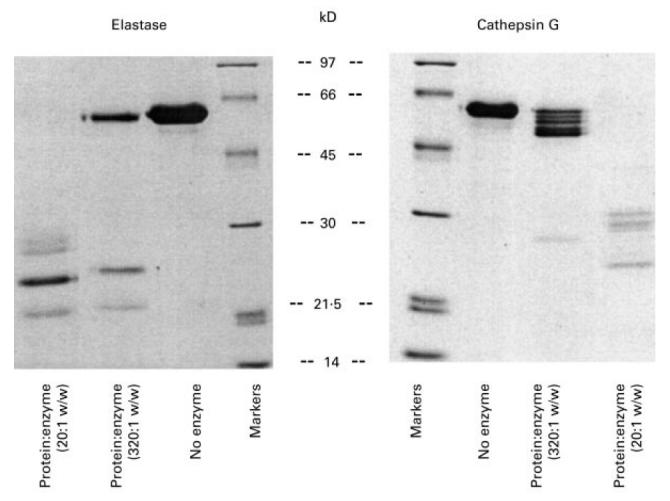
SDS–PAGE analysis of limited proteolytic treatment of full-length calreticulin (CRT) with elastase and cathepsin G after 24 h, demonstrating the existence of proteolytic resistant 20–30-kD N-terminal fragments of CRT.
DISCUSSION
A number of independent investigations have demonstrated that the full-length native [1,25] and recombinant N- and P-domains of CRT contain antigenic sites for antibodies from patients with unclassified SLE disease [2]. It has previously been reported that the first 24 amino acids (1–24) and the last 10 amino acids (170–180) of the N-domain of CRT elicit an immunogenic response in patients with autoimmune diseases [15,26], which suggests that a number of antigenic epitopes may exist. In this study, the linear antigenic epitopes of CRT in relation to clinically defined groups of patients were defined for the first time. Patients were selected for linear epitope mapping whose sera were initially positive for anti-CRT antibodies against the native protein, and in some cases SLE sera which were positive against CRT as judged by Western blotting [2] were used. Anti-CRT antibodies appeared to bind specifically to the immobilized CRT, as the raised ELISA readings were not due to the presence of non-specific rheumatoid factor. This was confirmed with the preincubation of fluid-phase CRT with sera from SLE and sSS containing anti-CRT antibodies that inhibited the anti-CRT antibodies binding to immobilized CRT.
Another consideration is the conformation of the protein sequence recognized by anti-CRT antibodies. Clearly, the use of linear peptides has limitations, as some of the antibodies reactive for the native molecule may recognize conformational epitopes on the surface of the molecule. Nevertheless, the majority of anti-CRT+ sera had antibodies which bound to one or more linear peptides. It is plausible that some of the antigenic epitopes recognized do assume a linear-like conformation of the surface of CRT. This will be known once the structure of CRT and other dominant autoantigens is resolved. Confirmation of reactivity against linear epitopes that are exposed on the surface of the molecule came from competitive inhibition assays. The binding of autoantibodies to native CRT could be blocked by at least 60% when sera mapped for reactivity for specific epitopes were incubated with the appropriate synthetic peptides. Antibodies against CRT are present in SLE patient sera whether they have active or inactive disease symptoms. Secondary, but not primary SS patients were identified with anti-CRT antibodies, reflecting the fact that SS patients with a history of SLE-like symptoms are more prone to develop anti-CRT antibodies than those with pSS.
Many autoantigens are intracellular proteins, and this is true of CRT which is primarily located in the lumen of the ER, where it functions in Ca2+ storage, signalling and protein processing [27]. The mechanisms of antigen release from intracellular organelles of cells are poorly understood. It is now known that during apoptosis, nucleosomes containing DNA and histones, as well as ER vesicles containing CRT, Ro and La proteins bleb off from the surface of dying cells [4,28]. Release of CRT from apoptotic blebs or from cells undergoing necrosis at a site of inflammation would expose CRT to proteolytic enzymes secreted from leucocytes. In addition, CRT would also be exposed to the serum proteins with which it is known to interact, such as the first component of complement C1q [2,23].
To characterize further the effect of proteolytic attack on CRT, limited digestion of full-length recombinant CRT by elastase and cathepsin G was carried out. We conclude that the N-terminal half of the protein has a highly organized, three-dimensional structure, as both enzymes failed to cleave this region of the protein, leaving a 25-kD intact fragment. Analysis of internally cleaved fragments by N-terminal amino acid sequencing indicated that the C-terminal end of the proteolytic-resistant domain lay between amino acids 205 and 215. This is in agreement with the predicted molecular weight of the 25-kD fragment. Two thirds of the antigenic epitopes mapped here are located in this region of the protein. There are three cysteines in the proteolytic-resistant region of CRT, two form a disulphide bond, while the third is free, but one would expect the free cysteine to be buried deep within the molecule to prevent it forming disulphide bonds with other proteins. The importance of the single disulphide bond in maintaining the structure being recognized or disguised by antibodies is uncertain. However, some SLE reactive sera recognized the Escherichia coli expressed N-domain of CRT, yet failed to react against the native protein. We know from our gel filtration studies that the N-domain of CRT (amino acids 1–180), expressed as an MBP-fusion protein, is not correctly folded. The N-domain forms high molecular weight aggregates under non-reducing conditions (data not shown). This suggests that antibodies recognize neoepitopes, which are not normally exposed in the correctly folded protein. The ability of the N-domain fragment to form aggregates with itself and interact with other autoantigens such as Ro52, Ro60 and C1q may facilitate epitope spreading between the host proteins [13]. Consequently, correctly folded protein partially digested by leucocyte proteases may alter the conformation of the N-domain and expose the free cysteine, leading to greater antigenicity and formation of aggregates. The greater antigenicity of incorrectly folded E. coli expressed N-domain of CRT is of concern given the recent interest in using this domain as an anticancer drug [29].
During the phenomenon of ‘epitope spreading’, inoculation of mice with the whole proteins, such as Ro60, results in the generation of antibodies against other common autoantigens such as La, Ro52 and CRT. It now appears that the whole molecule is not always required to elicit such a response. Specific self-peptides of SLE-related autoantigens can induce a diverse autoantibody response. For example, Ro60 self-peptides can generate antibodies against Ro52 and certain Sm and La peptides [30]. Since there is apparently very little structural homology between these various antigens, intermolecular determinant spreading may be due to the generation of subpopulations of antibodies to conformational epitopes shared amongst these autoantigens. Additional evidence in favour of this notion comes from analysis of autoantibodies against ribosomal-P proteins purified from the sera of lupus patients. Anti-ribosomal P protein antibodies bind to SmD and SmB/B′[31]. Whether proteolytic resistant calreticulin peptides drive T cell and B cell responses in a similar way requires further work. However, there do appear to be a number of immunogenic peptide regions within the N-terminal half of CRT which warrant further investigation, especially in light of examples where structurally similar epitopes on both CRT and gliadin cross-react with antigliadin antibodies found in the sera of patients with ceoliac disease [32].
In conclusion, multiple non-adjacent regions of the primary sequence of CRT are antigenic to sera from both clinically active and inactive SLE patients. The N-terminal half of the protein contains most of the antigenic sites and is also resistant to proteolytic degradation by leucocyte serine proteases. The same region of the molecule contains many of the CRT–protein and CRT–RNA binding sites [11,33], which might explain in part the diversification of the autoimmune response (epitope spreading) towards self components originating from intracellular and extracelluar locations. Work is now in progress to determine if there is a correlation between autoimmune sera positive for anti-CRT antibodies and other antigens known to interact with CRT such as Ro52 and C1q.
Acknowledgments
This work was supported by a grant (E0521) from the Arthritis Research Campaign of Great Britain (P.E. and K.B.M.R). P.E. is an ARC Research Fellow.
REFERENCES
- 1.Boehm J, Orth T, Van Nguyen P, et al. Systemic lupus erythematosus is associated with increased auto-antibody titres against calreticulin and grp94, but calreticulin is not the Ro/SS-A antigen. Eur J Clin Invest. 1994;24:248–57. doi: 10.1111/j.1365-2362.1994.tb01082.x. [DOI] [PubMed] [Google Scholar]
- 2.Kishore U, Sontheimer RD, Sastry KN, et al. The systemic lupus erythematosus (SLE) disease autoantigen-calreticulin can inhibit C1q association with immune complexes. Clin Exp Immunol. 1997;108:181–90. doi: 10.1046/j.1365-2249.1997.3761273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-Chetrit E. The molecular basis of the SSA/Ro antigens and the clinical significance of their autoantibodies. Br J Rheumatol. 1993;32:396–402. doi: 10.1093/rheumatology/32.5.396. [DOI] [PubMed] [Google Scholar]
- 4.Eggleton P, Reid KB, Kishore U, et al. Clinical relevance of calreticulin in systemic lupus erythematosus. Lupus. 1997;6:564–71. doi: 10.1177/096120339700600703. [DOI] [PubMed] [Google Scholar]
- 5.Karska K, Tuckova L, Steiner L, et al. Calreticulin—the potential autoantigen in celiac disease. Biochem Biophys Res Commun. 1995;209:597–605. doi: 10.1006/bbrc.1995.1542. [DOI] [PubMed] [Google Scholar]
- 6.Orth T, Dorner T, Meyer Zum Buschenfelde KH, et al. Complete congenital heart block is associated with increased autoantibody titres against calreticulin. Eur J Clin Invest. 1996;26:205–15. doi: 10.1046/j.1365-2362.1996.120270.x. [DOI] [PubMed] [Google Scholar]
- 7.Bonnefoy S, Attal G, Langsley G, et al. Molecular characterization of the heat shock protein 90 gene of the human malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 1994;67:157–70. doi: 10.1016/0166-6851(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 8.Rokeach LA, Zimmerman PA, Unnasch TR. Epitopes of the Onchocerca volvulus RAL1 antigen, a member of the calreticulin family of proteins, recognized by sera from patients with onchocerciasis. Infect Immun. 1994;62:3696–704. doi: 10.1128/iai.62.9.3696-3704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khalife J, Trottein F, Schacht AM, et al. Cloning of the gene encoding a Schistosoma mansoni antigen homologous to human Ro/SS-A autoantigen. Mol Biochem Parasitol. 1993;57:193–202. doi: 10.1016/0166-6851(93)90195-4. [DOI] [PubMed] [Google Scholar]
- 10.Sanders ML, Jaworski DC, Sanchez JL, et al. Antibody to a cDNA-derived calreticulin protein from Amblyomma americanum as a biomarker of tick exposure in humans. Am J Trop Med Hyg. 1998;59:279–85. doi: 10.4269/ajtmh.1998.59.279. [DOI] [PubMed] [Google Scholar]
- 11.Eggleton P, Llewellyn D. Pathophysiological roles of calreticulin in autoimmune disease. Scand J Immunol. 1999;49:466–73. doi: 10.1046/j.1365-3083.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- 12.Cheng ST, Nguyen TQ, Yang YS, et al. Calreticulin binds hYRNA and the 52-kDa polypeptide component of the Ro/SS-A ribonucleoprotein autoantigen. J Immunol. 1996;156:4484–91. [PubMed] [Google Scholar]
- 13.Kinoshita G, Keech CL, Sontheimer RD, et al. Spreading of the immune response from 52 kDaRo and 60 kDaRo to calreticulin in experimental autoimmunity. Lupus. 1998;7:7–11. doi: 10.1191/096120398678919606. [DOI] [PubMed] [Google Scholar]
- 14.Lieu TS, Newkirk MM, Capra JD, et al. Molecular characterization of human Ro/SS-A antigen. Amino terminal sequence of the protein moiety of human Ro/SS-A antigen and immunological activity of a corresponding synthetic peptide. J Clin Invest. 1988;82:96–101. doi: 10.1172/JCI113607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieu TS, Newkirk MM, Arnett FC, et al. A major autoepitope is present on the amino terminus of a human SS-A/Ro polypeptide. J Autoimmun. 1989;2:367–74. doi: 10.1016/0896-8411(89)90165-0. [DOI] [PubMed] [Google Scholar]
- 16.Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6:6–12. doi: 10.1038/sj.cdd.4400460. [DOI] [PubMed] [Google Scholar]
- 17.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 18.Petri M, Genovese M, Engle E, et al. Clinical definition, incidence, and clinical description of flare in systemic lupus erythematosus. Arthritis Rheum. 1991;34:937–44. doi: 10.1002/art.1780340802. [DOI] [PubMed] [Google Scholar]
- 19.Vitali C, Bombardieri S, Moutsopoulos HM, et al. Preliminary criteria for the classification of Sjogren's syndrome. Results of a prospective concerted action supported by the European Community. Arthritis Rheum. 1993;36:340–7. doi: 10.1002/art.1780360309. [DOI] [PubMed] [Google Scholar]
- 20.Harris EN. Antiphospholipid antibodies. Br J Haematol. 1990;74:1–9. doi: 10.1111/j.1365-2141.1990.tb02530.x. [DOI] [PubMed] [Google Scholar]
- 21.Baksh S, Burns K, Andrin C, et al. Interaction of calreticulin with protein disulphide isomerase. J Biol Chem. 1995;270:31338–44. doi: 10.1074/jbc.270.52.31338. [DOI] [PubMed] [Google Scholar]
- 22.Kishore U, Sontheimer RD, Sastry KN, et al. Release of calreticulin from neutrophils may alter C1q-mediated immune functions. Biochem J. 1997;322:543–50. doi: 10.1042/bj3220543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovacs H, Campbell ID, Strong P, et al. Evidence that C1q binds specifically to CH2-like immunoglobulin gamma motifs present in the autoantigen calreticulin and interferes with complement activation. Biochemistry. 1998;37:17865–74. doi: 10.1021/bi973197p. [DOI] [PubMed] [Google Scholar]
- 24.Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425–32. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 25.Sontheimer RD, Lieu TS, McCauliffe DP. Molecular characterization of the Ro/SS-A autoimmune response. Semin Dermatol. 1991;10:199–205. [PubMed] [Google Scholar]
- 26.Routsias JG, Tzioufas AG, Sakarellos-Daitsiotis M, et al. Calreticulin synthetic peptide analogues: anti-peptide antibodies in autoimmune rheumatic diseases. Clin Exp Immunol. 1993;91:437–41. doi: 10.1111/j.1365-2249.1993.tb05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meldolesi J, Krause K-H, Michalak M. Calreticulin: how many functions in how many cellular compartments? Cell Calcium. 1996;20:83–86. doi: 10.1016/s0143-4160(96)90053-6. [DOI] [PubMed] [Google Scholar]
- 28.Casciola Rosen L, Rosen A, Petri M, et al. Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc Natl Acad Sci USA. 1996;93:1624–9. doi: 10.1073/pnas.93.4.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pike SE, Yao L, Jones KD, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumour growth. J Exp Med. 1999;188:2349–56. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshmukh US, Lewis JE, Gaskin F, et al. Immune responses to Ro60 and its peptides in mice. I. The nature of the immunogen and endogenous autoantigen determine the specificities of the induced autoantibodies. J Exp Med. 1999;189:531–40. doi: 10.1084/jem.189.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caponi L, Bombardieri S, Migliorini P. Anti-ribosomal antibodies bind the Sm proteins D and B/B′. Clin Exp Immunol. 1998;112:139–43. doi: 10.1046/j.1365-2249.1998.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krupicková S, Tucková L, Flegelová Z, et al. Identification of common epitopes on gliadin, enterocytes, and calreticulin recognized by antigliadin antibodies of patients with coeliac disease. Gut. 1999;44:168–73. doi: 10.1136/gut.44.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sontheimer RD, Nguyen TQ, Cheng ST, et al. The unveiling of calreticulin—a clinically relevant tour of modern cell biology. J Invest Med. 1995;43:362–70. [PubMed] [Google Scholar]
- 34.Lieu TS, Reimer CB, Sontheimer RD. Immunoglobulin class and subclass profile of the Ro/SS-A autoantibody response. J Invest Dermatol. 1988;90:158–64. doi: 10.1111/1523-1747.ep12462142. [DOI] [PubMed] [Google Scholar]