

Following infection with intracellular pathogens like mycobacteria, listeria, toxoplasma and leishmania, mononuclear phagocytes and related antigen-presenting cells (APC), i.e. dendritic cells, secrete the heterodimeric cytokine IL-12. IL-12 comprises two disulphide-linked subunits, p40 and p35, which together form the biologically active p70 hetrodimer molecule (reviewed in [1,2]). IL-12 production by macrophages and dendritic cells can also be enhanced by a T cell-dependent pathway through interaction of CD40 on the surface of the APC with CD40-ligand expressed on activated T cells. The IL-12 receptor (IL-12R), which is expressed by both natural killer (NK) cells at certain stages of development and by activated T cells, is made up of two chains called IL-12Rβ1 and IL-12Rβ2, respectively. Both receptor chains have extracellular, transmembrane and intracellular segments. Each of these receptor proteins can only bind to IL-12 with low affinity, but when co-expressed can bind IL-12 with high affinity, initiating a physiological response to this cytokine [3]. A schematic representation of the IL-12 receptor-mediated intracellular signalling pathway is illustrated in Fig. 1.
Fig. 1.

Binding of IL-12 to the IL-12Rβ1 and β2 chains induces phosphorylation of the kinases Tyk2 and Jak2, which associate with the cytoplasmic tails of the β1 and β2 chains, respectively. Subsequently the signal transducing protein STAT4 binds to the cytoplasmic tail of the β2 receptor and in turn becomes phosphorylated. It then dissociates from the receptor, dimerizes and translocates to the nucleus, where it acts as a transcriptional activator by binding to specific DNA response elements (RE) in the promoter regions of IL-12 inducible genes [27,31].
Binding of IL-12 to activated CD4 T cells partitions them to develop and differentiate along the so-called Th1 pathway, crucially important for cell-mediated immunity against intracellular pathogens. Furthermore, acting at picomolar and subpicomolar levels on T cells and NK cells, IL-12 induces high-level production of the cytokine IFN-γ[2].
IFN-γ plays a central role in the resistance of mammalian hosts to pathogens, particularly bacteria and parasites capable of intramacrophage survival (reviewed in [4,5]). The main cells producing IFN-γ are activated Th1 cells, activated CD8 cytotoxic cells of the TC1 phenotype, and activated NK cells. Biologically active IFN-γ is a homodimer, which has a range of pleiotropic effects on a number of cell types, with the ability to modulate the function of over 200 genes. It is one of the principal macrophage-activating cytokines, and mice with disrupted IFN-γ or IFN-γ receptor (IFN-γR) genes show increased susceptibility to intracellular pathogens including Leishmania major, Listeria monocytogenes, mycobacteria and certain viruses, e.g. vaccinia virus. IFN-γ interacts with a specific cell surface receptor, which is widely expressed on most nucleated cells. The IFN-γR consists of two transmembrane proteins, namely IFN-γR1 which is a ligand-binding chain, and IFN-γR2, which is required for signal transduction. A schematic representation of the IFN-γ receptor-mediated intracellular signalling pathway is shown in Fig. 2.
Fig. 2.
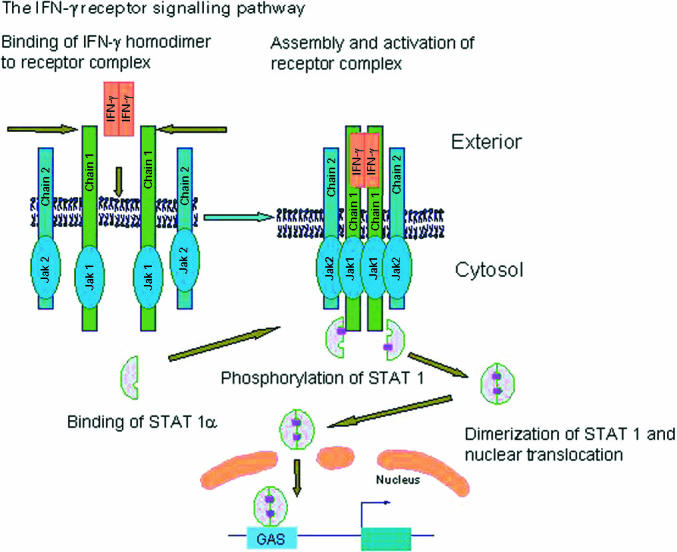
A schematic representation of the IFN-γ receptor-mediated intracellular signalling pathways (modified from [4,5]). Two IFN-γR1 chains dimerize on binding the IFN-γ homodimer and subsequently associate with two IFN-γR2 chains. Receptor subunit assembly leads to the trans-phosphorylation and activation of Jak1 and Jak2 kinases, which associate with the IFN-γR1 and IFN-γR2 chains, respectively. These phosphorylate a tyrosine residue on the intracytoplasmic domain of IFN-γR1, forming a paired set of docking sites for STAT1. Two STAT1 molecules then bind to these docking sites and subsequently become phosphorylated. Tyrosine phosphorylated STAT1 molecules then lose their affinity for IFN-γR1 chain docking sites, dissociate from the receptor and form homodimeric complexes. Phosphorylated STAT1 homodimers translocate to the nucleus and bind to specific sequences in the promoter region of early IFN-γ inducible genes, switching on gene transcription. Reprinted, with permission, from the Annual Review of Immunology, Vol. 15, © 1997 by Annual Reviews www.annualReviews.org
Key actions of IFN-γ include increased expression of MHC class I and class II proteins which enhance antigen processing and presentation, activation of mononuclear phagocytes through a multiplicity of effects, influencing IgG heavy-chain switching and modulating the production of cytokines such as IL-12, tumour necrosis factor-alpha (TNF-α) and IFN-γ itself.
Following IFN-γR ligation the receptor–ligand complexes recycle into an acidified subcellular compartment, where they dissociate. Free IFN-γ is then degraded by lysosomal enzymes. The uncoupled IFN-γR1 receptors eventually relocate to the cell surface via an intracellular pool. An amino acid motif present on the intracellular domain of the IFN-γR1 close to the Jak1 association site is required for normal recycling of this receptor (Fig. 2).
The above background information helps in the understanding of the clinical, pathological and immunological features of defects in the IL-12-dependent, high-output IFN-γ pathway and the laboratory methods required for identifying these defects.
MOLECULAR ASPECTS OF DEFECTS IN THE TYPE-1 CYTOKINE PATHWAY
The initial breakthrough arose from studies on a Maltese kindred who developed infection with poorly pathogenic non-tuberculous mycobacteria (NTM) [6]. Despite anti-mycobacterial chemotherapy three of the four affected patients died and the survivor had persistent infection. Peripheral blood mononuclear cells (PBMC) from the affected individuals failed to show up-regulation of endotoxin-induced TNF-α production on the addition of recombinant IFN-γ. Subsequently, a genome-wide search using microsatellite markers identified a region on 6q, where the affected children were homozygous for eight markers [7]. Investigations concentrating on the IFN-γR1 gene, which maps to 6q 23:24, revealed that the affected individuals had a point mutation at nucleotide 395 of this gene. This introduced a premature stop codon in the coding region for the extracellular domain of this receptor, preventing its expression, with complete abrogation of responses to IFN-γ[7]. Parallel work on idiopathic bacille Calmette–Guérin (BCG) infections in the absence of recognized immunodeficiency states [8] led to the simultaneous identification of a homozygous recessive mutation of the same gene as the cause of fatal disseminated BCG infection during infancy [9]. Subsequent investigation of patients with increased susceptibility to poorly pathogenic mycobacterial infections in the absence of known causes of primary and secondary immunodeficiency has led to the identification of three categories of gene defects. These comprise mutations in the IL12p40 subunit, IL-12Rβ1 as well as IFN-γR chains 1 and 2 [10].
COMPLETE IFN-γR1 DEFICIENCIES
Complete IFN-γR1 deficiency is caused by mutations resulting in the generation of premature stop codons in the DNA coding the extracellular domain of this protein. This prevents surface expression of the receptor, complete absence of binding of IFN-γ to the cell surface and consequent abrogation of the normal physiological responses to this cytokine [9,11–13]. These mutations have been seen in unrelated families from different ethnic backgrounds, each kindred having a unique mutation. A recessive mutation generating a premature stop codon in the extracellular domain of the IFN-γR2 chain resulted in the absence of STAT1 phosphorylation in response to IFN-γ, despite the normal expression of IFN-γR1 chains on the cells of the affected individual [14]. One patient has been described with complete absence of IFN-γR function because of compound heterozygous mutations in the IFN-γR1 chain [13]. A second type of complete IFN-γ receptor deficiency has recently been described which manifests itself not as a lack of receptor expression but by the failure of IFN-γ to bind to its receptor [15].
PARTIAL IFN-γR1 DEFICIENCIES
Partial IFN-γR1 deficiency resulting from a homozygous recessive missense mutation causing an isoleucine to threonine substitution at position 87 in the extracellular domain of the receptor (designated as I87T) has been described [16]. This probably reduced but did not abrogate binding of IFN-γ to the receptor. Cells from these patients showed signalling responses to high, but not low, in vitro concentrations of IFN-γ. A similar response phenotype has also been recently reported in a patient with partial IFN-γR2 deficiency [17]. This recessive mutation resulted from a nucleotide substitution in the IFN-γR2 gene causing an amino acid substitution in the extracellular region of the encoded receptor.
The commonest individual category of IFN-γR1 mutations comprise the heterozygous autosomal dominant mutation seen in nine individuals from three unrelated families and nine sporadic cases [18]. The mutation was either a one base-pair deletion (one kindred) or a four base-pair deletion (all others) at nucleotide 818 of the IFN-γR1. Thus there were 12 independent mutation events at a single mutation site indicating that this is a small deletion hot-spot in the human genome. The mutated allele is translated into stable mRNA, which encodes a truncated form of IFN-γR1 (illustrated in Fig. 3), that lacks the intracytoplasmic domain and thus has no Jak1 binding, recycling and STAT1 docking motifs. Due to these collective actions truncated receptors exert a dominant negative effect. The receptor deficiency however, is partial as weak signalling is detectable in vitro using supra-physiological levels of IFN-γ, and surprisingly clinical responses were obtained with IFN-γ augmentation of anti-mycobacterial chemotherapy (see below). The excessive accumulation of the abnormal receptors on the cell surface result in a 0·5–1 log increase in surface staining with MoAbs directed against IFN-γR1, observable by flow cytometry, which is diagnostically useful in screening for this defect.
Fig. 3.

A schematic representation of the IFN-γ receptor-mediated intracellular signalling pathways and its defective function within patients with heterozygous dominant negative IFN-γR1 deficiency. The truncated R1 chain paired with the wild-type R1 chain is able to bind the IFN-γ homodimer but is incapable of inducing signalling. Furthermore, unlike receptors comprising paired wild-type IFN-γR1 chains, these abnormal receptor complexes do not recycle following ligand binding, resulting in their accumulation at the cell surface.
COMPLETE IL-12p40 SUBUNIT DEFICIENCY
A child belonging to consanguineous Pakistani parents who presented with recurrent disseminated BCG infection (following neonatal immunization) and Salmonella enteritis septicaemia was found to have a 4·4-kb deletion spanning two coding exons of the IL-12p40 subunit [19]. The parents, the maternal grandmother and a healthy sibling were heterozygous for this defect. Monocytes and dendritic cells were unable to secrete IL-12 p40, or the biologically active IL-12 p70 cytokine, when appropriately activated. As a result, antigen-driven IFN-γ production by blood lymphocytes was markedly impaired, but could be reconstituted by the addition of exogenous recombinant IL-12. The child also showed a clinical response to IFN-γ complementation of anti-microbial chemotherapy, resulting in a complete cure of the BCG and salmonella infections, which has been sustained for 4 years after cessation of therapy.
In two additional reports of impaired IL-12 production, defects in the structural genes of IL-12 p40 and p35 have not been identified. The first report described a kindred with recurrent disseminated Mycobacterium avium infection, where the patient's cells had reduced, although not completely ablated, IL-12 p70 production [20]. The patient's lymphocytes had reduced in vitro phytohaemagglutinin (PHA)-driven IFN-γ production which could be enhanced by exogenous recombinant IL-12. The affected members of this kindred were all male and appeared to show abnormal regulation of IL-12 production, while the females showed an intermediate phenotype, implying putative X-linked inheritance of this defect. The second report documents a 3-year-old female with IL-12 deficiency associated with recurrent episodes of severe recurrent pneumococcal sepsis [21]. In this latter patient antibody production, including specific antibody levels to protein and polysaccharide antigens, was normal. The patient's mononuclear cells were however incapable of producing detectable IL-12 p70 or p40 when appropriately stimulated (with Staphylococcus aureus Cowan strain-1 (SAC)) even after supplementation with exogenous recombinant IFN-γ.
COMPLETE IL-12Rβ1 DEFICIENCY
Seven patients from six unrelated kindreds, affected by idiopathic severe disseminated mycobacterial and salmonella infection, were found to have homozygous recessive mutations of IL-12Rβ1 [22,23]. The mutations were unique to each kindred and affected the extracellular domain of the IL-12Rβ1 receptor. These null mutations prevented cell surface expression of the receptor on activated T cells, as detected by flow cytometry with a specific MoAb. These patients showed defective in vitro antigen-driven IFN-γ production that could not be augmented by exogenous recombinant IL-12. In one of these patients potentially fatal disseminated M. avium infection was cured by the augmentation of anti-mycobacterial chemotherapy with IFN-γ, suggesting that, as in the IL-12-deficient child, impaired IFN-γ secretion was probably responsible for the disease susceptibility of these patients.
CLINICAL FEATURES OF TYPE 1 CYTOKINE DEFICIENCY
All individuals with such defects so far reported presented with persistent salmonella and/or mycobacterial infections, with two exceptions. In one instance two young asymptomatic children have been identified with partial IFN-γR1 deficiency who have not been vaccinated with BCG [18]. In the second instance, a young adult sibling of an IL-12Rβ1-deficient patient was identified with the same genetic and functional defect. She had curable salmonella septicaemia as an infant but has not developed mycobacterial infection. She also had no reported problems following BCG vaccination in early infancy [23].
Fourteen patients with complete IFN-γR deficiency have developed mycobacterial infection in early childhood or infancy with a uniformly poor prognosis [24]. The infections were caused either by BCG following vaccination or by chance infection with poorly pathogenic environmental NTM. Mycobacterium avium-intracellulare is the commonest of the latter opportunistic pathogens, while there have been a smaller number of reports of infection caused by other atypical mycobacteria including M. chelonei, M. fortuitum, M. smegmatis, M. abscessus and M. kansasii [7,9,12,13]. All patients with complete IFN-γR deficiency vaccinated with BCG have developed severe disseminated disease. In patients with complete IFN-γR deficiency, mycobacterial granulomata are reminiscent of polar lepromatous leprosy, i.e. are poorly differentiated and comprise collections of macrophages which are multibacilliary. In the large majority of these patients, infections have been lethal despite anti-microbial chemotherapy, even when supplemented with IFN-γ therapy.
In patients with partial IFN-γR deficiency, BCG infection is associated with paucibacillary, tuberculoid-type granulomata and the infection is curable with anti-mycobacterial chemotherapy [16,18]. Two patients with partial dominant IFN-γR deficiency and two with IL-12Rβ1 deficiency have received BCG vaccination with no adverse effects [18,23]. In the IL-12 p40-deficient child, the disseminated BCG infection, although recurrent, was curable with IFN-γ supplementation of chemotherapy [19]. In IL-12Rβ1 deficiency, two patients who developed disseminated BCGosis with paucibacillary tuberculoid-type granulomata were cured with anti-mycobacterial chemotherapy alone [23]. Similarly, three patients with disseminated M. avium infections were also cured with prolonged anti-mycobacterial chemotherapy [22,23]. In another kindred with IL-12Rβ1 deficiency, one boy died of disseminated M. avium infection despite chemotherapy, while his brother, who developed disseminated M. avium infection at the age of 17 years, failed to respond to anti-mycobacterial chemotherapy but was cured by the addition of IFN-γ[23].
In patients with the dominant partial IFN-γR1 mutation [18], 13 out of 18 with poorly pathogenic mycobacterial infections recovered with anti-mycobacterial chemotherapy. However, most cases required prolonged courses of antibiotics, with frequent disease relapse being documented. Two females from one kindred who became infected with M. avium and BCG, respectively, did not respond to chemotherapy alone and were cured only by the addition of IFN-γ therapy. In a further kindred one patient died of disseminated M. avium infection, despite 5 years of continued chemotherapy with a range of anti-mycobacterial agents. Her identical twin sister had disseminated and recurrent M. avium infection, which was cured by prolonged courses of anti-mycobacterial chemotherapy. Both these patients had multibacillary disease with poor granuloma formation (lepromatous type). However, the daughter of the second patient had paucibacillary M. avium infection, with borderline tuberculoid-type granuloma formation, and was cured with 2 years of chemotherapy. This indicates that even in related individuals with the same molecular lesion, and probably the same strain of mycobacterial pathogen, there is variation in severity and outcome, presumably due to other compensatory immunological factors. The mother of two siblings with dominant negative IFN-γR1 mutation described in the above publication [18] had recurrent disseminated M. tuberculosis infection and died, despite chemotherapy, at the age of 33 years. While it is probable that she passed on these mutations to her children, no material from her was available for genetic analysis.
About a quarter of the patients with defects of the type-1 cytokine pathway also developed invasive salmonella infection; the majority due to non-typhi salmonella species (S. enteritidis; serogroup D or S. typhimurium; serogroup B or undetermined salmonella species). In addition, two other patients have had documented extra-intestinal disease due to S. typhi or paratyphi [18,22]. These infections have been curable with anti-microbial chemotherapy, although recurrences have been commonly observed. Interestingly, the father of the child with complete IL-12 p40 deficiency, who was heterozygous for this defect, had persistent systemic infection caused by S. bareilly between the ages of 2 and 4 years which was only cured with prolonged antibiotic therapy [19]. He has subsequently remained well. Other infections documented in type-1 cytokine-deficient patients include classical tuberculosis (n = 2) [16,18], histoplasmosis (n = 1) [18], meningitis due to listeriosis (n = 1) [25], and persistent oropharyngeal candidiasis (n = 1, Kumararatne, unpublished observations).
In contrast, these patients have not in general developed infections caused by extracellular bacteria or viruses causing respiratory infections, exanthematous infections or latent infections. Indeed, many of these patients have had unremarkable infections caused by common exanthematous viruses, including chicken pox and measles.
However, a recent report has documented the occurrence of severe viral infections in four patients with IFN-γR deficiency suffering from mycobacterial disease [26]. The viral pathogens included cytomegalovirus (CMV) (viraemia, pneumonia) Herpes simplex virus (gingivostomatitis, oesophagitis and skin lesions), Varicella zoster virus (prolonged illness or illness complicated with pneumonia) and respiratory syncytial virus (RSV) and parainfluenza virus type 3 (pneumonia). Hence, when caring for these patients the risk of severe viral infections should be considered. Moreover, the possibility of type-1 cytokine pathway deficiencies needs to be considered in cases of severe unexplained infections with such viruses.
Two further variations of the clinical picture in these patients deserve comment. First, two patients with partial dominant negative IFN-γR1 deficiency with osteolytic lesions were initially diagnosed as Histiocytosis-X [18]. Lesions in one patient contained S100+ cells, but had atypical pathological features. At initial diagnosis, no mycobacteria were detected by Ziehl–Nielson staining, or by culture. The patients did not respond to radiotherapy and chemotherapy used for treatment of Histiocytosis-X and were subsequently found to have multibacillary infection caused by BCG and M. avium, respectively. Both patients failed to respond to anti-mycobacterial chemotherapy, but were cured when their treatment was augmented with IFN-γ therapy (Edgar, Lammas, Novelli, Kumararatne, in preparation).
Second, NTM infections can be very difficult to diagnose in patients with underlying type-1 cytokine deficiencies [6,11]. For example, in two patients who had complete IFN-γR1 deficiency and were infected with M. chelonei and M. fortuitum, initial histology showed non-specific chronic inflammation with no visible acid-fast bacilli [6]. In addition, cultures for mycobacteria were repeatedly negative. Mycobacterial infection became evident only when the patients were treated with immunosuppressive therapy on the basis that they were suffering from an autoimmune disease. In another patient with IFN-γR1 deficiency, M. smegmatis infection was diagnosed with considerable difficulty after laparotomy and several liver biopsies [11]. Therefore, in children with unexplained persistent fever, weight loss, hepatosplenomegaly, lymphadenopathy and elevated inflammatory markers with or without chronic anaemia, defects of the type-1 cytokine pathway should be considered in the differential diagnosis with diligent searching for poorly pathogenic mycobacterial sepsis.
Th2 RESPONSES ARE NOT UP-REGULATED IN PATIENTS WITH DEFICIENT Th1 IMMUNITY
Because signalling by the major Th1 signature cytokine (IFN-γ), or its major inducer (IL-12), is disabled in type-1 cytokine pathway-deficient patients, these experiments of nature provided a unique opportunity to examine the functional relevance of the Th1/Th2 paradigm in man. It is generally accepted that Th2 immunity is down-regulated by Th1 cytokines, such as IFN-γ, and conversely that Th2-associated cytokines (IL-4, IL-5 and IL-13) are responsible for atopy. An increase of Th2 responses, with clinical and biological signs of atopy, may therefore have been predicted within patients with genetically abrogated IFN-γ- or IL-12-mediated responses. However, clinical observations on 19 patients with type-1 cytokine pathway deficiencies revealed that none of the patients showed signs of severe atopy, such as asthma or eczema, and only 6/19 exhibited mild allergies, such as seasonal rhino conjunctivitis. IgE levels were also within the normal range in 6/8 patients examined (Wood, Kumararatne, Casanova, Ottenhoff, in preparation). Overall, the clinical phenotype suggests that normal homeostasis of Th2 responses can occur in the absence of a fully functional Th1 pathway. These findings may have important implications for understanding the pathogenesis of atopic disease and for the development of rational approaches to immunotherapy for patients with severe allergies.
DIAGNOSIS OF DEFECTS IN THE TYPE-1 CYTOKINE CASCADE (TABLE 1)
Table 1.
Investigation of HIV− patients suffering from poorly pathogenic mycobacterial infections for type-1 cytokine/cytokine receptor pathway component deficiencies
| Deficiency | Subtype | Investigation a | Cell population (treatment) | Result | Investigation b | Cell population (treatment) | Result | Investigation c | Cell population (treatment) | Result (low dose/high dose)* |
|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γR1 | aComplete | IFN-γ ELISA | PBMC ± PHA† | Normal/low | Flow cytometry | PBMC | Hypo-expression | EMSA (STAT1) | PBMC | Negative/negative |
| PBMC ± PPD‡ | Very low/absent | Anti-CD119 | (nil) | (rhIFN-γ, 20 min/30°C) | ||||||
| IFN-γR1 | bDominant | IFN-γ ELISA | PBMC ± PHA† | Normal/low | Flow cytometry | PBMC | Hyper-expression | EMSA (STAT1) | PBMC | Negative/positive |
| IFN-γR1 | cPartial | IFN-γ ELISA | PBMC ± PHA† | Normal/low | Flow cytometry | PBMC | Normal expression | EMSA (STAT-1) | PBMC | Negative/positive |
| IFN-γR2 | Complete | IFN-γ ELISA | PBMC ± PHA† | Normal/low | Flow cytometry Anti-CD119 | PBMC | Normal expression | EMSA (STAT1) | PBMC (rhIFN-γ, 20 min/30°C) | Negative/negative |
| IL-12Rβ1 | Complete | IFN-γ ELISA | PBMC ± PHA† | Normal/low Low/very low | Flow cytometry Anti-IL-12Rβ1 | 3-day PHA blasts | Hypo-expression | EMSA (STAT4) | 3-day PHA blasts (rhIL-12, 20 min/37°C) | Negative§ |
| IL-12 p40 | Complete | IFN-γ ELISA | PBMC ± PHA† | Low/very low | ELISA IL-12 p70 | PBMC + SAC(o/n) | Low/absent production | ELISA IL-12 p40 | PBMC + SAC (o/n) | Low/absent production |
Low-dose IFN-γ = 101 IU/ml; high-dose IFN-γ = 105 IU/ml.
5 μg/ml phytohaemagglutinin (PHA) for 3 days.
5 μg/ml PPD for 7 days (PPD avium for Mycobacterium avium-intracellulare (MAI) patients, PPD Mtb for bacille Calmette–Guérin (BCG) patients).
Response to optimal dose of IL-12 (5 ng/ml).
SAC, Staphylococcus aureus Cowan strain suspension; EMSA, electromobility shift assay.
These defects must be sought and are most likely to be found in:
Patients with disseminated or recurrent infection due to poorly pathogenic mycobacteria (BCG or environmental NTM).
Patients with systemic infections caused by non-typhi salmonella species. Characteristically non-typhoid salmonella infections in these patients are persistent and recurrent despite antibiotic therapy.
In the following categories of patients defects in the type-1 cytokine axis should be considered in the differential diagnosis, but are likely to be rare:
Patients with extra-intestinal disease caused by S. typhi and paratyphii.
Patients with M. tuberculosis infection who are treatment-compliant, have drug-sensitive organisms but develop either recurrent or disseminated disease.
Patients with severe unexplained viral infection, especially due to herpes viruses (see [26]).
Children with unexplained persistent fever and night sweats, weight loss, lymphadenopathy, hepatosplenomegaly and raised acute-phase responses, despite the absence of obviously detectable mycobacterial infection (see [6,11]).
Children with Histiocytosis-X refractory to treatment or with atypical pathological features.
Finally, based on the limited observations in the current series of patients, but guided by information in mice with defects of the type-1 cytokine pathway, this differential diagnosis should be considered in patients with other infections characteristic of impaired cell-mediated immunity, i.e. toxoplasmosis, histoplasmosis, severe listeriosis, etc.
A characteristic feature of the type-1 cytokine pathway is the interdependence of its components on each other due to positive regulatory feedback. This is most clearly documented by Holland et al., who showed that patients with complete IFN-γR defects have reduced in vitro production of IFN-γ, IL-12, TNF-α, etc. [12]. Therefore, investigation of this pathway needs systematic assessment of all its components to build up a picture that will provide indication of a potential defect. Based on information derived from investigating patients documented to date, the following assessment steps may be undertaken to identify possible type-1 cytokine pathway deficiencies (outlined in Table 1).
The measurement of IFN-γ production is required following in vitro stimulation with mitogens (PHA) as well as relevant antigens, e.g. BCG, Mycobacterium avium-intracellulare (MAI), Salmonella. Virtually all the type-1 cytokine pathway-deficient patients demonstrate reduced in vitro IFN-γ production [6,12]. It is then important to investigate whether this deficit in antigen-dependent IFN-γ production can be corrected by supplementation with exogenous recombinant IL-12 [19,23].
IL-12 production should be measured using both unseparated mononuclear cell preparations as well as purified monocytes stimulated with S. aureus (Cowan strain). IL-12 p70 and p40 can be conveniently measured using commercially available ELISA kits [12,19,23]. TNF-α release from monocyte and macrophage cultures should also be assessed using lipopolysaccharide (LPS) stimulation with or without IFN-γ supplementation [6,12,19,23]. For the detection of impaired regulation of IL-12 production, IL-12 responses of patient monocyte cultures should be studied using S. aureus (Cowan strain) alone or with added exogenous IFN-γ[20]. Individuals with defective regulation of IL-12 production respond to S. aureus supplemented with IFN-γ but not with S. aureus alone [20].
Expression of cell-surface IFN-γR1 can be determined by flow cytometry using appropriate MoAbs [7,9,18]. Reduced or absent expression would indicate the likelihood of a mutation in the extracellular domain of the IFN-γR1 chain, while enhanced expression would suggest heterozygous dominant-negative IFN-γR1 mutation. IFN-γR2 expression can be similarly assessed by flow cytometry [27], although the MoAb cited is not commercially available. Unfortunately, IFN-γR dysfunction may occur despite normal surface expression, hence it is important to assess functional responses to exogenous IFN-γ by means of the electromobility shift assay (EMSA) (see below) or by investigating up-regulation of MHC class I or CD40 on monocytes, or MHC class II molecules on fibroblast cell lines derived from skin biopsies from patients [9,18]. For assessment of signal transduction by EMSA, following specific cytokine stimulation, isolated cell nuclei are lysed, proteins separated by gel electrophoresis and probed with radio-labelled oligonucleotides complementary to STAT factors or with an antibody capable of detecting phosphorylated STATs. If IFN-γ-induced signalling is intact, phosphorylated STAT1 will be detected within the nuclear lysates. These methods are described in detail elsewhere [9,13,16]. By studying the dose response required for nuclear translocation of phosphorylated STAT1 (pSTAT1) induced by IFN-γ, it is possible to discriminate between complete and partial forms of receptor dysfunction [16,18]. Alternatively, flow cytometry may be used to assess IFN-γR function by detection of intracellular phosphorylated STAT1 within IFN-γ-stimulated cells using a specific anti-human pSTAT1 MoAb [28].
Assessment of IL-12Rβ1 and IL-12Rβ2 expression is determined by flow cytometry following staining of mitogen-induced T cell blasts with a MoAb against IL-12Rβ1 [22,23] or IL-12Rβ2 [29]. Although an anti-IL-12Rβ1 MoAb is commercially available for expression screening, the cited anti-IL-12Rβ2 MoAb is not. The integrity of IL-12-induced signalling pathways can be assessed using EMSA for the detection of the translocation of phosphorylated STAT4 [22,23]. To date, about half to a third of patients with idiopathic disseminated poorly pathogenic mycobacteria/salmonella infection have been identified to have mutations of the type-1 cytokine cascade described above.
Mutations identified to date in the type-1 cytokine pathway are listed in the Mendelian Inheritance in Man (OMIM) database (see below). To date, the genome-scanning approach has only been successful in identifying a relevant gene defect in the original Maltese family, described by Newport et al. [7]. This approach is not ideally suited to small families and cannot be applied to sporadic patients. Immunological analysis that allows the investigator to target genes of interest has been more productive. Once a candidate gene has been identified it is possible to apply single‐strand conformational polymorphism (SSCP) to screen for potential gene mutations or polymorphisms which can be confirmed by gene sequencing. Gene complementation assays to determine the functional relevance of such mutations may be useful [16,18].
PROGNOSIS OF DEFECTS IN THE TYPE-1 CYTOKINE PATHWAY
The outcome in patients with complete IFN-γR defects who develop BCG or NTM infections is poor. Bone marrow transplantation (BMT) is the only potentially curative therapeutic option for these patients [25]. To date, experience of BMT is limited, but two patients have been successfully transplanted with HLA-identical intrafamily donors (M. Levin and W. Friedrich, personal communications). A haplo-identical bone marrow graft was rejected by another patient (J.-L.C., unpublished).
The main treatment for mycobacterial infection is chemotherapy directed towards the mycobacterial species identified. These therapeutic regimes should include at least four drugs and should be governed, where possible, by in vitro susceptibility data. The poor correlation between in vitro antibiotic sensitivity and clinical response in NTM infection is well known and has been discussed elsewhere. Supplementary measures like drainage of collections of pus and attention to nutrition are important. For initial empirical therapy of patients with a history of BCG vaccination, the choice of drugs should include rifampicin, INAH, ethambutol and clofazimine. Unvaccinated patients should be considered to be affected with NTM and receive a combination including rifampicin or rifabutin, clarithromycin, or azithromycin and ciprofloxacin or another 4-quinolone. These regimes should be altered depending on the mycobacterial species identified and antibiotic susceptibility data. Anti-mycobacterial therapy may have to be continued for longer periods than average, possibly even for life. Due to the emerging experience of the value of supplementation with IFN-γ, this should be considered early except in patients with complete IFN-γR deficiency [26,30]. The initial regimen should use a dose of 30–50 μg/m2 administered subcutaneously, three times a week [30]. In one patient who failed to respond to this regimen, IFN-γ was increased step-wise at monthly intervals until a response was finally observed at a dose of 400 μg/m2, three times a week (Edgar, Lammas, Kumararatne, in preparation). The main side-effect observed with IFN-γ therapy is a febrile response within 24 h of each dose of IFN-γ that can be easily controlled with paracetamol. Those patients with a therapeutic response to IFN-γ showed early evidence of this in terms of falling acute-phase markers like C-reactive protein, weight gain, and later on radiological evidence of healing (Edgar, Lammas, Kumararatne, in preparation).
Prophylactic measures should include avoidance of exposure to salmonella and listeria. Tuberculosis would obviously be a risk to these patients, but avoiding exposure may not be straightforward. BCG immunization is obviously contraindicated and it would be prudent to avoid certain other live vaccines, e.g. yellow fever and typhoid vaccines. Based on experience in HIV-infected children with the measles vaccine, the MMR vaccine is probably safe and may be preferable to the risk of measles infection. For those individuals living in typhoid-endemic areas, inactivated typhoid vaccine (e.g. Typhoid Vi vaccine) should be considered in addition to sanitation measures. Genetic counselling is an important component of patient support as in other primary immune-deficiency diseases.
Detailed information on these disorders is available via the Mendelian Inheritance in Man database (OMIM) available at http://http://www.ncbi.nlm.nih.gov/omim/
REFERENCES
- 1.Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83–243. doi: 10.1016/s0065-2776(08)60387-9. [DOI] [PubMed] [Google Scholar]
- 2.Gately MX, Renzetti LM, Magram J, et al. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 3.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gatety MX, Gubler U. A functional interleukin 12 receptor complex is composed of two β type cytokine receptor subunits. Proc Natl Acad Sci USA. 1996;93:14002–7. doi: 10.1073/pnas.93.24.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 5.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-γ. Annu Rev Immunol. 1997;15:749–95. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 6.Levin M, Newport M, D'souza S, et al. Familial disseminated atypical mycobacterial infection in childhood: a human mycobacterial susceptibility gene? Lancet. 1995;345:79–83. doi: 10.1016/s0140-6736(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 7.Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;335:1941–9. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 8.Casanova JL, Jouanguy E, Lamhamedi S, Blanche S, Fischer A. Immunological conditions of children with BCG disseminated infection. Lancet. 1995;346:581–2. doi: 10.1016/s0140-6736(95)91421-8. [DOI] [PubMed] [Google Scholar]
- 9.Jouanguy E, Altare F, Lamhamedi S, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette–Guerin infection. N Engl J Med. 1996;335:1956–61. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 10.Casanova JL, Ochs H. Interferon-gamma receptor deficiency: an expanding clinical phenotype? [Editorial; Comment] J Pediatr. 1999;135:543–5. doi: 10.1016/s0022-3476(99)70050-8. [DOI] [PubMed] [Google Scholar]
- 11.Pierre-Audigier C, Jouanguy E, Lamhamedi S, et al. Fatal disseminated Mycobacterium smegmatis infection in a child with inherited interferon gamma receptor deficiency. Clin Infect Dis. 1997;24:982–4. doi: 10.1093/clinids/24.5.982. [DOI] [PubMed] [Google Scholar]
- 12.Holland SM, Dorman SE, Kwon A, et al. Abnormal regulation of interferon-gamma, interleukin-12, and tumor necrosis factor-alpha in human interferon-gamma receptor 1 deficiency. J Infect Dis. 1998;178:1095–104. doi: 10.1086/515670. [DOI] [PubMed] [Google Scholar]
- 13.Altare F, Jouanguy E, Lamhamedi-Cherradi S, et al. A causative relationship between mutant IFNγRI alleles and impaired cellular response to IFNγ in a compound heterozygous child [letter] Am J Hum Genet. 1998;62:723–6. doi: 10.1086/301750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–9. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jouanguy E, Dupuis S, Pallier A, et al. In a novel form of IFNγ receptor 1 deficiency, cell surface receptors fail to bind IFNγ. J Clin Invest. 2000;105:1429–36. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jouanguy E, Lamhamedi-Cherradi S, Altare F, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette–Guerin infection and a sibling with clinical tuberculosis. J Clin Invest. 1997;100:2658–64. doi: 10.1172/JCI119810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Döffinger R, Jouanguy E, Dupuis S, et al. Partial interferon-γ receptor signaling chain deficiency in a patient with Bacille Calmette-Guérin and Mycobacterium abscessus infection. J Infect Dis. 2000;181:379–84. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- 18.Jouanguy E, Lamhamedi-Cherradi S, Lammas D, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21:370–8. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- 19.Altare F, Lammas D, Revy P, et al. Inherited interleukin 12 deficiency in a child with bacille Calmette–Guerin and Salmonella enteritidis disseminated infection. J Clin Invest. 1998;102:2035–40. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frucht D, Holland S. Defective monocyte costimulation for IFNγ production in familial disseminated Mycobacterium avium complex infection. J Immunol. 1996;157:411–6. [PubMed] [Google Scholar]
- 21.Haraguchi S, Day N, Nelson RJ, et al. Interleukin 12 deficiency associated with recurrent infections. Proc Natl Acad Sci USA. 1998;95:13125–9. doi: 10.1073/pnas.95.22.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Jong R, Altare F, Haagen IA, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280:1435–8. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- 23.Altare F, Durandy A, Lammas D, et al. Impairment of mycobacterial immunity in human interleukin- 12 receptor deficiency. Science. 1998;280:1432–5. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- 24.Jouanguy E, Altare F, Lamhamedi-Cherradi S, Casanova JL. Infections in IFNγR-1-deficient children. J Interferon Cytokine Res. 1997;17:583–7. doi: 10.1089/jir.1997.17.583. [DOI] [PubMed] [Google Scholar]
- 25.Roesler J, Kofink B, Wendisch J, et al. Listeria monocytogenes and recurrent mycobacterial infections in a child with complete interferon-gamma-receptor (IFNγR1) deficiency: mutational analysis and evaluation of therapeutic options. Exp Hematol. 1999;27:1368–74. doi: 10.1016/s0301-472x(99)00077-6. [DOI] [PubMed] [Google Scholar]
- 26.Dorman SE, Uzel G, Roester J, et al. Viral infections in interferon-gamma receptor deficiency. J Pediatr. 1999;135:640–3. doi: 10.1016/S0022-3476(99)70064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novelli F, Bernabei L, Ozmen L, Rigamonti A, Allione S, Pestka G, Garotta G, Forni G. Switching on of the proliferation or apoptosis of activated human T lymphocytes by IFNγ is correlated with the differential expression of the alpha and beta chains of the receptor. J Immunol. 1996;157:1935–8. [PubMed] [Google Scholar]
- 28.Fleisher TA, Dorman SE, Anderson JA, Vail M, Brown MR, Holland SM. Detection of intracellular phosphorylated STAT-1 by flow cytometry. Clin Immunol. 1999;90:1–6. doi: 10.1006/clim.1998.4654. [DOI] [PubMed] [Google Scholar]
- 29.Rogge L, Barberis-Maino L, Presky DH, et al. Antibodies to the interleukin 12 receptor β2 chain mark human Th1 but not Th2 cells in vitro and in vivo. J Immunol. 1999;162:3926–32. [PubMed] [Google Scholar]
- 30.Holland SM, Eisenstein EM, Kuhns DB, et al. Treatment refractory disseminated nontuberculous mycobacterial infection with interferon gamma: a preliminary report. N Engl J Med. 1994;330:1348–55. doi: 10.1056/NEJM199405123301904. [DOI] [PubMed] [Google Scholar]
- 31.Yao BB, Niu P, Surowy CS, Faltynek CR. Direct interaction of STAT4 with the IL-12 receptor. Arch Biochem Biophys. 1999;368:147–55. doi: 10.1006/abbi.1999.1302. [DOI] [PubMed] [Google Scholar]