

INTRODUCTION
A clinical syndrome characterized by recurrent life-threatening Staphylococcus aureus, Proteus or Pseudomonas, hypergammaglobulinaemia, and widespread chronic granulomatous infiltration was first recognized in the paediatric literature between 1954 and 1960 [1–3]. The pathological mechanisms responsible for this condition became evident when it was demonstrated that neutrophils collected from a male patient were unable to kill S. aureus in vitro, and that there was a primary abnormality of neutrophil function [4]. In the same year, it was shown that neutrophils from patients with this familial granulomatosis, now called chronic granulomatous disease (CGD), failed to exhibit a characteristic increase of oxidative metabolism, called the ‘respiratory burst’, during phagocytosis [5].
IDENTIFICATION OF THE NAPDH–OXIDASE
The enhanced ‘respiration’ of leucocytes was first described as a small but significant increase in the oxygen consumption of canine neutrophils during phagocytosis of bacteria [6]. This metabolic response was attributed to increased generation of energy during phagocytosis, and was known as ‘the extra respiration of phagocytosis’, but was later shown to be resistant to conventional inhibitors of mitochondrial respiration [7]. It was also shown that the necessary energy for phagocytosis and cytoplasmic degranulation was provided by the glycolytic pathway. The function of the ‘respiratory burst’ remained obscure until it became apparent that the ability of phagocytic cells to kill certain bacteria in vitro was markedly diminished under anaerobic conditions, and that cells obtained from patients with CGD, which were unable to mount this metabolic response, exhibited the same microbicidal deficiency in the presence of oxygen [4]. The identity of the substrate for the reaction was also the subject of considerable speculation [8,9], but the sharp increase in oxidation of glucose via the hexose monophosphate shunt (the purpose of which is to maintain cellular NADPH levels, and the activity of which is controlled by the rate of oxidation of NADPH) coincident with neutrophil activation, suggested that NADPH was the most likely candidate molecule.
COMPONENTS OF THE NAPDH–OXIDASE
The NAPDH–oxidase catalyses the formation of superoxide, which is a precursor for the generation of potent oxidant compounds, by transmembrane passage of electrons from NADPH to molecular O2. It is most abundant in phagocytic cells, particularly neutrophils, eosinophils and cells of the monocyte/macrophage lineage, consisting of a membrane-bound flavocytochrome b558 and four cytosolic factors, p47phox, p67phox, p40phox and p21rac, which translocate to the membrane on activation of the cell (the suffix phox represents ph agocyte ox idase) [10]. Activation is initiated classically by opsonized particles, but also by many soluble inflammatory mediators. More recently, components of the NAPDH–oxidase and low level enzymatic activity have been detected in other cell types, although their biological functions are unclear and will not be discussed further here.
The redox centre of the oxidase is the flavocytochrome b558, the midpoint potential for which is sufficiently low to induce direct reduction of oxygen to superoxide. The flavocytochrome consists of two proteins with apparent molecular weights of 23 kD (p22phox, α-subunit) and 76–92 kD (gp91phox, β-subunit), respectively, and are arranged as a 1:1 heterodimer [11–14]. The larger β-subunit migrates on SDS–PAGE as a broad band, an electrophoretic property characteristic of glycoproteins, and comprises about 21% carbohydrate, predominantly of the N-linked high-lactosamine complex type [15]. Both p22phox and gp91phox are missing in cells derived from most CGD patients with a molecular lesion of either subunit, indicating that mutual interaction is necessary for assembly of the mature complex [16–18]. A 65-kD biosynthetic gp91phox intermediate precursor with high-mannose type oligosaccharide side chains has been detected in membrane fractions (which includes endosomal compartments) derived from patients with p22phox-deficient CGD, which is processed to a mature terminally glycosylated form only if the deficiency of the smaller subunit protein is restored [19]. Biosynthesis of gp91phox glycoprotein is also dependent on the incorporation of two non-identical haem groups for each heterodimer within the membrane-spanning α helices, one predicted to lie near the inner face, and the other towards the outer face of the cell [18]. The locations of the binding sites for the substrate NADPH, and for the electron carrier flavin adenine dinucleotide (FAD), are now known to lie within the C-terminal region of gp91phox itself (although an additional NADPH-binding site may exist on p67phox). Final transfer of electrons across the cell membrane is probably mediated by the two associated haem groups. The flavocytochrome b558 therefore comprises the complete electron transporting apparatus of the NAPDH–oxidase. The membrane-spanning N-terminal region of the flavocytochrome has recently been identified as the site of a charge-compensating H + conductance during activation of the respiratory burst, and may also therefore be responsible for maintaining intracellular (preventing deleterious acidification) and phagosomal pH (for optimal activity of proteolytic enzymes) [20–26].
Fig. 1.
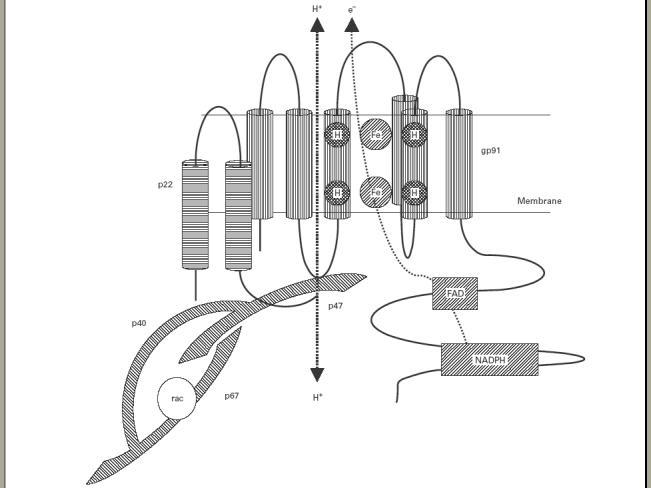
Schematic representation of the active NAPDH–oxidase complex. The flavocytochrome b558 consists of two components, gp91phox and p22phox, which reside in the membrane as a heterodimer. On activation of the cell, a complex of cytosolic components (see text) assembles at the membrane and induces electron transport from NADPH to molecular oxygen via flavin adenine dinucleotide (FAD) and haem groups. gp91phox has also recently been shown to act as a voltage-gated H + channel which could participate in charge compensation. Termination of activity may be regulated by the GTPase, p21rac.
The first abnormalities defined for autosomal recessive CGD (AR-CGD) were deficiencies of two cytosolic proteins p47phox and p67phox[27]. Phosphorylation of both proteins coincides with activation of the NAPDH–oxidase, and is likely to be necessary for regulated assembly of the functional complex in vivo, as it is not necessary for activation in cell-free systems [28–34]. p40phox exists in a complex with p47phox and p67phox in resting cytosol, and translocates to the membrane on cell activation, although the function of this component is not yet known. All three cytosolic phox proteins contain src-homology 3 (SH3) domains which provide considerable opportunity for intramolecular and intermolecular interaction between themselves, with the cytoplasmic tail of p22phox, and with other signalling or cytoskeleton-associated molecules (reviewed in [36]). The Rho family GTPase p21rac was co-purified with rho-GDI (GDP-dissociation inhibitor) from a cytosolic fraction prepared from guinea pig macrophages, and found to be essential for cell-free activation of the NAPDH–oxidase [37]. In contrast to p21rac1, which is ubiquitously expressed, p21rac2 is restricted to myeloid lineage cells. However, studies in p21rac2-deficient mice generated by gene targeting indicate that activation of the NAPDH–oxidase by p21rac2 is stimulus-specific [38]. This raises the possibility that other GTPases, including p21rac1, could activate the system in other circumstances. Another GTP-binding protein, Rap1A, has been shown to co-purify with the flavocytochrome b558, although its role in NAPDH–oxidase function has not been clarified [39,40].
ACTIVATION OF ELECTRON TRANSPORT
The flavocytochrome b558 almost certainly comprises the complete electron transporting system and forms the membrane docking site for the cytosolic components. In resting neutrophils, the plasma membrane is devoid of flavocytochrome b558 which resides almost exclusively in specialized light density intracellular vesicles and within the membranes of specific granules [41,42]. When the cell is activated the plasma membrane invaginates to form the phagocytic vacuole with which vesicles containing flavocytochrome b558 fuse. The cytosolic components form an activation complex which translocates to the membrane to associate with the flavocytochrome b558. Cytosolic residues on both p22phox and gp91phox appear to be important for this interaction, which is thought to be mediated predominantly by p47phox[43,44]. p67phox probably localizes by virtue of interaction with p47phox, as it does not do so in its absence. p21rac has been shown to interact directly with p67phox, and they may therefore translocate together [45–47]. In contrast, translocation of p21rac2 has been shown to occur in the absence of either p47phox or p67phox, suggesting that alternative mechanisms exist for oxidase activation [48]. Assembly of the complete NAPDH–oxidase complex may induce conformational changes in flavocytochrome b558 which permit binding of the substrate NADPH, and which are energetically favourable for electron transport. Other proteins, for example p40phox, are not essential in cell-free systems, and are therefore more likely to be important for the stabilization of individual component proteins or for their initial assembly into an activation complex at the cell membrane.
The assembled NAPDH–oxidase transfers electrons to molecular oxygen, which is reduced to a free radical superoxide anion O2− within the phagocytic vacuole. O2− participates in a series of reactions that result in generation of hydrogen peroxide, hypohalous acids, and possibly other more reactive oxygen radicals, including those derived from nitric oxide. The discovery of an intrinsic NAPDH–oxidase hydrogen ion conductance also lends support to the idea that a major function of the system is to regulate phagosomal pH, and therefore the activity of secreted granule proteins [20–26,49]. NAPDH–oxidase activity occurs transiently after phagocytosis and is dependent upon continued receptor occupancy, and continued association of oxidase components at the membrane. Termination of the response is not simply mediated by the release of p47phox and p67phox into the cytosol, as these remain in association with the membrane well after the burst is over [34]. One potential regulator of termination is the GTP-binding protein p21rac, the activity of which is governed by the phosphorylation state of the guanine nucleotide.
ANIMAL MODELS
Murine models have been developed for CGD (gp91phox and p47phox) by gene targeting [50,51]. Affected mice lack phagocyte superoxide production, and are susceptible to infection with S. aureus and Aspergillus fumigatus, reflecting a similar phenotype to that seen in human CGD. These have therefore become useful models for study of NAPDH–oxidase function in both phagocytic and non-phagocytic cells, and for development of novel therapies. To investigate the contribution of mechanisms other than generation of reactive oxygen or nitrogen species to the killing of bacteria, mice have been generated that are deficient in both gp91phox and inducible nitric oxide synthase (NOS2) [52]. These mice develop massive abscesses containing mainly commensal enteric bacteria, even when reared under pathogen-free conditions. Interestingly, though susceptible to virulent Listeria, doubly deficient animals retained partial killing activity against S. typhimurium, Escherichia coli and attenuated Listeria, suggesting that alternative mechanisms exist. More recently, mice deficient in the granule serine proteases, elastase and cathepsin G, have been shown to be susceptible to fungal infection, indicating the importance of non-oxidative effector mechanisms in host defence [53]. Neutrophils obtained from mice deficient in the Rho family GTPase Rac2 display significant defects of chemotaxis, adhesion, superoxide production, and increased mortality when challenged with A. fumigatus [38]. The suggestion that expression of other genes may be important for the development of inflammatory complications is supported by studies showing that administration of sterilized aspergillus hyphae to X-CGD lungs in vivo is characterized by exaggerated production of IL-1β, tumour necrosis factor-alpha (TNF-α), and the chemokine KC [54]. Therefore, for reasons unrelated to active infection, the cytokine response in X-CGD mice is proinflammatory, and may reflect the development of sterile granulomatous inflammation seen in patients.
MOLECULAR PATHOLOGY
The genes encoding both subunits of the flavocytochrome b558, and four cytosolic factors, p40phox, p47 phox, p67phox, and p21rac2, have been cloned, and molecular lesions identified in all but p40phox[55–58]. The distribution of genetic lesions within CGD patients is shown in Table 1. Molecular lesions on the X chromosome at CYBB account for the majority of cases of CGD (X910CGD). The mutations are heterogeneous, and are unique to individual families in over 90% of cases [59–62]. As expected, missense mutations result in considerable heterogeneity of phenotype and in some cases expression of a mutant flavocytochrome b558 with residual biochemical activity (variant CGD). In rare cases, extreme patterns of X-inactivation can result in an X-CGD phenotype in girls. Molecular lesions at CYBA are also heterogeneous. The second most common cause of CGD is A470CGD. In contrast to other forms of CGD, a GT dinucleotide deletion at a GTGT repeat at the boundary between the first intron and second exon is found in the majority of mutant alleles, resulting in a chain terminator at amino acid residue 51 [63]. This has now been found to arise from recombination events between the p47phox gene and highly homologous pseudogenes, which contain the GT deletion [64]. For A470CGD, carrier testing is therefore problematic as all normal individuals possess the deleted pseudogenes. Most recently, two patients with recurrent bacterial infections, and associated abnormalities of cell migration, degranulation, and O2-production have been reported to have a dominant-negative mutation in p21rac2 [38].
Table 1.
Characteristics and distribution of gene defects in chronic granulomatous disease (CGD)
| Component | gp91phox | p22phox | p47phox | p67phox | p40phox | p21rac2 |
|---|---|---|---|---|---|---|
| Disease | X-linked | Autosomal | Autosomal | Autosomal | Not known | Autosomal |
| X91 CGD* | recessive | recessive | recessive | dominant | ||
| A22 CGD* | A47 CGD* | A67 CGD* | ||||
| Results from USA and European studies of a total of 122 families†: | X91069 (56%) | A2207 (6%) | A47028 (23%) | A6707 (6%) | Not described | |
| Numbers of affected | X91−8 (7%) | A22+1 (1%) | ||||
| families, and incidence | X91+2 (2%) | |||||
| Genetic locus | CYBB | CYBA | NCF-1 | NCF-2 | ||
| Chromosomal location | Xp21.1 | 16q24 | 7q11.23 | 1q25 | 22q13.1 | 22q12 |
| Gene/mRNA size | 30 kb/4·7 kb | 8·5 kb/0·8 kb | 15·2 kb/1·4 kb | 37 kb/2·4 kb | 18 kb/1·2 kb | 18 kb/1·5 kb |
| Exons | 13 | 6 | 11 | 16 | 10 | 7 |
| Tissue specificity | Myeloid Low levels in mesangial cells, and some B lymphocytes. Pulmonary neuroepithelial bodies. | mRNA ubiquitous Protein expression only in presence of gp91phox | Myeloid | Myeloid | Myeloid | p21rac2,myeloid |
Accepted classification of CGD, in which A or X denote inheritance pattern. This is followed by the molecular weight of the affected component in kD. The superscript refers to the level of detectable immunoreactive protein: (0) indicates no protein, (−) indicates diminished protein, and (+) indicates normal levels of defective protein.
Adapted from reference [140].
In a recent study of 129 patients with CGD, genetic modifiers (single nucleotide polymorphisms (SNP)) of disease severity were identified and linked to the development of inflammatory complications [65]. In particular, physiologically relevant polymorphisms were identified in myeloperoxidase and Fcγ RIIIb, and were strongly associated with an increased risk of gastrointestinal inflammation. The development of other inflammatory complications was also shown to be associated with polymorphisms in the mannose binding lectin gene (MBL), and less so with Fcγ RIIa genotype.
CLINICAL PRESENTATION
Estimates of the incidence of CGD vary geographically and range from 1 in 200 000 in the USA [66] and 1 in 287 000 in Japan [67] to 1 in 450 000 in Sweden [68]. It is likely that the disease continues to be underdiagnosed. The hallmark of the clinical presentation of CGD is recurrent infections at epithelial surfaces in direct contact with the environment such as the skin, lungs and gut. The majority of affected individuals are diagnosed before the age of 2 years [69,70], although patients may remain undiagnosed until adult life despite the early onset of symptoms [71]. Lymphadenitis is the most common presenting feature [72,73], followed by skin abscesses, pneumonia and hepatomegaly. Diarrhoea and sepsis syndromes may also be a presenting feature and CGD may be misdiagnosed as Crohn's disease when diarrhoea and colitis are the initial findings [74]. A registry established in the USA contains data on 368 patients and has documented the clinical complications [66]. The most commonly described complications in this cohort are pneumonia (79%), followed by lymphadenitis (53%), subcutaneous abscess (42%), liver abscess (27%), osteomyelitis (25%) and sepsis (18%). Long-term follow up of CGD has revealed that with improved survival or increasing age, symptoms of obstruction in hollow organs or inflammation not obviously associated with infection may become prominent [70]. These include colitis, gastric outlet obstruction secondary to granulomatous involvement of the stomach wall, urinary tract obstruction secondary to granulomatous cystitis, and oesophageal obstruction secondary to granulomatous involvement of the oesophageal wall. Other rare complications of unknown aetiology such as pericardial effusion [75] and chorioretinitis [76] are well described. Individuals with X-CGD are said to have a more severe clinical phenotype and increased mortality than those with A470CGD[66].
The pathogens responsible for the majority of infections in CGD are characteristic bacteria and fungi. Catalase-positive bacteria are the most important and include S. aureus and the Gram-negative enterobacteriacea including Salmonella, Klebsiella, Aerobacter and Serratia. Pseudomonas (Burkholderia) cepacia is increasingly being recognized as an important pathogen [77]. Catalase-negative bacteria such as streptococci rarely cause problems, probably because small amounts of H2O2 are produced by the bacteria within the phagocytic vacuole. Aspergillus (predominantly fumigatus) is the fungus most commonly implicated in CGD, although reports of infections with other members of the Aspergillus family such as A. nidulans [78–79] and other fungi such as Candida albicans, Scedosporium apiospernum [80] and Chyrosporium zonatum [81] are prevalent.
Patients are often anaemic with an iron-deficient pattern, although this may remain stubbornly resistant to iron supplementation. Some patients with bowel disease may also malabsorb vitamin B12. A raised erythrocyte sedimentation rate (ESR) can be found, even in the apparently uninfected patients, and probably reflects ongoing, subclinical inflammation. The level of C-reactive protein is rarely raised when the patient is apparently infection-free and thus remains a better marker of bacterial sepsis in the acutely ill patient.
DIAGNOSIS
The functional diagnosis of CGD can be made by demonstrating the inability of phagocytes from affected individuals to produce a normal respiratory burst. This is conveniently done by the phorbol myristate acetate-stimulated nitroblue tetrazolium (NBT) test [82]. In this test, incubation of activated neutrophils with the yellow dye NBT results in the accumulation of dark blue pigment, formazan, within normal phagocytes, although proper interpretation relies on an experienced observer. For X-CGD, carrier status can be determined by observing a mixed population of NBT-positive and NBT-negative cells (assuming that X-inactivation is random). A flow cytometric test for superoxide production was first described in 1985 [83], being subsequently refined by many groups [84], and is now available in a kit form (Bursttest (Phagoburst®); Orpogen, Heidelberg, Germany) [85]. It relies on the reduction of dihydrorhodamine by stimulated phagocytes in heparinized whole blood and provides a quick and convenient method for semiquantitatively determining NAPDH–oxidase function. It can also accurately detect carrier status in X-CGD. Immunoblotting for individual components of the NAPDH–oxidase can help identify the defective protein in the majority of cases (remembering that mutations in either subunit of flavocytochrome b558 usually result in the absence of both), while confirmation of the molecular defect can be obtained by sequencing of the relevant gene. A database of CGD mutations has been set up [62] and can be accessed via the internet (http://http://www.uta.fi/laitokset/imt/bioinfo/XCGDbase/).
Prenatal diagnosis can be made on tissue obtained by chorionic villus sampling in the first trimester. This strategy is dependent on identification of specific family based mutations or on informative polymorphisms [86–88]. It may also be possible to detect the presence of individual NAPDH–oxidase components in chorion-derived macrophages with specific antibody [89]. Alternatively, prenatal diagnosis can be reliably determined by measurement of NAPDH–oxidase activity in fetal blood samples taken during the second trimester.
CLINICAL MANAGEMENT
Prophylactic treatments
Live bacterial vaccines such as bacille Calmette–Guérin (BCG) should be avoided. The mainstay of therapy is adequate anti-microbial prophylaxis. The most common antibiotic used for chemoprophylaxis is cotrimoxazole, which has broad activity against the pathogens encountered in CGD, is lipophilic and is thus concentrated inside cells, and is well tolerated because it does not affect anaerobic gut flora [90–92]. No randomized controlled trials of cotrimoxazole have been performed but a number of small studies have shown a reduction in the incidence of serious infections in patients on regular prophylaxis [93–96]. Anti-fungal prophylaxis can be achieved with itraconazole, a triazole anti-fungal with good activity against Aspergillus species. While no double blind, randomized, placebo controlled trials of itraconazole in CGD have been performed, a number of studies suggest it is efficacious in the prevention of Aspergillus infection [97,98]. Many anecdotal reports of its efficacy in the treatment of established Aspergillus infection have been published [99–101]. Interferon-gamma (IFN-γ) is an immunomodulatory cytokine which was shown to partially restore NAPDH–oxidase activity in the neutrophils and monocytes of selected patients with X-linked CGD [102–105]. On the basis of these preliminary results, IFN-γ was evaluated in a large multicentre study for its efficacy and potential toxicity in the prevention of infection in CGD [106]. While overall the study demonstrated significant efficacy in the IFN-γ arm, there was a powerful centre effect with the European study sites demonstrating similar rates of infection in the IFN-γ and control arms. Other European centres subsequently demonstrated incidence rates of infection using antibiotic prophylaxis that were lower than those seen in the IFN-γ arm in the USA sites [107]. In addition, there was a failure to demonstrate restoration of NAPDH–oxidase activity in patients enrolled in the clinical study [108,109]. While ongoing studies in Europe and the USA have demonstrated the safety of IFN-γ prophylaxis [110,111], it is not used universally.
Treatment of acute infection
Infections in CGD need to be managed promptly and aggressively with appropriate intravenous antibiotics or anti-fungal agents where necessary. In many sepsis syndromes, therapy must be empirical as a pathogen is rarely isolated. Ciprofloxacin is a useful first line agent with an appropriate spectrum of activity and an advantage in being lipophilic, thus achieving a high intracellular concentration [112,113]. Together with the Gram-positive cover provided by flucloxacillin, they form a useful first line empirical strategy for bacterial sepsis. Amphotericin is the mainstay of anti-fungal treatment; liposomal amphotericin at high dose (5 mg/kg) may be required to eradicate established Aspergillus infection [114] although itraconazole is also useful [99,100,115]. IFN-γ has been used in the treatment of deep seated infections (such as liver abscesses) that are refractory to conventional therapy, although efficacy remains anecdotal [99,115–119]. Leukophoresed normal granulocytes (matched for ABO and Rhesus antigens) are also useful in refractory situations [120,121], although the risk of alloimmunization remains significant [122].
The mainstay of therapy for inflammatory complications in CGD, including colitis and those leading to hollow organ obstruction, is immunomodulatory. Topical and systemic steroids have been used to treat colitis and obstructive complications [123] such as those encountered in the oesophagus [124] and bladder [125]. Some patients with mild enteritis or cystitis may be exquisitely sensitive to steroids (which may provide relief within the first 12 h of treatment), although relapse once off steroids may occur. A significant number of patients unfortunately remain dependent on steroids. In these cases, a variety of additional agents including sulphasalazine, cyclosporin [126], thalidomide [127] and granulocyte colony-stimulating factor [128] have been used.
OUTCOME
In 1967 only 21% of children with CGD were said to survive beyond 5 years of age, although the actuarial survival to 8 years had improved to 70% in a cohort born between 1967 and 1977 [93]. In this same study a cohort born after 1978 demonstrated an improved survival to 8 years of 92%. In a report by Finn et al. of CGD patients born over a 32-year period, 50% were alive through to the third decade [69]. A recent report from Japan has demonstrated an increase in the mean age of survivors from 8 years in 1985 to 16 years in 1998, although the overall mortality remained unchanged at 23·1% over the 13 years of the study [67]. The reasons for the failure to improve overall mortality are multifactorial, but probably include the problem of compliance with medication in adolescents with CGD.
CURATIVE THERAPY
Bone marrow transplantation has been shown to be potentially curative in CGD, although it has been associated with unacceptably high rates of morbidity, mortality and graft failure except in patients with an HLA-identical sibling donor [129]. Attempts to reduce conditioning-related toxicity are continuing using non-myeloablative regimens [130]. For patients without an HLA-identical donor, gene therapy may soon be a possibility. A number of preclinical studies have established the potential efficacy of this strategy [131–138]. However, in clinical studies reconstitution of NAPDH–oxidase activity in peripheral blood neutrophils has been transient [139]. It is therefore likely that some form of conditioning of the patients will be required for successful engraftment of corrected stem cells.
PATIENT SUPPORT
The CGD Research Trust and Support Group is an active group based in the UK who also raise money for, and fund research into CGD (http://www.cgd.org.uk). They are part of the International Patient Organization for Primary Immunodeficiency (IPOPI). Their website has useful links to other organizations (http://www.ipopi.org). In the USA, a number of websites provide information about CGD, including one at http://www.healthlinkusa.com/72ent.htm, which has links to a variety of other CGD-related websites.
Acknowledgments
Wellcome Trust, CGD Trust, PIA.
REFERENCES
- 1.Janeway CA, Craig J, Davidson M, Dwoney W, Gitlin D, Sullivan JC. Hypergammaglobulinaemia associated with severe recurrent and chronic non-specific infection. Am J Dis Child. 1954;88:388–92. [Google Scholar]
- 2.Kanding BH, Shirkey HS. A syndrome of recurrent infection and infiltration of viscera by pigmented lipid histiocytes. Pediatrics. 1957;20:431–8. [PubMed] [Google Scholar]
- 3.Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood. Am J Dis Child. 1959;97:387–408. [PubMed] [Google Scholar]
- 4.Quie PG, White JG, Holmes B, Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest. 1967;46:668–79. doi: 10.1172/JCI105568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes B, Quie PG, Windhorst DB, Good RA. Fatal granulomatous disease of childhood. An inborn abnormality of phagocytic function. Lancet. 1966;1:1225–8. doi: 10.1016/s0140-6736(66)90238-8. [DOI] [PubMed] [Google Scholar]
- 6.Baldridge CW, Gerard RW. The extra respiration of phagocytosis. Am J Physiol. 1933;103:235–6. [Google Scholar]
- 7.Sbarra AJ, Karnovsky MJ. The biochemical basis of phagocytosis: metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234:1355–62. [PubMed] [Google Scholar]
- 8.Baehner RL, Karnovsky ML. Deficiency of reduced nicotinamide-adenine dinucleotide oxidase in chronic granulomatous disease. Science. 1968;162:1277–9. doi: 10.1126/science.162.3859.1277. [DOI] [PubMed] [Google Scholar]
- 9.Segal AW, Peters TJ. Characterisation of the enzyme defect in chronic granulomatous disease. Lancet. 1976;1:1363–5. doi: 10.1016/s0140-6736(76)93021-x. [DOI] [PubMed] [Google Scholar]
- 10.Babior BM. NADPH. oxidase: an update. Blood. 1999;93:1464–76. [PubMed] [Google Scholar]
- 11.Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest. 1987;80:732–42. doi: 10.1172/JCI113128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shatwell KP, Segal AW. NADPH oxidase. Int J Biochem Cell Biol. 1996;28:1191–5. doi: 10.1016/s1357-2725(96)00084-2. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, Hitt ND, Kleinberg ME. Stoichiometry of p22-phox and gp91-phox in phagocyte cytochrome b558. Biochemistry. 1995;34:16753–7. doi: 10.1021/bi00051a024. [DOI] [PubMed] [Google Scholar]
- 14.Wallach TM, Segal AW. Stoichiometry of the subunits of flavocytochrome b558 of the NADPH oxidase of phagocytes. Biochem J. 1996;320:33–38. doi: 10.1042/bj3200033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallach TM, Segal AW. Analysis of glycosylation sites on gp91phox, the flavocytochrome of the NADPH oxidase, by site-directed mutagenesis and translation in vitro. Biochem J. 1997;321:583–5. doi: 10.1042/bj3210583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segal AW. Absence of both cytochrome b-245 subunits from neutrophils in X-linked chronic granulomatous disease. Nature. 1987;326:88–91. doi: 10.1038/326088a0. [DOI] [PubMed] [Google Scholar]
- 17.Parkos CA, Dinauer MC, Jesaitis AJ, Orkin SH, Curnutte JT. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood. 1989;73:1416–20. [PubMed] [Google Scholar]
- 18.Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558. Role of heme incorporation and heterodimer formation in maturation and stability of gp91phox and p22phox subunits. J Biol Chem. 1997;272:27288–94. doi: 10.1074/jbc.272.43.27288. [DOI] [PubMed] [Google Scholar]
- 19.Porter CD, Parkar MH, Verhoeven AJ, Levinsky RJ, Collins MK, Kinnon C. p22-phox-deficient chronic granulomatous disease: reconstitution by retrovirus-mediated expression and identification of a biosynthetic intermediate of gp91-phox. Blood. 1994;84:2767–75. [PubMed] [Google Scholar]
- 20.Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290:406–9. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 21.Nanda A, Grinstein S, Curnutte JT. Abnormal activation of H+ conductance in NADPH oxidase-defective neutrophils. Proc Natl Acad Sci USA. 1993;90:760–4. doi: 10.1073/pnas.90.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanda A, Curnutte JT, Grinstein S. Activation of H+ conductance in neutrophils requires assembly of components of the respiratory burst oxidase but not its redox function. J Clin Invest. 1994;93:1770–5. doi: 10.1172/JCI117162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson LM, Banting G, Chappell JB. The arachidonate-activable, NADPH oxidase-associated H+ channel. Evidence that gp91-phox functions as an essential part of the channel. J Biol Chem. 1995;270:5909–16. [PubMed] [Google Scholar]
- 24.Henderson LM, Meech RW. Evidence that the product of the human X-linked CGD gene, gp91-phox, is a voltage-gated H (+) pathway. J Gen Physiol. 1999;114:771–86. doi: 10.1085/jgp.114.6.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson LM, Thomas S, Banting G, Chappell JB. The arachidonate-activatable, NADPH oxidase-associated H+ channel is contained within the multi-membrane-spanning N-terminal region of gp91-phox. Biochem J. 1997;325:701–5. doi: 10.1042/bj3250701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banfi B, Schrenzel J, Nusse O, et al. A novel H (+) conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J Exp Med. 1999;190:183–94. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volpp BD, Nauseef WM, Clark RA. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science. 1988;242:1295–7. doi: 10.1126/science.2848318. [DOI] [PubMed] [Google Scholar]
- 28.Heyworth PG, Segal AW. Further evidence for the involvement of a phosphoprotein in the respiratory burst oxidase of human neutrophils. Biochem J. 1986;239:723–31. doi: 10.1042/bj2390723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal AW, Heyworth PG, Cockcroft S, Barrowman MM. Stimulated neutrophils from patients with autosomal recessive chronic granulomatous disease fail to phosphorylate a Mr-44,000 protein. Nature. 1985;316:547–9. doi: 10.1038/316547a0. [DOI] [PubMed] [Google Scholar]
- 30.Caldwell SE, McCall CE, Hendricks CL, Leone PA, Bass DA, McPhail LC. Coregulation of NADPH oxidase activation and phosphorylation of a 48-kD protein(s) by a cytosolic factor defective in autosomal recessive chronic granulomatous disease. J Clin Invest. 1988;81:1485–96. doi: 10.1172/JCI113480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer IM, van der Bend RL, Verhoeven AJ, Roos D. The 47-kDa protein involved in the NADPH:O2 oxidoreductase activity of human neutrophils is phosphorylated by cyclic AMP-dependent protein kinase without induction of a respiratory burst. Biochim Biophys Acta. 1988;971:189–96. doi: 10.1016/0167-4889(88)90191-7. [DOI] [PubMed] [Google Scholar]
- 32.Okamura N, Curnutte JT, Roberts RL, Babior BM. Relationship of protein phosphorylation to the activation of the respiratory burst in human neutrophils. Defects in the phosphorylation of a group of closely related 48-kDa proteins in two forms of chronic granulomatous disease. J Biol Chem. 1988;263:6777–82. [PubMed] [Google Scholar]
- 33.Bolscher BG, van Zwieten R, Kramer IM, Weening RS, Verhoeven AJ, Roos D. A phosphoprotein of Mr 47,000, defective in autosomal chronic granulomatous disease, copurifies with one of two soluble components required for NADPH:O2 oxidoreductase activity in human neutrophils. J Clin Invest. 1989;83:757–63. doi: 10.1172/JCI113954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dusi S, Rossi F. Activation of NADPH oxidase of human neutrophils involves the phosphorylation and the translocation of cytosolic p67phox. Biochem J. 1993;296:367–71. doi: 10.1042/bj2960367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPhail LC. SH3-dependent assembly of the phagocyte NADPH oxidase. J Exp Med. 1994;180:2011–5. doi: 10.1084/jem.180.6.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grogan A, Reeves E, Keep N, et al. Cytosolic phox proteins interact with and regulate the assembly of coronin in neutrophils. J Cell Sci. 1997;110:3071–81. doi: 10.1242/jcs.110.24.3071. [DOI] [PubMed] [Google Scholar]
- 37.Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–70. doi: 10.1038/353668a0. [DOI] [PubMed] [Google Scholar]
- 38.Roberts AW, Kim C, Zhen L, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–96. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 39.Quinn MT, Parkos CA, Walker L, Orkin SH, Dinauer MC, Jesaitis AJ. Association of a Ras-related protein with cytochrome b of human neutrophils. Nature. 1989;342:198–200. doi: 10.1038/342198a0. [DOI] [PubMed] [Google Scholar]
- 40.M'rabet L, Coffer P, Zwartkruis F, et al. Activation of the small GTPase rap1 in human neutrophils. Blood. 1998;92:2133–40. [PubMed] [Google Scholar]
- 41.Garcia RC, Segal AW. Changes in the subcellular distribution of the cytochrome b-245 on stimulation of human neutrophils. Biochem J. 1984;219:233–42. doi: 10.1042/bj2190233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wientjes FB, Panayotou G, Reeves E, Segal AW. Interactions between cytosolic components of the NADPH oxidase: p40phox interacts with both p67phox and p47phox. Biochem J. 1996;317:919–24. doi: 10.1042/bj3170919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leusen JH, Bolscher BG, Hilarius PM, et al. 156Pro–>Gln substitution in the light chain of cytochrome b558 of the human NADPH oxidase (p22-phox) leads to defective translocation of the cytosolic proteins p47-phox and p67-phox. J Exp Med. 1994;180:2329–34. doi: 10.1084/jem.180.6.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biberstine-Kinkade KJ, Yu L, Dinauer MC. Mutagenesis of an arginine- and lysine-rich domain in the gp91 (phox) subunit of the phagocyte NADPH-oxidase flavocytochrome b558. J Biol Chem. 1999;274:10451–7. doi: 10.1074/jbc.274.15.10451. [DOI] [PubMed] [Google Scholar]
- 45.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 46.Prigmore E, Ahmed S, Best A, et al. A 68-kDa kinase and NADPH oxidase component p67phox are targets for Cdc42Hs and Rac1 in neutrophils. J Biol Chem. 1995;270:10717–22. doi: 10.1074/jbc.270.18.10717. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed S, Prigmore E, Govind S, et al. Cryptic Rac-binding and p21 (Cdc42Hs/Rac)-activated kinase phosphorylation sites of NADPH oxidase component p67 (phox) J Biol Chem. 1998;273:15693–701. doi: 10.1074/jbc.273.25.15693. [DOI] [PubMed] [Google Scholar]
- 48.Heyworth PG, Bohl BP, Bokoch GM, Curnutte JT. Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. Evidence for its interaction with flavocytochrome b558. J Biol Chem. 1994;269:30749–52. [PubMed] [Google Scholar]
- 49.Qu AY, Nanda A, Curnutte JT, Grinstein S. Development of a H (+)-selective conductance during granulocytic differentiation of HL-60 cells. Am J Physiol. 1994;266:C1263–C1270. doi: 10.1152/ajpcell.1994.266.5.C1263. [DOI] [PubMed] [Google Scholar]
- 50.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–8. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollock JD, Williams DA, Gifford MA, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–9. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 52.Shiloh MU, MacMicking JD, Nicholson S, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 53.Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–10. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 54.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med. 1997;185:207–18. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Royer-Pokora B, Kunkel LM, Monaco AP, et al. Cloning the gene for the inherited disorder chronic granulomatous disease on the basis of its chromosomal location. Cold Spring Harb Symp Quant Biol. 1986;51:177–83. doi: 10.1101/sqb.1986.051.01.021. [DOI] [PubMed] [Google Scholar]
- 56.Volpp BD, Nauseef WM, Donelson JE, Moser DR, Clark RA. Cloning of the cDNA and functional expression of the 47-kilodalton cytosolic component of human neutrophil respiratory burst oxidase [published erratum appears in Proc Natl Acad Sci USA 1989 86:9563] Proc Natl Acad Sci USA. 1989;86:7195–9. doi: 10.1073/pnas.86.18.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dinauer MC, Pierce EA, Bruns GA, Curnutte JT, Orkin SH. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J Clin Invest. 1990;86:1729–37. doi: 10.1172/JCI114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Francke U, Hsieh CL, Foellmer BE, Lomax KJ, Malech HL, Leto TL. Genes for two autosomal recessive forms of chronic granulomatous disease assigned to 1q25 (NCF2) and 7q11.23 (NCF1) Am J Hum Genet. 1990;47:483–92. [PMC free article] [PubMed] [Google Scholar]
- 59.Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 60.Weening RS, Wever R, Roos D. Quantitative aspects of the production of superoxide radicals by phagocytizing human granulocytes. J Lab Clin Med. 1975;85:245–52. [PubMed] [Google Scholar]
- 61.Roos D, de Boer M, de Klein A, Bolscher BG, Weening RS. Chronic granulomatous disease: mutations in cytochrome b558. Immunodeficiency. 1993;4:289–301. [PubMed] [Google Scholar]
- 62.Roos D, de Boer M, Kuribayashi F, et al. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood. 1996;87:1663–81. [PubMed] [Google Scholar]
- 63.Casimir CM, Bu-Ghanim HN, Rodaway AR, Bentley DL, Rowe P, Segal AW. Autosomal recessive chronic granulomatous disease caused by deletion at a dinucleotide repeat. Proc Natl Acad Sci USA. 1991;88:2753–7. doi: 10.1073/pnas.88.7.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roesler J, Curnutte JT, Rae J, et al. Recombination events between the p47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood. 2000;95:2150–6. [PubMed] [Google Scholar]
- 65.Foster CB, Lehrnbecher T, Mol F, et al. Host defense molecule polymorphisms influence the risk for immune-mediated complications in chronic granulomatous disease. J Clin Invest. 1998;102:2146–55. doi: 10.1172/JCI5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winkelstein JA, Marino K, Johnston RB, et al. Chronic granulomatous disease: report on a registry of 368 patients. Mol Immunol. 1998;35:RE10. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Hasui M. Chronic granulomatous disease in Japan: incidence and natural history. The Study Group of Phagocyte Disorders of Japan. Pediatr Int. 1999;41:589–93. doi: 10.1046/j.1442-200x.1999.01129.x. [DOI] [PubMed] [Google Scholar]
- 68.Ahlin A, de Boer M, Roos D, et al. Prevalence, genetics and clinical presentation of chronic granulomatous disease in Sweden. Acta Paediatr. 1995;84:1386–94. doi: 10.1111/j.1651-2227.1995.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 69.Finn A, Hadzic N, Morgan G, Strobel S, Levinsky RJ. Prognosis of chronic granulomatous disease. Arch Dis Child. 1990;65:942–5. doi: 10.1136/adc.65.9.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cale CM, Jones AM, Goldblatt D. Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clin Exp Immunol. 2000;120:1–6. doi: 10.1046/j.1365-2249.2000.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liese JG, Jendrossek V, Jansson A, et al. Chronic granulomatous disease in adults. Lancet. 1996;347:220–3. doi: 10.1016/s0140-6736(96)90403-1. [DOI] [PubMed] [Google Scholar]
- 72.Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, Gahr M, Belohradsky B. Chronic granulomatous disease (CGD): incidence of infections and complications in 39 patients. Mol Med. 1998;35:CL65. [Google Scholar]
- 73.Gomez L. Outcome of CGD patients. Mol Immunol. 1998;35:CL64. [Google Scholar]
- 74.Mitomi H, Mikami T, Takahashi H, et al. Colitis in chronic granulomatous disease resembling Crohn's disease: comparative analysis of CD68-positive cells between two disease entities. Dig Dis Sci. 1999;44:452–6. doi: 10.1023/a:1026643609944. [DOI] [PubMed] [Google Scholar]
- 75.Macedo F, McHugh K, Goldblatt D. Pericardial effusions in two boys with chronic granulomatous disease. Pediatr Radiol. 1999;29:820–2. doi: 10.1007/s002470050704. [DOI] [PubMed] [Google Scholar]
- 76.Goldblatt D, Butcher J, Thrasher AJ, Russell-Eggitt I. Chorioretinal lesions in patients and carriers of chronic granulomatous disease. J Pediatr. 1999;134:780–3. doi: 10.1016/s0022-3476(99)70299-4. [DOI] [PubMed] [Google Scholar]
- 77.Dorman SE, Gill VJ, Gallin JI, Holland SM. Burkholderia pseudomallei infection in a Puerto Rican patient with chronic granulomatous disease: case report and review of occurrences in the Americas. Clin Infect Dis. 1998;26:889–94. doi: 10.1086/513928. [DOI] [PubMed] [Google Scholar]
- 78.Kim M, Shin JH, Suh SP, et al. Aspergillus nidulans infection in a patient with chronic granulomatous disease. J Korean Med Sci. 1997;12:244–8. doi: 10.3346/jkms.1997.12.3.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Segal BH, DeCarlo ES, Kwon-Chung KJ, Malech HL, Gallin JI, Holland SM. Aspergillus nidulans infection in chronic granulomatous disease. Medicine (Baltimore) 1998;77:345–54. doi: 10.1097/00005792-199809000-00004. [DOI] [PubMed] [Google Scholar]
- 80.Jabado N, Casanova JL, Haddad E, et al. Invasive pulmonary infection due to Scedosporium apiospermum in two children with chronic granulomatous disease. Clin Infect Dis. 1998;27:1437–41. doi: 10.1086/515015. [DOI] [PubMed] [Google Scholar]
- 81.Roilides E, Sigler L, Bibashi E, Katsifa H, Flaris N, Panteliadis C. Disseminated infection due to Chrysosporium zonatum in a patient with chronic granulomatous disease and review of non-Aspergillus fungal infections in patients with this disease. J Clin Microbiol. 1999;37:18–25. doi: 10.1128/jcm.37.1.18-25.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Repine JE, Rasmussen B, White JG. An improved nitroblue tetrazolium test using phorbol myristate acetate-coated coverslips. Am J Clin Pathol. 1979;71:582–5. doi: 10.1093/ajcp/71.5.582. [DOI] [PubMed] [Google Scholar]
- 83.Taga K, Seki H, Miyawaki T, et al. Flow cytometric assessment of neutrophil oxidative metabolism in chronic granulomatous disease on small quantities of whole blood: heterogeneity in female patients. Hiroshima J Med Sci. 1985;34:53–60. [PubMed] [Google Scholar]
- 84.Perticarari S, Presani G, Banfi E. A new flow cytometric assay for the evaluation of phagocytosis and the oxidative burst in whole blood. J Immunol Methods. 1994;170:117–24. doi: 10.1016/0022-1759(94)90251-8. [DOI] [PubMed] [Google Scholar]
- 85.Hirt W, Nebe T, Birr C. Phagotest and Bursttest (Phagoburst), test kits for study of phagocyte functions. Wien Klin Wochenschr. 1994;106:250–2. [PubMed] [Google Scholar]
- 86.Muhlebach TJ, Robinson W, Seger RA, Machler M. A second NsiI RFLP at the CYBB locus. Nucl Acids Res. 1990;18:4966. doi: 10.1093/nar/18.16.4966-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pelham A, O'reilly MA, Malcolm S, Levinsky RJ, Kinnon C. RFLP and deletion analysis for X-linked chronic granulomatous disease using the cDNA probe: potential for improved prenatal diagnosis and carrier determination. Blood. 1990;76:820–4. [PubMed] [Google Scholar]
- 88.Kenney RT, Leto TLA. HindIII polymorphism in the human NCF2 gene. Nucl Acids Res. 1990;18:7193. doi: 10.1093/nar/18.23.7193-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nakamura M, Imajoh-Ohmi S, Kanegasaki S, et al. Prenatal diagnosis of cytochrome-deficient chronic granulomatous disease. Lancet. 1990;336:118–9. doi: 10.1016/0140-6736(90)91635-n. [DOI] [PubMed] [Google Scholar]
- 90.Seger RA, Baumgartner S, Tiefenauer LX, Gmunder FK. Chronic granulomatous disease: effect of sulfamethoxazole/trimethoprim on neutrophil microbicidal function. Helv Paediatr Acta. 1981;36:579–88. [PubMed] [Google Scholar]
- 91.Jacobs RF, Wilson CB. Activity of antibiotics in chronic granulomatous disease leukocytes. Pediatr Res. 1983;17:916–9. doi: 10.1203/00006450-198311000-00016. [DOI] [PubMed] [Google Scholar]
- 92.Gmunder FK, Seger RA. Chronic granulomatous disease: mode of action of sulfamethoxazole/trimethoprim. Pediatr Res. 1981;15:1533–7. doi: 10.1203/00006450-198112000-00017. [DOI] [PubMed] [Google Scholar]
- 93.Mouy R, Fischer A, Vilmer E, Seger R, Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114:555–60. doi: 10.1016/s0022-3476(89)80693-6. [DOI] [PubMed] [Google Scholar]
- 94.Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. 1990;162:723–6. doi: 10.1093/infdis/162.3.723. [DOI] [PubMed] [Google Scholar]
- 95.Weening RS, Kabel P, Pijman P, Roos D. Continuous therapy with sulfamethoxazole-trimethoprim in patients with chronic granulomatous disease. J Pediatr. 1983;103:127–30. doi: 10.1016/s0022-3476(83)80798-7. [DOI] [PubMed] [Google Scholar]
- 96.Kobayashi Y, Amano D, Ueda K, Kagosaki Y, Usui T. Treatment of seven cases of chronic granulomatous disease with sulfamethoxazole-trimethoprim (SMX-TMP) Eur J Pediatr. 1978;127:247–54. doi: 10.1007/BF00493540. [DOI] [PubMed] [Google Scholar]
- 97.Petropoulou T, Liese J, Tintelnot K, Gahr M, Belohradsky BH. Long-term treatment of patients with itraconazole for the prevention of Aspergillus infections in patients with chronic granulomatous disease (CGD). Langzeitbehandlung mit Itraconazol zur Prophylaxe von Aspergillus-Infektionen bei Patienten mit chronischer Granulomatose (CGD) Mycoses. 1994;37(Suppl. 2):64–69. [PubMed] [Google Scholar]
- 98.Mouy R, Veber F, Blanche S, et al. Long-term itraconazole prophylaxis against Aspergillus infections in thirty-two patients with chronic granulomatous disease. J Pediatr. 1994;125:998–1003. doi: 10.1016/s0022-3476(05)82023-2. [DOI] [PubMed] [Google Scholar]
- 99.Spencer DA, John P, Ferryman SR, Weller PH, Darbyshire P. Successful treatment of invasive pulmonary aspergillosis in chronic granulomatous disease with orally administered itraconazole suspension. Am J Respir Crit Care Med. 1994;149:239–41. doi: 10.1164/ajrccm.149.1.8111588. [DOI] [PubMed] [Google Scholar]
- 100.Kloss S, Schuster A, Schroten H, Lamprecht J, Wahn V. Control of proven pulmonary and suspected CNS aspergillus infection with itraconazole in a patient with chronic granulomatous disease. Eur J Pediatr. 1991;150:483–5. doi: 10.1007/BF01958428. [DOI] [PubMed] [Google Scholar]
- 101.‘t-Wout JW, Raven EJ, van der Meer JW. Treatment of invasive aspergillosis with itraconazole in a patient with chronic granulomatous disease. J Infect. 1990;20:147–50. doi: 10.1016/0163-4453(90)93418-r. [DOI] [PubMed] [Google Scholar]
- 102.Ezekowitz RA, Dinauer MC, Jaffe HS, Orkin SH, Newburger PE. Partial correction of the phagocyte defect in patients with X-linked chronic granulomatous disease by subcutaneous interferon gamma. N Engl J Med. 1988;319:146–51. doi: 10.1056/NEJM198807213190305. [DOI] [PubMed] [Google Scholar]
- 103.Newburger PE, Ezekowitz RA, Whitney C, Wright J, Orkin SH. Induction of phagocyte cytochrome b heavy chain gene expression by interferon gamma. Proc Natl Acad Sci USA. 1988;85:5215–9. doi: 10.1073/pnas.85.14.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Newburger PE, Ezekowitz RA. Cellular and molecular effects of recombinant interferon gamma in chronic granulomatous disease. Hematol Oncol Clin North Am. 1988;2:267–76. [PubMed] [Google Scholar]
- 105.Ezekowitz RA, Orkin SH, Newburger PE. Recombinant interferon gamma augments phagocyte superoxide production and X-chronic granulomatous disease gene expression in X-linked variant chronic granulomatous disease. J Clin Invest. 1987;80:1009–16. doi: 10.1172/JCI113153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.The International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med. 1991;324:509–16. doi: 10.1056/NEJM199102213240801. [DOI] [PubMed] [Google Scholar]
- 107.Mouy R, Seger R, Bourquin JP, et al. Interferon gamma for chronic granulomatous disease. N Engl J Med. 1991;325:1516–7. doi: 10.1056/NEJM199111213252115. [DOI] [PubMed] [Google Scholar]
- 108.Muhlebach TJ, Gabay J, Nathan CF, et al. Treatment of patients with chronic granulomatous disease with recombinant human interferon-gamma does not improve neutrophil oxidative metabolism, cytochrome b558 content or levels of four anti-microbial proteins. Clin Exp Immunol. 1992;88:203–6. doi: 10.1111/j.1365-2249.1992.tb03062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Woodman RC, Erickson RW, Rae J, Jaffe HS, Curnutte JT. Prolonged recombinant interferon-gamma therapy in chronic granulomatous disease: evidence against enhanced neutrophil oxidase activity. Blood. 1992;79:1558–62. [PubMed] [Google Scholar]
- 110.Weening RS, Leitz GJ, Seger RA. Recombinant human interferon-gamma in patients with chronic granulomatous disease—European follow up study. Eur J Pediatr. 1995;154:295–8. doi: 10.1007/BF01957365. [DOI] [PubMed] [Google Scholar]
- 111.Gallin JI. Interferon-gamma in the treatment of the chronic granulomatous diseases of childhood. Clin Immunol Immunopathol. 1991;61:S100–S105. doi: 10.1016/s0090-1229(05)80044-3. [DOI] [PubMed] [Google Scholar]
- 112.Peman J, Canton E, Hernandez MT, Gobernado M. Intraphagocytic killing of gram-positive bacteria by ciprofloxacin. J Antimicrob Chemother. 1994;34:965–74. doi: 10.1093/jac/34.6.965. [DOI] [PubMed] [Google Scholar]
- 113.Canton E, Peman J, Cabrera E, et al. Killing of gram-negative bacteria by ciprofloxacin within both healthy human neutrophils and neutrophils with inactivated O2-dependent bactericidal mechanisms. Chemotherapy. 1999;45:268–76. doi: 10.1159/000007196. [DOI] [PubMed] [Google Scholar]
- 114.Ozsahin H, von Planta M, Muller I, et al. Successful treatment of invasive aspergillosis in chronic granulomatous disease by bone marrow transplantation, granulocyte colony-stimulating factor-mobilized granulocytes, and liposomal amphotericin-B. Blood. 1998;92:2719–24. [PubMed] [Google Scholar]
- 115.Pasic S, Abinun M, Pistignjat B, et al. Aspergillus osteomyelitis in chronic granulomatous disease: treatment with recombinant gamma-interferon and itraconazole. Pediatr Infect Dis J. 1996;15:833–4. doi: 10.1097/00006454-199609000-00021. [DOI] [PubMed] [Google Scholar]
- 116.Rosh JR, Tang HB, Mayer L, Groisman G, Abraham SK, Prince A. Treatment of intractable gastrointestinal manifestations of chronic granulomatous disease with cyclosporine. J Pediatr. 1995;126:143–5. doi: 10.1016/s0022-3476(95)70519-8. [DOI] [PubMed] [Google Scholar]
- 117.Lekstrom-Himes JA, Holland SM, DeCarlo ES, et al. Treatment with intralesional granulocyte instillations and interferon-gamma for a patient with chronic granulomatous disease and multiple hepatic abscesses. Clin Infect Dis. 1994;19:770–3. doi: 10.1093/clinids/19.4.770. [DOI] [PubMed] [Google Scholar]
- 118.Hague RA, Eastham EJ, Lee RE, Cant AJ. Resolution of hepatic abscess after interferon gamma in chronic granulomatous disease. Arch Dis Child. 1993;69:443–5. doi: 10.1136/adc.69.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Malmvall BE, Follin P. Successful interferon-gamma therapy in a chronic granulomatous disease (CGD) patient suffering from Staphylococcus aureus hepatic abscess and invasive Candida albicans infection. Scand J Infect Dis. 1993;25:61–66. doi: 10.1080/00365549309169671. [DOI] [PubMed] [Google Scholar]
- 120.Quie PG. The white cells: use of granulocyte transfusions. Rev Infect Dis. 1987;9:189–93. doi: 10.1093/clinids/9.1.189. [DOI] [PubMed] [Google Scholar]
- 121.von Planta M, Ozsahin H, Schroten H, Stauffer UG, Seger RA. Greater omentum flaps and granulocyte transfusions as combined therapy of liver abscess in chronic granulomatous disease. Eur J Pediatr Surg. 1997;7:234–6. doi: 10.1055/s-2008-1071100. [DOI] [PubMed] [Google Scholar]
- 122.Stroncek DF, Leonard K, Eiber G, Malech HL, Gallin JI, Leitman SF. Alloimmunization after granulocyte transfusions. Transfusion. 1996;36:1009–15. doi: 10.1046/j.1537-2995.1996.36111297091747.x. [DOI] [PubMed] [Google Scholar]
- 123.Chin TW, Stiehm ER, Falloon J, Gallin JI. Corticosteroids in treatment of obstructive lesions of chronic granulomatous disease. J Pediatr. 1987;111:349–52. doi: 10.1016/s0022-3476(87)80452-3. [DOI] [PubMed] [Google Scholar]
- 124.al Tawil YS, Abramson SL, Gilger MA, Paul ME. Steroid-responsive esophageal obstruction in a child with chronic granulomatous disease (CGD) J Pediatr Gastroenterol Nutr. 1996;23:182–5. doi: 10.1097/00005176-199608000-00015. [DOI] [PubMed] [Google Scholar]
- 125.Collman RJ, Dickerman JD. Corticosteroids in the management of cystitis secondary to chronic granulomatous disease. Pediatrics. 1990;85:219–21. [PubMed] [Google Scholar]
- 126.Barton LL, Moussa SL, Villar RG, Hulett RL. Gastrointestinal complications of chronic granulomatous disease: case report and literature review. Clin Pediatr (Phila) 1998;37:231–6. doi: 10.1177/000992289803700403. [DOI] [PubMed] [Google Scholar]
- 127.Nielsen H, Valerius NH. Thalidomide enhances superoxide anion release from human polymorphonuclear and mononuclear leukocytes. Acta Pathol Microbiol Immunol Scand [C ] 1986;94:233–7. doi: 10.1111/j.1699-0463.1986.tb02117.x. [DOI] [PubMed] [Google Scholar]
- 128.Myrup B, Valerius NH, Mortensen PB. Treatment of enteritis in chronic granulomatous disease with granulocyte colony stimulating factor. Gut. 1998;42:127–30. doi: 10.1136/gut.42.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leung T, Chik K, Li C, Shing M, Yuen P. Bone marrow transplantation for chronic granulomatous disease: long-term follow-up and review of literature. Bone Marrow Transplant. 1999;24:567–70. doi: 10.1038/sj.bmt.1701932. [DOI] [PubMed] [Google Scholar]
- 130.Horwitz ME, Barrett AJ, Childs R, et al. Nonmyeloablative, T-cell depleted allogeneic peripheral blood stem cell (PBSC) transplantation for patients with chronic granulomatous disease. American Society of Haematology. 1999:710a. [Google Scholar]
- 131.Sekhsaria S, Gallin JI, Linton GF, Mallory RM, Mulligan RC, Malech HL. Peripheral blood progenitors as a target for genetic correction of p47phox-deficient chronic granulomatous disease. Proc Natl Acad Sci USA. 1993;90:7446–50. doi: 10.1073/pnas.90.16.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li F, Linton GF, Sekhsaria S, et al. CD34+ peripheral blood progenitors as a target for genetic correction of the two flavocytochrome b558 defective forms of chronic granulomatous disease. Blood. 1994;84:53–58. [PubMed] [Google Scholar]
- 133.Weil WM, Linton GF, Whiting-Theobald N, et al. Genetic correction of p67phox deficient chronic granulomatous disease using peripheral blood progenitor cells as a target for retrovirus mediated gene transfer. Blood. 1997;89:1754–61. [PubMed] [Google Scholar]
- 134.Becker S, Wasser S, Hauses M, et al. Correction of respiratory burst activity in X-linked chronic granulomatous cells to therapeutically relevant levels after gene transfer into bone marrow CD34+ cells. Hum Gene Ther. 1998;9:1561–70. doi: 10.1089/hum.1998.9.11-1561. [DOI] [PubMed] [Google Scholar]
- 135.Iwata M, Nunoi H, Matsuda I, Kanegasaki S, Tsuruo T, Sugimoto Y. Drug-selected complete restoration of superoxide generation in Epstein–Barr virus-transformed B cells from p47phox-deficient chronic granulomatous disease patients by using a bicistronic retrovirus vector encoding a human multi-drug resistance gene (MDR1) and the p47phox gene. Hum Genet. 1998;103:419–23. doi: 10.1007/s004390050844. [DOI] [PubMed] [Google Scholar]
- 136.Bjorgvinsdottir H, Ding C, Pech N, Gifford MA, Li LL, Dinauer MC. Retroviral-mediated gene transfer of gp91phox into bone marrow cells rescues defect in host defense against Aspergillus fumigatus in murine X-linked chronic granulomatous disease. Blood. 1997;89:41–48. [PubMed] [Google Scholar]
- 137.Mardiney M, III, Jackson SH, Spratt SK, Li F, Holland SM, Malech HL. Enhanced host defense after gene transfer in the murine p47phox- deficient model of chronic granulomatous disease. Blood. 1997;89:2268–75. [PubMed] [Google Scholar]
- 138.Dinauer MC, Li LL, Bjorgvinsdottir H, Ding C, Pech N. Long-term correction of phagocyte NADPH oxidase activity by retroviral- mediated gene transfer in murine X-linked chronic granulomatous disease. Blood. 1999;94:914–22. [PubMed] [Google Scholar]
- 139.Malech HL, Maples PB, Whiting-Theobald N, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA. 1997;94:12133–8. doi: 10.1073/pnas.94.22.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roos D, Curnutte JT. Chronic granulomatous disease. In: Ochs HD, Edvard Smith CI, Puck JM, editors. Primary immunodeficiency diseases. Oxford: Oxford University Press; 1999. [Google Scholar]