

Abstract
Many cell surface proteins attached to the membrane by GPI are involved in cell signalling. However, the role of the GPI membrane anchor itself remains poorly understood. GPI-defective cells from patients with paroxysmal nocturnal haemoglobinuria (PNH) are relatively resistant to apoptosis induction. We developed a Jurkat T cell model for GPI deficiency by isolating a GPI-negative mutant, which is defective in the GPI biosynthetic gene PIG-A. Using retroviral PIG-A gene transfer along with the transfer of a vector control, we obtained two genetically identical cell lines, distinguished only by expression of the PIG-A gene and, thus, their ability to produce GPI. Cell proliferation and survival were not affected by this difference. Apoptotic stimuli such as serum starvation and camptothecin exposure elicited similar responses. In contrast, GPI-defective Jurkat cells were more susceptible to Fas-mediated apoptosis than GPI-positive cells. These results indicate that a deficiency in GPI-anchored proteins, as is found in PNH, does not confer resistance to apoptosis.
Keywords: GPI membrane anchor, paroxysmal nocturnal haemoglobinuria, PIG-A, T lymphocytes, apoptosis
INTRODUCTION
A fundamental issue in immunology concerns the molecular mechanisms of cell activation, by which proliferation, differentiation and functioning of cells is controlled. In lymphocytes, a complex network of signals is delivered by the T cell receptor complex (TCR) and numerous costimulatory molecules to regulate positive and negative selection during T cell development as well as mature T cell activities [1,2]. A striking number of cell surface molecules implicated in T cell signalling are attached to the plasma membrane by covalently linked GPI [3,4]. These include the leucocyte antigens CD48, CD55 (DAF), CD58 (LFA-3), CD59 (MIRL), CD90 (Thy-1) and members of the Ly-6 family. They are found in specific, detergent-resistant subdomains of the plasma membrane, which are probably kept together by the hydrophobic interaction of their lipid constituents [5,6]. Cytoplasmic protein kinases of the Src family as well as some transmembrane proteins, including CD4 and CD8, associate with these membrane islets [7,8]. Cross-linking of GPI-anchored molecules can lead to cell activation by triggering diverse signalling pathways. For example, CD59 cross-linking can induce a time-dependent activation of p56lck as well as activation via ZAP-70, eliciting the synthesis of IL-2 [9]. In contrast, apoptotic cell death of murine thymocytes can be induced by antibodies directed against Thy-1, which is abundantly expressed on haematopoietic stem cells, lymphoid progenitors and mouse T cells [10].
Despite these and many other indications for a role of GPI-anchored molecules in T cell physiology, there is no clear evidence for the importance of the GPI anchor itself. Studies comparing GPI-deficient T cell lines with normal controls indicate some stimulatory defect in the response to phytohaemagglutinin (PHA) [11]. In contrast, TCR-specific stimulation elicited a comparable response [11]. However, the GPI-deficient T cell lines used in this and other studies were isolated from patients with paroxysmal nocturnal haemoglobinuria (PNH). This disorder of haematopoiesis is characterized by GPI deficiency on a subset of all blood cell lineages. The defect is due to an acquired mutation of the X-linked, GPI biosynthetic gene PIG-A in early haematopoietic progenitors [12]. Since PNH is a clonal disorder of haematopoiesis and is associated with a relative growth advantage of the GPI-deficient clone, results from ‘activation’ studies must be interpreted with caution. Although yet unidentified, there might be one or more additional genetic alterations causing the GPI-deficient clone to expand. Thus, GPI-deficient bone marrow cells from PNH cells have been reported to be resistant to apoptosis induction [13]. Others found that both GPI-positive and -negative peripheral blood leucocytes from PNH patients are relatively resistant to apoptosis induction [14,15]. It is unclear to what extent the GPI‐anchoring defect itself contributes to these phenomena. Therefore, we established a new model of GPI deficiency in T cells by isolating a GPI-negative Jurkat T cell clone. This clone, which is characterized here, will be particularly useful in studying the role of GPI anchors in T cell biology.
MATERIALS AND METHODS
Cells
Jurkat E.6-1 cells (ATCC, Rockville, MD) were grown in standard RPMI 1640 medium (Seromed, Berlin, Germany) supplemented with 10% fetal calf serum (FCS; PAA Laboratories, Ling, Austria), 2 mm l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all Seromed). Cell mutagenization and isolation of GPI-deficient mutants was carried out as described [16]. Briefly, cells were cultured at a density of 0·5 × 106/ml in ethyl-methansulfonate at a concentration of 200 μg/ml for 18 h. Cells were then washed and allowed to recover for 5 days. Using CD55, CD58 and CD59 MoAbs and non-toxic rabbit complement (Behring, Marburg, Germany), cells expressing GPI-anchored proteins were eliminated by three rounds of negative selection. After labelling with CD59 MoAb H19 (PharMingen, Hamburg, Germany), CD59− cells were sorted using a fluorescence activated cell sorter (FACStar; Becton Dickinson, Mountain View, CA). The absence of surface expression of GPI-anchored proteins was checked by flow cytometry.
Antibodies
The following MoAbs were used: OKT3 (CD3, unconjugated, mouse; Ortho Diagnostics, Krefeld, Germany); SK3 (CD4, unconjugated, mouse; Becton Dickinson); B9.11 (CD8, FITC-conjugated, mouse; Immunotech, Hamburg, Germany); IOT28 (CD28, mouse: Immunotech); MEM102 (CD48, unconjugated, mouse; Dianova, Hamburg, Germany); YTH66.9HL (CD52, FITC-conjugated, rat; Serotech, Eching, Germany); BRIC110 (CD55, unconjugated, mouse; Integra Biosciences, Fernwald, Germany); MEM43 (CD59, unconjugated, mouse; Dianova); H19 (CD59, unconjugated, mouse; PharMingen); and G254-274 (CD95, unconjugated, mouse; PharMingen). FITC-conjugated goat anti-mouse serum was purchased from PharMingen. FACS analysis was carried out using standard techniques and equipment (FACScan cytometer; Becton Dickinson).
In vitro analysis of GPI anchors
Cells (2 × 107) were subjected to hypotonic lysis after pretreatment with 5 μg/ml tunicamycin for 2 h. GPI biosynthetic intermediates were labelled with 2 μCi UDP-3H-GlcNAc [17]. Following butanol/water extraction, lipids were resolved by thin layer chromatography (TLC) in chloroform/methanol/water (10:10:3). TLC plates were scanned using a Tracemaster 20 linear scanner (Chroma 2D; Berthold, Bad Wildbad, Germany).
Analysis of PIG-A
Total RNA was isolated using RNAzol B (Biozol, Eching, Germany). Reverse transcription was performed with M-MLV reverse transcriptase (Boehringer, Mannheim, Germany) using 30 pmol of a specific antisense primer (5′-AATGATATAGAGGTAGCATAA). Polymerase chain reaction (PCR) amplification was performed in duplicate assays using Taq polymerase (Boehringer) and the following printers: sense 5′-CCGTCGCTCGAGTCTTACAATCTAGGCTTCC, antisense 5′-GGGGTACCAGAACTGATGTCTAAACCG. Cycling parameters were: 60 s 90°C, 50 s 55°C, 70 s 72°C, 32 cycles. PCR fragments were cloned into the pGEM T vector (Promega, Mannheim, Germany) and sequenced in both directions (MWG Biotech, Ebersberg, Germany). Genomic DNA was prepared using the Qiamp Tissue Kit (Qiagen, Hilden, Germany). Amplification of PIG-A exon 2 was carried out with 5 μg gDNA and the following set of primers: sense 5′-GAGCTGAGATCCTGTTTTACTCT, antisense 5′-GCCAAACAATCATTATATACAAG. PCR conditions, cloning and sequencing were carried out as described above.
Retroviral PIG-A expression vectors
The complete PIG-A coding region was PCR-amplified from cDNA using the following primers: sense 5′-CCGTCGCTCGAGTCTTACAATCTAGGCTTCC, antisense 5′-CAGGCACTCGAGAACTGATGTCTAAACCG. PCR products were cloned into the G418-selectable retroviral vector pLXSN [18] using Xho I restriction sites engineered into PCR primers. The resulting vector is termed pL-PIG-A-SN.
Transient expression of PIG-A
pL-PIG-A-SN plasmid (20 μg) was transfected into 5 × 106 cells by electroporation. Cells were incubated in standard medium overnight and subsequently selected by incubation with 750 μg/ml G418.
Generation of retrovirus producer cells
A subconfluent monolayer of ecotropic GP+E86 cells was transfected with 20 μg of the retroviral vector plasmid with the calcium precipitation method. Twenty-four hours after transfection, cells were selected with 600 μg/ml G418 for 2 weeks. After an additional incubation period of 24 h in G418-free medium, virus-containing supernatant was removed and filtered (0·45 μm). A subconfluent monolayer of GALV pseudotyped PG13 or amphotropic PA317 packaging cells was layered with virus in the presence of 5 μg/ml protamine sulfate. After 24 h, cells were selected with 600 μg/ml G418. Subsequent to subcloning, cells were screened for virus production. Titres ranged between 5 × 104 and 2 × 105 colony-forming units (CFU)/ml.
Retroviral infection of Jurkat-7 cells
GPI-deficient Jurkat-7 cells were transduced with filtered viral supernatant of PG13 producer cells at a multiplicity of infection (moi) of 1 in the presence of 5 μg/ml protamine sulfate. Cells were cultured for 24 h in the presence of 750 μg/ml G418. Restoration of GPI anchoring was tested by fluorimetric analysis of CD48, CD55 and CD59 expression.
Apoptosis assays
Jurkat-7 cells transfected with pL-PIG-A-SN or pLXSN (vector control) were subjected to apoptosis induction by the following methods: (i) serum removal for 1–4 days; (ii) addition of 200 ng/ml camptothecin (Sigma, Munich, Germany) for 2–6 h; (iii) incubation with CD95 MoAb for 1 h. To measure apoptosis, 106 washed cells were resuspended in 1 ml binding buffer (10 mm HEPES/NaOH pH 7·4, 140 mm NaCl, 2·5 mm CaCl2) and incubated in buffer alone or buffer with 5% (v/v) annexin V–PE (PharMingen), 5% 7-AAD or 5% annexin plus 5% 7-AAD for 15 min. After addition of 4 volumes of binding buffer, cells were analysed by fluorimetry. Cells positive for annexin but negative for 7-AAD were regarded as apoptotic. In some experiments, 7-AAD was replaced by propidium iodide.
RESULTS
Jurkat-7 cells are deficient in GPI biosynthesis
Jurkat E.6-1 cells were mutagenized in vitro. After enrichment and subcloning of CD59− cells, these were analysed for expression of other GPI-anchored proteins. One clone, termed Jurkat-7, was negative for CD48, CD55 and CD59 (not shown). This clone was used for further analysis. In vitro labelling of GPI biosynthesis demonstrated abolished formation of its first intermediate, N-acetyl-glucosaminylphosphatidylinositol (GlcNAc-PI, Fig. 1). Among the four genes involved in this reaction, only PIG-A is linked to the X chromosome ([19–23], reviewed in [24]). Therefore, we suspected a mutation in the PIG-A gene to be responsible for GPI deficiency in this clone.
Fig. 1.
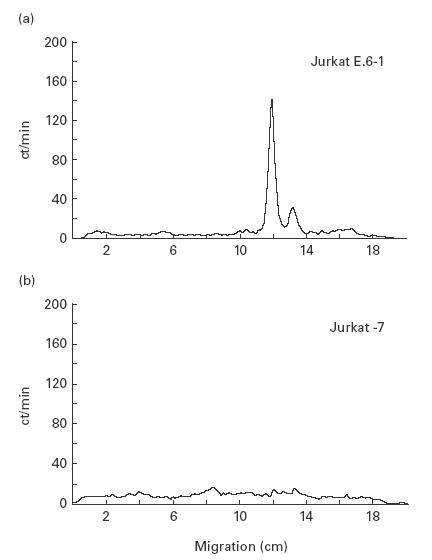
Jurkat-7 cells are deficient in the first step of GPI biosynthesis. Cell lysates of (a) wild-type Jurkat E.6-1 and (b) Jurkat-7 cells were labelled with UDP-3H-GlcNAc. Lipids were extracted in butanol/water and resolved by thin layer chromatography. The two labelled species observed in Jurkat E.6-1 cells are the first two intermediates of the GPI biosynthetic pathway, GlcNAc-PI (right) and GlcN-PI (left). These intermediates are not present in Jurkat-7 cells due to a mutation in the GPI biosynthetic gene PIG-A.
As anticipated by the amount of reverse transcriptase (RT)-PCR product, the PIG-A expression level in Jurkat-7 cells was comparable to the parental cell line. Cloning and sequencing of the cDNA revealed a guanine to adenine transition in exon 2 (position 228 in the cDNA sequence reported by Miyata et al. [19]). This finding was verified by sequencing a PCR product of PIG-A exon 2, amplified from Jurkat-7 genomic DNA. The 288 G→A mutation causes substitution of glycine to aspartate at position 48 of the deduced amino acid sequence. No other alteration of the cDNA or genomic sequence was observed.
Phenotypic characterization of Jurkat-7 cells in comparison with the Jurkat E.6-1 parental cell line revealed no differences in the expression of CD2 (positive), CD3 (positive), CD4 (positive), CD8 (negative) and CD28 (positive). In contrast, GPI-anchored antigens CD48, CD55 and CD59 were negative on Jurkat-7 cells, but positive on Jurkat E.6-1 cells. GPI-anchored CD52 was negative on Jurkat-7 and Jurkat E.6-1 cells. As the parental cell line, Jurkat-7 cells have unlimited expansion potential, independent of medium conditioning or supplementation with IL-2.
Retroviral gene transfer of PIG-A into Jurkat-7 cells completely restores GPI anchoring
In order to demonstrate that GPI deficiency in the isolated Jurkat-7 mutant is solely due to the observed PIG-A mutation, transient transfection of Jurkat-7 mutants with the L-PIG-A-SN plasmid resulted in a re-expression of the CD48 antigen in 20% of cells. In contrast, using the LXSN plasmid for transfection, no CD48 re-expressing cells could be detected (data not shown).
Retroviral gene transfer conditions were optimized to effectively transduce Jurkat-7 cells with pL-PIG-A-SN and pLXSN. In brief, ecotropic GP+E86 packaging cells were transfected with both vectors by electroporation. Then, virus-containing supernatant was used to transduce GALV pseudotyped PG13 packaging cells. Packaging cells were subcloned and screened for virus titres, which reached a maximum of 1 × 105 CFU/ml for both pL-PIG-A-SN and pLXSN. Virus-containing supernatant of these cells was used to transduce Jurkat-7 cells. After 2 weeks of selection for G418 resistance, cells were analysed for restoration of CD48 expression (Fig. 2). Wild-type levels of CD48 expression were found in pL-PIG-A-SN-transduced cells (now termed Jurkat-7.P); in contrast, no expression of CD48 was found in pLXSN-transduced cells (Jurkat-7.X). Likewise, expression of other GPI-anchored proteins (CD55, CD59) was restored in Jurkat-7.P, but not in Jurkat-7.X cells (data not shown). Therefore, PIG-A had been efficiently introduced into Jurkat-7.P to restore its capability of GPI anchoring. After removal of G418 selective pressure, Jurkat-7.P cells continued to express GPI-anchored protein for at least 6 months, indicating that PIG-A expression is stable in this cell line.
Fig. 2.
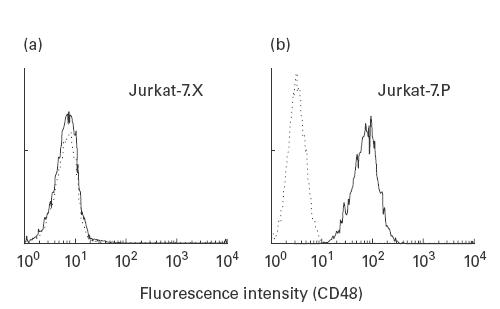
Stable retroviral PIG-A transduction of Jurkat-7 cells. (a) No CD48 expression in Jurkat-7.X cells transduced with an empty pLXSN vector (–––––, CD48 expression; - - - - -, isotype control). (b) Completely restored CD48 expression in Jurkat-7.P cells transduced with pL-PIG-A-SN.
Susceptibility to apoptotic stimuli
GPI-positive Jurkat-7.P and GPI-negative Jurkat-7.X cells showed no significant differences in morphology, proliferative capacity or rate of spontaneous cell death as judged by trypan-blue exclusion. To induce apoptosis, Jurkat-7.P and Jurkat-7.X cells were placed in serum-free medium. In five independent experiments, cultures were followed up for 4 days. As shown in Fig. 3, the number of living cells was reduced by 40% after 4 days of starvation. No significant difference was observed when comparing Jurkat-7.P and Jurkat-7.X cells. We therefore exposed both lines to the topoisomerase inhibitor camptothecin for 2 h to induce apoptotic cell death by a different mechanism. Numbers of apoptotic cells were determined by annexin V-PE and 7-AAD staining (Fig. 4). Statistical analyses from four independent experiments failed to show any significant differences in the number of camptothecin-induced apoptotic cells between Jurkat-7.P and Jurkat-7.X cells (40·4% and 42·4%, respectively). To induce Fas-mediated apoptosis, cells were incubated with CD95 MoAb. As measured by annexin V-PE and 7-AAD staining, GPI-defective Jurkat-7.X cells were more susceptible to this stimulus than GPI-positive Jurkat-7.P cells. In 13 separate experiments, the number of apoptotic Jurkat-7.X cells was 1·3-fold higher than the number of apoptotic Jurkat-7.P cells (P < 0·001 using Student's matched pair t-test, Fig. 5). To rule out the possibility that this difference resulted from different levels of Fas expression, Jurkat-7.X and Jurkat-7.P cells were stained with CD95 MoAb. There was no detectable difference in the expression of Fas (not shown).
Fig. 3.

GPI-anchoring has no impact on serum starvation-induced apoptosis in Jurkat cells. Jurkat-7.X and Jurkat-7.P cells were incubated in serum-free medium for 4 days. ▪, Base-line cell titres (day 0, arbitrarily set at 1·0), □, living cells after 4 days of starvation.
Fig. 4.
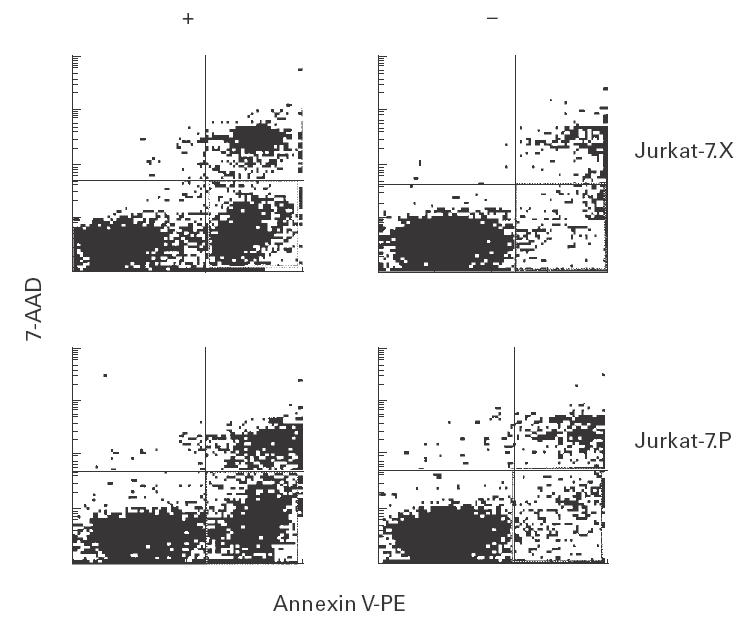
GPI-anchoring has no impact on camptothecin-induced apoptosis in Jurkat cells. Jurkat-7.X and Jurkat-7.P cells were incubated with camptothecin for 2 h and subsequently stained with annexin V-PE (abscissa, apoptotic and dead cells) and 7-AAD (ordinate, dead cells). Cells positive for annexin, but negative for 7-AAD were regarded as apoptotic (right lower quadrant). In four separate experiments, these were not significantly different for Jurkat-7.X and Jurkat-7.P cells.
Fig. 5.
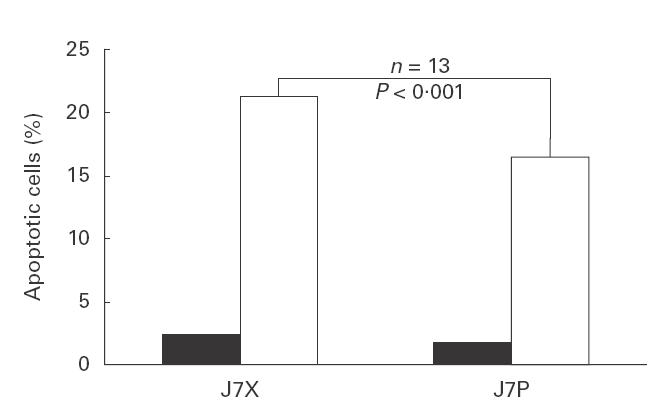
Fas-mediated apoptosis is more pronounced in GPI-deficient Jurkat-7.X cells. Cells were incubated with CD95 MoAb for 1 or 2 h. Apoptosis was measured as indicated in Fig. 4. The number of apoptotic bodies in Jurkat-7.X cells is higher by a factor of 1·3 compared with Jurkat-7.P cells (P < 0·001 using Student's matched pair t-test). ▪, Mock; □, anti-Fas.
DISCUSSION
In this study, we introduce a GPI-deficient Jurkat T cell line as a model for studying the role of GPI anchoring in T cells. The Jurkat-7 clone is the first human T cell line with a defined GPI biosynthetic defect, which is not derived from a PNH patient. Transduction of this clone with a functional copy of the PIG-A gene, along with a vector negative control, produced two stable cell lines that are genetically identical, but distinguished by their ability to express functional PIG-A and, in consequence, GPI-anchored proteins.
Earlier results indicated that GPI deficiency might cause resistance to apoptosis induced by serum removal or ionizing irradiation [13]. However, cells from PNH patients were used in this study, which might not be acceptable as a general model for GPI deficiency. Thus, bone marrow stem cells from many PNH patients are capable of spontaneous colony formation in vitro without erythropoietin or colony-stimulating factor [25]. This feature, which is not observed in cells from normal individuals, is just one example among others indicating alterations of cell growth regulation in PNH, which could also be responsible for the observed resistance to apoptosis. It is generally accepted that GPI-defective stem cell(s) in PNH must harbour some form of growth advantage over their normal counterparts in the same individual. However, the GPI biosynthetic defect itself is not sufficient to cause the disease, since it does not confer a growth advantage to the GPI-detective clone [26,27]. It has been suggested that this growth advantage might result from an additional, yet unidentified genetic disorder. Alternatively, immune selection against GPI-positive cells might play a role (for recent review, see [28]).
It is important to dissect the different mechanisms proposed for PNH pathogenesis. The Jurkat T cell model introduced here allows the analysis of the role of GPI anchoring under exclusion of other, potentially intervening pathogenic factors. When comparing Jurkat-7.P and Jurkat-7.X cells, we found, to our surprise, no difference in the susceptibility to apoptosis induction by serum removal or camptothecin. In contrast, we found a subtle, yet reproducible difference in the cells' ability to undergo Fas-mediated apoptosis. Thus, GPI-defective Jurkat cells are more susceptible to this stimulus than their GPI-positive counterparts. At present, we have no definite explanation for this difference. However, it may be postulated that the presence of GPI-linked surface proteins at the cell surface may specifically interfere with elements of Fas-mediated apoptosis.
Many GPI-anchored cell surface proteins have been implicated in cell signalling, especially in the activation of T lymphocytes. In general, GPI-anchored proteins serve as costimulatory receptors and it appears possible that their signals might also interfere with apoptotic pathways. However, the GPI anchor itself might not always be required, since transmembrane variants of GPI-anchored proteins were often found to also stimulate the cells [29–31]. Moreover, there are examples of GPI-anchored molecules suspected to play a key role in T cell biology, which did not cause any aberration in T cell development or function when absent from the cell. For instance, GPI-anchored Thy-1 was believed to be crucial for thymocyte-negative selection. Surprisingly, Thy-1 knock-out mice have no apparent defect in thymocyte development as assessed by measurements of thymocyte subpopulations, T cell MHC restriction as well as antigen- and antibody-dependent negative selection [32]. Recently, mice with a targeted disruption of PIG-A in their T cell precursors were generated [33]. Again, these mice have no apparent deficiency in T cell development. Measurements of reactivity in response to TCR stimulation were normal in vitro as well as in vivo.
The discrepancy between the expected effect due to the action of GPI-anchored molecules as measured by immunological methods and the minor defects, which result from their genetic inactivation, might be explained by redundancy in function. Structures that are able to substitute for GPI-linked molecules might be involved in the same function under normal circumstances. In addition, the functional measurements employed today might be inadequate or insufficiently sensitive to document defects in the absence of GPI anchors. Therefore, the development of new functional assays might be a key step in further research on the role of these molecules. The introduced Jurkat model for GPI deficiency might help to make this progress an attainable goal.
Acknowledgments
This work was supported in part by BMBF OIGE9616 and DFG IIIGK-GRK139/3. We would like to thank Dr M. Exton, University of Essen, for carefully revising the manuscript.
REFERENCES
- 1.Kupfer A, Singer SJ. Cell biology of cytotoxic and helper T cell functions: immunofluorescence microscopic studies of single cells and cell couples. Annu Rev Immunol. 1989;7:309–37. doi: 10.1146/annurev.iy.07.040189.001521. [DOI] [PubMed] [Google Scholar]
- 2.Elliot JI. Thymic selection reinterpreted. Immunol Rev. 1993;135:227–42. [PubMed] [Google Scholar]
- 3.Schubert J, Ostendorf T, Schmidt RE. Biology of GPI anchors and pathogenesis of paroxysmal nocturnal hemoglobinuria. Immunol Today. 1994;15:299–301. doi: 10.1016/0167-5699(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 4.Malek TR, Fleming TJ, Codias EK. Regulation of T lymphocyte function by glycosylphosphatidylinositol (GPI)-anchored proteins. Semin Immunol. 1994;6:105–13. doi: 10.1006/smim.1994.1015. [DOI] [PubMed] [Google Scholar]
- 5.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–44. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 6.Stulnig TM, Berger M, Sigmund T, Stockinger H, Horejsi V, Waldhausl W. Signal transduction via glycosyl phosphatidylinositol-anchored proteins in T cells is inhibited by lowering cellular cholesterol. J Biol Chem. 1997;272:19242–7. doi: 10.1074/jbc.272.31.19242. [DOI] [PubMed] [Google Scholar]
- 7.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1991;254:1016–9. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 8.Cinek T, Hilgert I, Horejsi V. An alternative way of CD4 and CD8 association with protein kinases of the Src family. Immunogenetics. 1995;41:110–6. doi: 10.1007/BF00182321. [DOI] [PubMed] [Google Scholar]
- 9.Deckert M, Ticchioni M, Mari B, Mary D, Bernard A. The glycosylphosphatidylinositol-anchored CD59 protein stimulates both T cell receptor zeta/ZAP-70-dependent and -independent signaling pathways in T cells. Eur J Immunol. 1995;25:1815–22. doi: 10.1002/eji.1830250704. [DOI] [PubMed] [Google Scholar]
- 10.Hueber AO, Raposo G, Pierres M, He HT. Thy-1 triggers mouse thymocyte apoptosis through a bcl-2-resistant mechanism. J Exp Med. 1994;179:785. doi: 10.1084/jem.179.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schubert J, Uciechowski P, Zielinska-Skowronek M, Tietjen C, Leo R, Schmidt RE. Differences in activation of normal and glycosylphosphatidylinositol-negative lymphocytes derived from patients with paroxysmal nocturnal hemoglobinuria. J Immunol. 1992;148:3814–9. [PubMed] [Google Scholar]
- 12.Rosse WF, Ware RE. The molecular basis of paroxysmal nocturnal hemoglobinuria. Blood. 1995;86:3277–86. [PubMed] [Google Scholar]
- 13.Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to apoptosis caused by PIG-A gene mutations in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1997;94:8756–60. doi: 10.1073/pnas.94.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horikawa K, Nakakuma H, Kawaguchi T, Iwamoto N, Nagakura S, Kagimoto T, Takatsuki K. Apoptosis resistance of blood cells from patients with paroxysmal nocturnal hemoglobinuria, aplastic anemia, and myelodysplastic syndrome. Blood. 1997;90:2716–22. [PubMed] [Google Scholar]
- 15.Ware RE, Nishimura J, Moody MA, Smith C, Rosse WF, Howard TA. The PIG-A mutation and absence of glycosylphosphatidylinositol-linked proteins do not confer resistance to apoptosis in paroxysmal nocturnal hemoglobinuria. Blood. 1998;92:2541–50. [PubMed] [Google Scholar]
- 16.Hyman R. Cell-surface-antigen mutants of haematopoietic cells. Tools to study differentiation, biosynthesis and function. Biochem J. 1985;225:27. doi: 10.1042/bj2250027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidugiriene J, Menon AK. Biosynthesis of glycosylphosphatidylinositol anchors. Methods Enzymol. 1995;250:513–35. doi: 10.1016/0076-6879(95)50094-4. [DOI] [PubMed] [Google Scholar]
- 18.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–90. [PMC free article] [PubMed] [Google Scholar]
- 19.Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259:1318–20. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 20.Inoue N, Watanabe R, Takeda J, Kinoshita T. PIG-C, one of the three human genes involved in the first step of glycosylphosphatidylinositol biosynthesis is a homologue of Saccharomyces cerevisiae GPI2. Biochem Biophys Res Commun. 1996;226:193–9. doi: 10.1006/bbrc.1996.1332. [DOI] [PubMed] [Google Scholar]
- 21.Kamitani T, Chang HM, Rollins C, Waneck GL, Yeh ET. Correction of the class H defect in glycosylphosphatidylinositol anchor biosynthesis in Ltk-cells by a human cDNA clone. J Biol Chem. 1993;268:20733–6. [PubMed] [Google Scholar]
- 22.Tiede A, Schubert J, Nischan C, Jensen I, Westfall B, Taron CH, Orlean P, Schmidt RE. Human and mouse Gpi1p homologues restore glycosylphosphatidylinositol membrane anchor biosynthesis in yeast mutants. Biochem J. 1998;334:609–16. doi: 10.1042/bj3340609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe R, Inoue N, Westfall B, Taron CH, Orlean P, Takeda J, Kinoshita T. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998;17:877–85. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiede A, Bastisch I, Schubert J, Orlean P, Schmidt RE. Biosynthesis of glycosylphosphatidylinositols in mammals and unicellular microbes. Biol Chem. 1999;380:503–23. doi: 10.1515/BC.1999.066. [DOI] [PubMed] [Google Scholar]
- 25.Issaragrisil S, Piankijagum A, Chinprasertsuk S, Kruatrachue M. Growth of mixed erythroid-granulocytic colonies in culture derived from bone marrow of patients with paroxysmal nocturnal hemoglobinuria without addition of exogenous stimulator. Exp Hematol. 1986;14:861–6. [PubMed] [Google Scholar]
- 26.Kawagoe K, Kitamura D, Okabe M, Taniuchi I, Ikawa M, Watanabe T, Kinoshita T, Takeda J. Glycosylphosphatidylinositol-anchor-deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:3600–6. [PubMed] [Google Scholar]
- 27.Rosti V, Tremml G, Soares V, Pandolfi PP, Luzzatto L, Bessler M. Murine embryonic stem cells without pig-a gene activity are competent for hematopoiesis with the PNH phenotype but not for clonal expansion. J Clin Invest. 1997;100:1028–36. doi: 10.1172/JCI119613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunn DE, Ware RE, Parker CJ, Mishoe HO, Young NS. Research directions in paroxysmal nocturnal hemoglobinuria. Immunol Today. 1999;20:168–71. doi: 10.1016/s0167-5699(98)01424-8. [DOI] [PubMed] [Google Scholar]
- 29.Itzhaky D, Raz N, Hollander N. The glycosylphosphatidylinositol-anchored form and the transmembrane form of CD58 associate with protein kinases. J Immunol. 1998;160:4361–6. [PubMed] [Google Scholar]
- 30.Saitoh S, Kosugi A, Noda S, Yamamoto N, Ogata M, Minami Y, Miyake K, Hamaoka T. Modulation of TCR-mediated signaling pathway by thymic shared antigen-1 (TSA-1)/stem cell antigen-2 (Sca-2) J Immunol. 1995;155:5574–81. [PubMed] [Google Scholar]
- 31.Resta R, Hooker SW, Laurent AB, Shuck JK, Misumi Y, Ikehara Y, Koretzky GA, Thompson LF. Glycosyl phosphatidylinositol membrane anchor is not required for T cell activation through CD73. J Immunol. 1994;153:1046–53. [PubMed] [Google Scholar]
- 32.Page DM, Tokugawa Y, Silver J, Stewart CL. Role of Thy-1 in T cell development. J Immunol. 1997;159:5285–92. [PubMed] [Google Scholar]
- 33.Takahama Y, Ohishi K, Tokoro Y, Sugawara T, Yoshimura Y, Okabe M, Kinoshita T, Takeda J. Functional competence of T cells in the absence of glycosylphosphatidylinositol-anchored proteins caused by T cell-specific disruption of the pig-a gene. Eur J Immunol. 1998;28:2159–66. doi: 10.1002/(SICI)1521-4141(199807)28:07<2159::AID-IMMU2159>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]