

Abstract
Adhesion mechanisms play a major role in the recruitment of peripheral blood lymphocytes (PBL) which characteristically infiltrate rheumatoid arthritis (RA) synovium and other chronically inflamed tissues. Through a sequential series of complex integrated adhesion and signalling events, ‘multistep model of migration’, specific subsets of PBL are recruited into inflamed tissues. In this process both leucocyte receptors and microvascular endothelial (MVE) counter-receptors play a critical role. The MVE in particular, during an inflammatory state, is the target of various inflammatory mediators that cause the up-regulation of several cell adhesion molecules (CAM). One of the most important factors known to be a powerful inducer of MVE CAM is TNF-α. Conversely, blocking TNF-α causes a down-modulation of CAM expression. To test directly the capacity of TNF-α to induce cell migration into RA synovium we adapted a model in which synovial grafts were implanted into SCID mice subcutaneously. Using this model we demonstrate that: (i) transplants remain viable and become vascularized and fed by mouse subdermal vessels; (ii) the mouse vasculature connects to the transplant vasculature which maintains the ability to express human CAM; (iii) intragraft injections of TNF-α up-regulate the expression of human CAM, following the down-regulation which occurred 4 weeks post-transplantation; and (iv) the up-regulation of graft CAM is associated with increased human PBL migration into the transplants. This study provides direct evidence in vivo of the capacity of TNF-α to induce cell migration. In addition, it provides the experimental background for the optimal use of this model.
Keywords: adhesion molecules, in vivo SCID model, lymphocyte migration, tumour necrosis factor-alpha, rheumatoid arthritis
INTRODUCTION
Adhesion mechanisms play a major role in the pathogenesis of rheumatoid arthritis (RA) synovitis [1,2]. The RA synovitis is characterized by new blood vessel formation, thickening of the lining layer and an inflammatory infiltrate constituted mainly of mononuclear cells (MNC). The MNC consist mainly of strongly HLA-DR+ antigen-presenting cells (APC) in close contact with T lymphocytes, the majority of which express the helper/memory phenotype (CD4+CD45RO+) [3–6]. The crucial factor in the generation of this characteristic cellular infiltrate, typical of most inflammatory conditions, is represented by the interactions of circulating leucocytes with the microvascular endothelium (MVE). Through a sequential series of complex integrated adhesion and signalling events, ‘multistep model of migration’, specific subsets of MNC are recruited into various tissues [7–10]. In this process both leucocyte receptors and MVE counter-receptors play a critical role. The MVE in particular, during an inflammatory state, is the target of various inflammatory mediators which cause the up-regulation of several cell adhesion molecules (CAM) [2,11]. These include E- and P-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) which under non-inflamed/resting conditions are expressed at very low level [2,11]. Therefore, while in physiological situations the endothelial luminal surface is typically non-adhesive to circulating cells, during inflammation the increased expression of endothelial CAM is instrumental in facilitating the adhesion cascade which leads to transendothelial migration and exudation of inflammatory cells into the affected tissues.
One of the most important factors known to be a powerful inducer of MVE CAM is TNF-α. TNF-α is a multifunctional proinflammatory cytokine whose effects are initiated by interaction with two distinct cell-surface receptors of 55 kD (TNFRp55) and 75 kD (TNFRp75), respectively [12]. In human umbilical vein endothelial cells (HUVEC) stimulation of the TNFRp55 results in strong induction of ICAM-1, E-selectin, and VCAM-1, whereas TNFRp75-mediated signals do not influence the expression of these three molecules [12]. The essential role of TNFRp55 is further confirmed by studies using TNFRp55−/− mice in which TNF-α could not induce endothelial CAM, and as a consequence there was a decreased leucocyte extravasation in inflamed organs [13]. Furthermore, human TNF-α transgenic animals spontaneously develop a destructive arthritis with a massive leucocytic infiltration of the affected joints, while the administration of anti-TNF-α monoclonal antibody in this model completely prevented development of the arthritis [14]. Similar results were obtained in collagen-induced arthritis (CIA) in rats and mice, where intra-articular administration of TNF-α either prior to or after the induction of CIA led to an accelerated onset and more severe course of the disease [15].
The critical role of TNF-α in the development of chronic arthritis is also universally accepted in humans [16–18]. This has led to a series of clinical trials in RA using both MoAbs and soluble TNFR which unequivocally showed a very significant clinical benefit [19–21]. Interestingly, in one of the early studies using the MoAb cA2, a rise in the number of circulatory peripheral blood lymphocytes led to the suggestion that an important consequence of blocking TNF-α was to inhibit cell migration to the joint [22]. This was indeed confirmed by analysing synovial biopsies obtained from RA patients pre- and post-cA2 therapy. Following cA2 treatment, a significant reduction in the number of infiltrating T cells was demonstrated to be associated with a decreased expression of vascular VCAM-1 and E-selectin [22].
Therefore, this provides strong indirect evidence for the hypothesis that an important way in which TNF-α exerts its proinflammatory effects is by potentiating those mechanisms that increase cell adhesion and migration. However, in humans direct support for this modality of action is still lacking. The main reason for this is that there are obvious technical difficulties and ethical considerations in injecting TNF-α in humans in order to perform studies on MVE physiology and lymphocyte migration. For this reason, to test the direct functional role of this important cytokine we have adapted a model in which human RA synovium was transplanted into SCID mice subcutaneously [23,24]. This model has been validated using both autologous and allogeneic human lymphocytes, where both cell types were shown to migrate equally effectively to the human transplants but not to murine tissues [25]. In our modified model TNF-α can be injected directly into the synovial grafts, allowing examination of its effects on human synovial MVE. In addition, as the animals were injected intravenously with human peripheral blood lymphocytes (PBL) we could examine the resulting effects of TNF-α in modulating leucocyte migration into this ‘newly inflamed’ living human tissue.
MATERIALS AND METHODS
Tissue collection, preparation and storage
Synovial samples were obtained from surgical specimens of patients requiring joint replacement. Procedures were performed after informed consent approved by the Ethical Committee. Samples were divided into two parts. One part was used for immunohistology and the second for transplantation. The part assigned for immunohistology was embedded in Optimal Cutting Temperature compound (OCT; Agar Scientific Ltd, UK), snap-frozen in liquid nitrogen-cooled isopentane (BDH, Poole, UK) and stored at −70°C until analysis. The second part, assigned for transplantation, was cut into 0·5-cm3 pieces, frozen in 20% DMSO (Sigma, Poole, UK) in heat-inactivated fetal calf serum (FCS; PAA Labs GmbH, Linz, Austria) and stored in liquid nitrogen until engraftment.
Preparation and labelling of human peripheral blood lymphocytes
Lymphocytes were isolated from peripheral blood using Ficoll–Hypaque (Lymphoprep; Nycomed, Oslo, Norway) density gradient centrifugation as described previously (C. Pitzalis et al. J Rheumatol 1987; 14:660). The isolated human peripheral blood mononuclear cells (PBMC) were suspended in tissue culture medium (10% heat-inactivated FCS (PAA) in RPMI 1640 (Gibco, Paisley, UK) and then incubated overnight in plastic flasks in order to deplete adherent monocytes. The remaining, non-adherent cells, mostly lymphocytes, were then washed and resuspended in serum-free PBS ready for labelling.
Double cell labelling—PKH26 labelling
Separated huPBL were incubated with PKH26 dye (Sigma) at room temperature, 10 μl dye/20 × 106 cells for 2 min before stopping the reaction with heat-inactivated FCS. The cells were then washed twice to remove unbound dye. Cell viability was determined via trypan blue exclusion and always found to be >95%.
Double cell labelling—111indium labelling
PKH26-labelled huPBL (PKH26-huPBL) were washed and resuspended in serum-free PBS. 111Indium oxinate solution (Mallinckrodt Medical B.V., Petten, The Netherlands) was added at 0·2 mCi/50 × 106 cells. After 15 min of incubation with occasional shaking at room temperature, the cells were washed twice to remove unbound 111indium. The supernatant and cell-bound activity was measured to calculate the percentage efficiency of labelling. Viability of the cells was determined via trypan blue exclusion and, as above, was always found to be >95%.
In vivo migration assay
Animals
A colony of Beige SCID C.B-17 mice was maintained under pathogen-free conditions in the biological facilities of Kings College (St Thomas' Campus).
Tissue transplantation
Samples of human synovium were thawed from liquid nitrogen storage immediately before surgery, washed in saline and kept in saline-moistened gauze until transplantation. The mice were anaesthetised with 0·2 ml Domitor (Smithkline Beecham, UK) and 0·1 ml Ketamine (SKB). A small incision was made in the dorsal aspect behind the ear of each SCID mouse and the synovial tissue inserted subcutaneously. The wound was closed with soluble suture material (Ethicon, Congleton, UK).
In vivo cell migration assay
Double-labelled cells (111In-PKH26-huPBL), as described above, were injected intravenously into the tail vein of the synovium transplanted SCID mice. TNF-α (Genzyme, West Malling, UK) was injected intragraft at the same time. HuPBL migration into the grafts was assessed (i) by calculating the ratio of graft radioactivity (as a percentage of total injected dose calculated as total body mouse radioactivity)/graft weight, and (ii) histologically by immunofluorescence, where the results were expressed as the number of PKH26+ cells identified/mm2 of tissue (see quantification of immunoperoxidase staining). Total body mouse radioactivity was determined by gamma camera scintigraphy using a light weight mobile gamma camera (Apex 215M; Elscint, Crawley, UK). Following the migration study, animals were killed, grafts removed and graft radioactivity measured by γ counter (1282 Compugamma CS; Wallac, Milton Keynes, UK). In order to be able to compare the results from the γ counter and the γ camera the data were normalized. A standard of known radioactivity (in MBq) was measured on the γ camera, then a 1:1000 dilution of this standard was measured on the γ counter. The total mouse and individual transplant counts were then normalized against their respective standards. Once normalized, the results could be compared directly using the equation described above.
Grafts were weighed and then rapidly snap-frozen in liquid nitrogen for immunohistological analysis after radioactive decay.
Assessment of graft viability
In order to determine whether human synovium grafting was feasible and grafts remained viable in the SCID mouse, 4 weeks post-transplantation, prior to the migration studies, one animal from each batch was killed. Transplants were assessed macroscopically and microscopically. Macroscopically transplants were assessed by observing the viable appearance of the tissue and the degree of neovascularization of murine vasculature into the human grafts (see Fig. 1). Microscopically, viability was based on the criteria that in transplant cryostat sections the extracellular matrix (ECM) was intact, cells were not apoptotic, and immunoperoxidase staining did not result in diffuse non-specific stain.
Fig. 1.

A representative example of a SCID mouse transplanted with human rheumatoid synovium. Transplants look viable and vascularized by murine subdermal vessels indicated by the arrows.
Assessment of human vasculature within grafts
In order to establish whether human vasculature adhesion molecule expression was preserved in the grafts, we assessed the expression of human ICAM-1, VCAM-1 and E-selectin pre- and post-transplantation using species-specific MoAbs (murine clones 6.5B5, 4B9, 1.2B6, respectively, produced by Professor D. Haskard, British Heart Foundation, Hammersmith Hospital, London, UK). Cryostat sections (10 μm) of the grafts were immunostained using indirect immunoperoxidase methods as described below. In addition, in order to determine whether the human transplant vasculature remained patent and connected with the murine vasculature infiltrating the grafts, transplanted mice were injected intravenously with either anti-human ICAM-1-biotinylated or an isotype-matched control antibody (MOPC21-biotinylated). Ten minutes later mice were killed and the transplants embedded in OCT and snap-frozen. Cryostat sections were then incubated with avidin-biotin complex conjugated with horseradish peroxidase (ABC–HRP) alone (no primary antibody) for 30 min and then developed using diaminobenzidine (DAB; Sigma). Sequential sections were incubated with anti-human VWFVIII (Dako, Ely, UK), in order to identify human blood vessels and therefore determine the site of localization of the anti-ICAM-1 and control antibodies.
Assessment of murine vasculature within the grafts
Murine vasculature was identified using species-specific rat anti-mouse MoAbs against either ICAM-1 (HB233; ATCC, Rockville, MD) or CD31 (PharMingen, San Diego, CA) and standard immunoperoxidase or immunofluorescence methods were used as described below.
Quantification of human and murine vasculature
Cryostat sections of the transplants were immunostained for both human vasculature (VWFVIII; Dako) and murine vasculature (CD31; PharMingen) using either single or double immunoperoxidase methods as described below. Digital colour images of the immunostained tissue sections were obtained using a microcomputer-based image analysis system attached to a video camera-equipped Olympus BX-60 light microscope (KS300; Zeiss Inc., Olympus Optical Co., London, UK). Digitized images were analysed by adaptive thresholding to render binary images of the positively stained areas from the original colour image data. A custom-designed macro was then used to analyse the fractional area of stained versus unstained tissue for each field and calculate the volume fraction (vV) of murine and human vasculature.
Assessment of huPBL circulatory time and non-specific tissue localization in the SCID mouse
In order to determine the length of time that the huPBL persist in the murine circulation, non-transplanted SCID mice were injected with 10 × 106 PKH26-huPBL at time (t) = 0. At t = 10 min, 30 min, 1 h, 24 h, 48 h, 72 h, 100 μl blood samples were extracted from the tail vein. The erythrocytes were lysed using Lysis Buffer (Becton Dickinson, Cowley, UK) and the PKH26-huPBL were analysed by FACScan analysis (Becton Dickinson). Control huPBL were labelled with an irrelevant PE control antibody.
To determine the tissue localization of huPBL in the mouse, transplanted animals, injected intravenously with 111In-huPBL, were killed after 48 h. The grafts as well as spleen, thymus and liver were extracted and their radioactivity measured before snap freezing them for histological analysis (see below).
Immunohistological analysis of human synovial grafts, murine tissues and original human tissues
The synovial samples and transplants were embedded in OCT compound (Miles Labs) and snap-frozen in isopentane-cooled liquid nitrogen. Samples were stored at −70°C until sectioned for immunohistologic staining. Sections (10 μm thick) were cut with a cryostat (Leica; CM1900) at −22°C. Sequential sections were mounted on Poly L-lysine (Sigma)-coated slides and dried overnight at room temperature. Sections for immunohistologic analysis were fixed in acetone at 4°C for 10 min, wrapped in aluminium foil and stored at −70°C until use. Sections for analysis of PKH26+ cells were counterstained with anti-human VWFVIII–FITC (Dako) and then fixed in 2% formaldehyde for 30 min. The VWFVIII–FITC provided a reference for orientation within the tissue. Coverslips were mounted using Immunofluor (ICN Pharmaceuticals, Basingstoke, UK). Sections were analysed using a fluorescence microscope (Olympus). Cell surface antigens within the tissues were identified by standard immunoperoxidase technique (see below).
Immunoperoxidase staining
Indirect immunoperoxidase staining was performed using standard techniques as previously described [26]. Briefly, after incubating with serum for 20 min to block non-specific binding, the sections were incubated with primary antibody for 1 h. The sections were washed and incubated with the secondary antibody for 30 min. Where the secondary antibody was conjugated to biotin, the sections were washed and incubated with either the ABC–HRP (Dako) or avidin-biotin–alkaline phosphatase complex (ABC–AP; Dako). Again the sections were washed and then developed with DAB (Sigma), the substrate for HRP, or Vector red (Sigma), the substrate for AP, and counterstained with haematoxylin. In the case of double-staining the AP method was employed first, and then before applying the substrate the HRP method was carried out. Following development of the HRP method using DAB, Vector red was used to develop the AP method.
Finally, sections were dehydrated by passing through ethanol and xylene. The coverslips were mounted using DPex (BDH) and analysed using a microscope (Olympus) attached to an image analysis system (Keiss 300; Imaging Associates, Thame, UK).
Quantification
To obtain an accurate representation of (i) the number of PKH26+ cells present in the synovial grafts, (ii) the murine/human vascularity in the grafts, and (iii) relative adhesion molecule expression, three whole sections were analysed, each from a different cutting level (i.e. from the top, bottom and middle) per graft without any field selection. In the case of the PKH26 cells, sections were analysed using a fluorescence microscope (Olympus). The results were expressed as the average of the total number of cells identified in each section in which, on average, more than 100 high power fields were counted. The vascularity within each section was analysed using an image analysis system (Keiss 300; Imaging Associates).
The relative expression of E-selectin, ICAM-1 and VCAM-1 was quantified in terms of intensity of staining using an arbitrary scale from 0 to 3, where 0 indicated no staining and 3 indicated maximal staining. Results were analysed by a single observer followed by random sample analysis by a second observer. The interobserver error was found to be <5%.
FACS analysis
Single labelling immunofluorescence staining and FACS analysis were performed as previously described [27]. Briefly, 0·5–1 × 106 huPBL were incubated for 10 min with 10 μl of normal human serum for 10 min. After washing, the cells were incubated for 20 min with the primary antibody anti-CD3 (Becton Dickinson), -CD19 (Becton Dickinson), -LFA-1 (HB203; ATCC), -CLA and -L-selectin (Heca 452 and Dreg 200, respectively; gifts from Dr M. Robinson, Celltech, Slough, UK). The cells were washed again and then incubated with the secondary antibody, goat anti-mouse Ig–FITC (Becton Dickinson) for another 20 min, except where the primary antibodies were directly conjugated. The cells were washed twice and then fixed with 1% paraformaldehyde in PBS–azide. Control samples were incubated with an irrelevant Ig–FITC. Samples were analysed by flow cytometry using a FACS analyser (FACScan; Becton Dickinson). Debris and dead cells were excluded by setting the appropriate SSc and FSc gates, while the positive cells for the various markers were considered those with a fluorescence signal >10−1.
Statistical analysis
Results are expressed as mean ±s.e.m. unless otherwise indicated. Statistical analyses were performed using Kruskall–Wallis and/or multiple comparisons, one-way anova (Dunnett's method).
RESULTS
Transplant viability and vascularization
In order to determine whether human synovial transplantation was successful, grafts were assessed at 4 weeks prior to performing migration studies. One animal from each batch was killed and transplants examined macroscopically and microscopically as described in the methods. As seen in Fig. 1, mouse subdermal vessels are clearly feeding the grafts, which look macroscopically healthy. By this method of assessment transplantation was successful in >95% of cases. Microscopic analysis confirmed that transplant viability was > 90%.
In order to assess whether the human vasculature was preserved in the grafts following transplantation, we analysed the grafts for human and murine vessel markers using species-specific reagents and double immunoperoxidase. As can be seen in Fig. 2A, both human (red staining, anti-human VWFVIII) and mouse (brown staining, anti-murine CD31) blood vessels were present in synovial grafts 4 weeks post-transplantation. In some instances the blood vessels of the two species appeared to be directly connected (indicated by the black arrow). To determine whether such connections were functional, transplanted mice were injected intravenously into the tail vein with either anti-human-specific ICAM-1-biotinylated or an isotype-matched control antibody-biotinylated (MOPC21) as described in Materials and methods. As shown in Fig. 2C, detection of anti-human ICAM-1 by using the detection substrate alone (ABC complex), showed a discrete staining confined to the luminal surface of the human vessels in the grafts but not in the perivascular tissue. This was taken as an indication that the antibody localization in the grafts did not occur by simple diffusion from the mouse circulation feeding the transplants, but via a direct anastomosis of the mouse–human vasculature. In the animal injected with control biotinylated MoAb there was no staining when applying ABC complex only (Fig. 2E), although blood vessels could be clearly visualized when counterstained with anti-human VWFVIII–FITC (Fig. 2F). The functionality of the mouse–human vascular connection was further confirmed by the injection of PKH26-labelled huPBL intravenously into synovial transplanted SCID mice. As it can be seen in Fig. 2G, PKH26+ cells (red fluorescence) localize to the grafts via the human vasculature (counterstained with anti-human VWFVIII–FITC) as indicated by their perivascular distribution. Figure 2H illustrates control sections from synovial transplanted SCID mice not injected with huPBL.
Fig. 2.
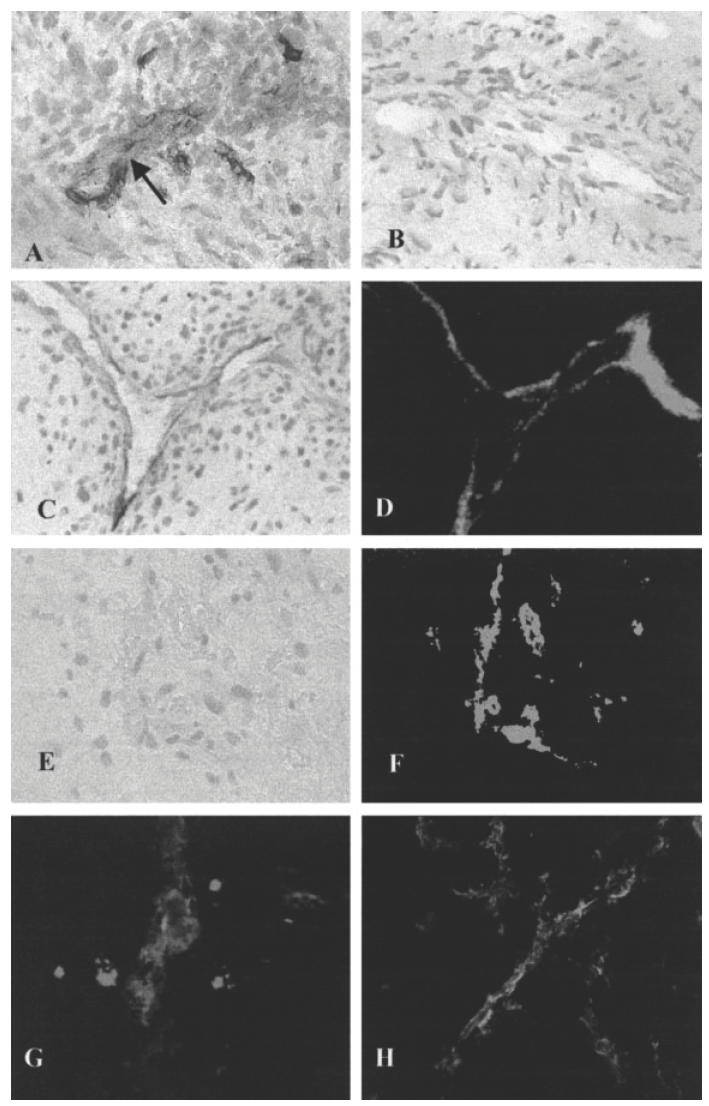
Representative examples of assessment of graft human–murine vasculature and localization of huPBL to synovial transplants. (A) The red stain denotes human blood vessels (anti-human VWFVIII), while the brown stain denotes murine vasculature (anti-murine CD31). The black arrow indicates a possible connection between the human and murine blood vessels. (B) Negative control. (C) Localization of anti-human intercellular adhesion molecule (ICAM)–biotinylated MoAb, following i.v. injection, in the blood vessels of synovium transplanted into SCID mice. The MoAb was detected in the human grafts using avidin-biotin complex (ABC) alone. (D) A sequential section of (C) counterstained with anti-human FITC–VWFVIII. (E,F) Control mice injected with MOPC21 biotinylated MoAb and stained with anti-human FITC–VWFVIII, respectively. (G) PKH26-labelled (red fluorescence) huPBL localization to synovial transplants. The section was counterstained with VWFVIII–FITC (green fluorescence). (H) Grafts from control animals not injected with PKH26–huPBL. This section was also stained with anti-human VWFVIII–FITC.
HuPBL kinetics of circulation and localization in murine organs
In order to determine the fate of huPBL injected into the mouse we first analysed the proportion of huPBL in the SCID mouse circulation at various time points (Fig. 3). It can be seen that at 24 h there is a significant drop in huPBL present in the mouse circulation. To establish whether the majority of the huPBL disappear from the circulation following sequestration in the reticuloendothelial system (RES), we injected 111In-huPBL intravenously into synovium transplanted animals and at 24 h measured the activity in the organs using a γ counter. As expected, most of the radioactivity was found in murine liver and spleen (data not shown).
Fig. 3.
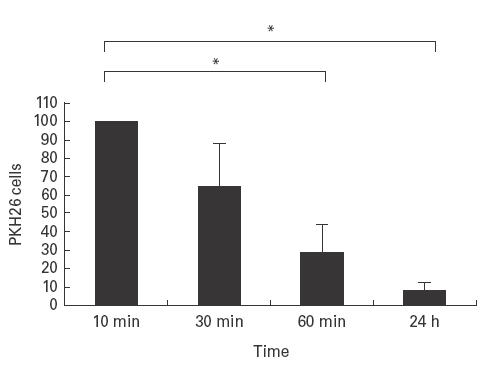
Kinetics of circulation of PKH26-huPBL in murine circulation. PKH26-HuPBL (10 × 106) were injected intravenously into SCID mice and blood samples were taken at t = 10 min and analysed by FACScan. This value was taken as 100% of the circulating cells. The cells obtained via blood samples taken at 30 min, 60 min and 24 h were calculated as a percentage of that obtained at t = 10 min for each animal. n = 5 animals; mean ± s.d.; P = < 0·001.
HuPBL localization into synovial transplants
To examine accurately the capacity of huPBL to localize into human synovial grafts, two methods (using huPBL double-labelled with PKH26 and 111In) were utilized in the experiments described below unless specified. Before labelling, the cells were phenotyped and it was demonstrated that in all the samples utilized the majority of the donor cells were CD3+ (83·6 ± 4·3%), CD19+ (8·8 ± 2·0%), LFA-1+ (MIF 349·9 ± 60·0), CLA+ (13·6 ± 1·2%), L-selectin+ (71·5 ± 4·3%) and CD14+ (< 1%), confirming that the cells were mainly of the CD3+, non-adherent phenotype.
In preliminary experiments, the optimal number of huPBL required for maximal migration to the grafts was determined by injecting varying numbers (1-, 5-, 10-, 20 × 106) of double-labelled cells into the tail vein of synovium-transplanted animals. The results showed that optimal migration was achieved with 5 × 106 huPBL with no further increase observed following an increase in the number of cells injected (data not shown). Similarly, in order to determine whether the degree of migration into the synovial grafts was TNF-α dose-dependent, varying concentrations (200–800 ng/graft) were injected intragraft prior to i.v. injection of double-labelled 111In-PKH26-huPBL. Both methods showed that intragraft injection of TNF-α (200 ng/graft) caused more than two-fold increase in migration of huPBL to the synovial grafts. No further increase was noted augmenting the TNF-α dose/graft, indicating that the first dose of 200 ng was already maximally stimulating the system (data not shown).
In order to determine the optimal time point at which huPBL migrate to the grafts, animals were injected intravenously with single-labelled 111In huPBL (5 × 106) and killed at different time points. In addition, to investigate basal versus stimulated migration, transplants were injected with TNF-α (200 ng/graft). As can be seen in Fig. 4, the degree of migration to the transplants gradually increased, reaching a plateau over a 48-h period (probably cumulative migration). Of relevance, as expected, migration in response to TNF-α was greater than in the control saline-treated group at each time point.
Fig. 4.
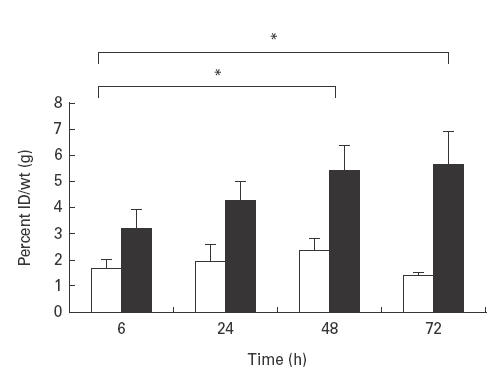
Time course to determine optimal time of migration of 111In-huPBL into synovial grafts. 111In-huPBL (10 × 106) were injected intravenously into synovium-transplanted SCID mice which had received intragraft injections of either 200 ng TNF-α (▪) or saline (□). At various time points mice were killed and the grafts extracted and radioactivity levels determined by γ counter. Migration is expressed as percentage of injected dose (ID)/wt (g). n = 6 transplants/condition; mean ±s.e.m.; P = 0·007.
We then investigated the potential mechanism of action of TNF-α by examining the variation in CAM expression in the transplants and the relationship of this to the degree of migration of huPBL into the grafts. In addition, in order to ascertain whether these results were influenced by the level of graft vascularization, we also examined human and murine vascular surface area (see below).
Intragraft injections of TNF-α cause an up-regulation in expression of human ICAM-1 and VCAM-1
Transplant sections from grafts injected with TNF-α (200 ng/graft) or control-PBS were immunostained for human ICAM-1, VCAM-1 and E-selectin. As the samples were analysed at 48 h post-injection (time at which migration experiments were terminated), E-selectin expression was negligible (data not shown). This is in keeping with the described kinetics of E-selectin expression in vitro [28,29]. On the other hand, as can be seen in a representative example in Fig. 5 and all samples in Fig. 6, TNF-α (200 ng/graft) significantly up-regulated the expression of ICAM-1 and VCAM-1 in comparison with PBS-treated control samples. It is interesting to note that in these latter samples there was a significantly lower CAM expression in comparison with the original pretransplantation tissue, indicating that in the absence of the inflammatory stimuli present in the RA joint (including TNF-α), synovial transplants revert to a ‘resting state’ in the animals. Following stimulation with a single injection of TNF-α, grafts regained pretransplantation CAM expression levels, suggesting, like other groups before, that TNF-α plays a pivotal role in the regulation of vascular CAM [30]. The TNF-α-induced increase in MVE CAM expression was demonstrated to be functional, as it was paralleled by the increase in huPBL migration to the grafts.
Fig. 5.
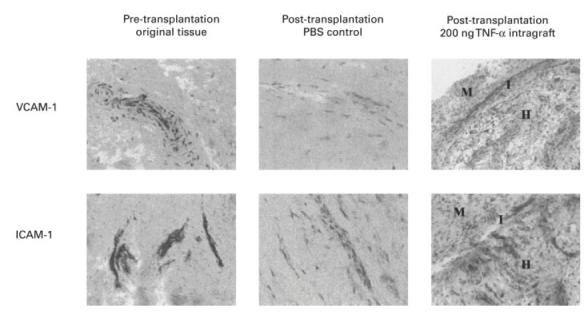
Representative samples of the original and grafted synovial tissue, pre- and post-transplantation. Transplants were injected intragraft with PBS (control) or with TNF-α (200 ng/graft). Cryostat sections (10 μm) were stained for anti-human intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) using standard immunoperoxidase technique. The intensity of staining was scored using an arbitrary scale shown graphically in Fig. 6. M, Murine tissue; H, human tissue; I, interface.
Fig. 6.
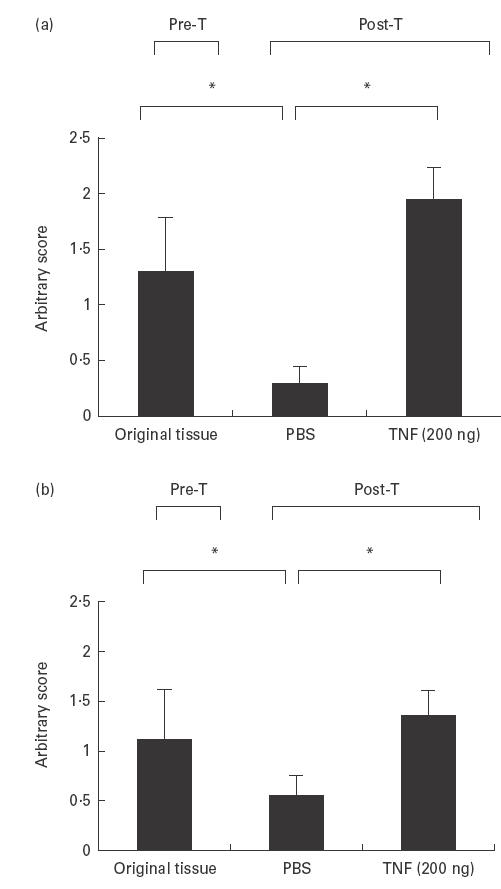
(a) Intercellular adhesion molecule-1 (ICAM-1) and (b) vascular cell adhesion molecule-1 (VCAM-1) expression in the original and grafted synovial tissue, pre- (Pre-T) and post-transplantation (Post-T). Transplants were injected with PBS (control) or TNF-α (200 ng/graft). Cryostat sections (10 μm) were stained for anti-human ICAM-1 and VCAM-1 using a standard immunoperoxidase technique. The intensity of staining was assessed using an arbitrary score 0–3 with increasing intensity. (a) P = < 0·001. (b) P = < 0·003. Original tissue n = 3 patient samples. Pre-T and Post-T n = 6 transplants/condition. Mean ± s.e.m.
Finally, in order to determine whether the up-regulation in migration induced by intragraft injections of TNF-α was simply due to an increase in the vascular beds feeding the graft, cryostat sections of the transplants were immunostained for human and mouse vasculature. The analysis of the human blood vessel area, expressed as a volume fraction, showed no statistical difference between the TNF-α-treated and untreated samples (PBS group = 0·37 ± 0·12; TNF-α group = 0·32 ± 0·06; P > 0·05). Similar results were obtained when the murine vasculature was analysed (PBS group = 0·79 ± 0·12; TNF-α group = 1·12 ± 0·16; P > 0·05). In addition, there was no correlation between human or murine vasculature and huPBL migration into the grafts (r values −0·157 and 0·0889, respectively).
DISCUSSION
In this study we provide evidence that direct intragraft injection of TNF-α up-regulates the expression of human MVE CAM in RA synovial membrane (SM) transplanted into SCID mice. Although it has been known for some time that TNF-α can induce the expression of CAM on endothelial cells (EC) in vitro, in humans the in vivo evidence is mainly indirect. The principal reason for this is the obvious ethical issue of injecting TNF-α in humans in order to perform studies on MVE physiology and leucocyte migration. For this reason, we have adapted a model in which human tissues grafted into SCID mice can be studied under controlled experimental conditions. Using this model we have demonstrated that synovial transplantation is successful in almost 100% of cases, with grafts remaining viable following re-vascularization by mouse subdermal vessels. This was confirmed both macro- and microscopically, as analysis of the grafts 4 weeks post-transplantation revealed vasculature of both human and murine origin. Furthermore, the two vascular systems were demonstrated to be connected both by double immunoperoxidase, using species-specific MoAb, and by analysing the localization of an anti-human ICAM-1 MoAb to the grafts following injection into the SCID mouse tail vein. Using the latter technique, the anti-human ICAM-1 MoAb was found to bind to the luminal surface of human blood vessels, confirming that the murine vasculature not only anastomoses with the human vasculature, but also maintains the capacity of expressing human CAM. Even more importantly, we demonstrated that this is a viable delivery system of huPBL to the human grafts in vivo. The degree of huPBL localization to the grafts was quantified by two methods and both demonstrated a significantly increased migration in response to TNF-α. We then explored the mechanisms by which TNF-α up-regulated this phenomenon. First we considered the likely possibility that TNF-α was acting via an up-regulation of CAM. Indeed it was demonstrated that while 4 weeks post-transplantation there was a lower expression of graft CAM in comparison with the original tissue, intragraft injection of TNF-α massively up-regulated CAM expression. This correlated well with the increased migration of huPBL, and therefore these results were explained on the basis that the increased density of luminal CAM would facilitate the interactions of huPBL surface receptors with MVE counter-receptors. An alternative explanation was to attribute the increased degree of migration after intragraft injection of TNF-α to the degree of transplant vascularization. Although TNF-α is known to induce neovascularization via vascular endothelial growth factor (VEGF)[31,32], we thought this was unlikely to be the case in our model, as TNF-α was injected only 48 h prior to the termination of the migration experiment. Neovascularization of the grafts clearly took place at some stage, as demonstrated by the survival of the transplants, and by the evidence of anastomosis between the mouse and human vasculature, but this was not significantly different between the TNF-α-treated and untreated groups. In addition, there was no correlation between the number of huPBL infiltrating the grafts and the level of human or mouse vascularity expressed as total endothelial surface. Taken together, these observations indicate that the level of huPBL localization to the transplant relates to the increased leucocyte–EC interactions secondary to the up-regulation of CAM induced by TNF-α, and not to the variation in the degree of graft vascularization.
Another important problem that we addressed in this study was the proportion of human cells injected into mice which actually reached the transplants. Radioactive studies estimated 1–5% of the injected huPBL concentration reached the transplants. We believe that this is partially due to the small size of the transplants and the low number of huPBL circulating in the SCID mice after 24 h. The reasons for this will include at least two non-mutually exclusive possibilities. First, huPBL could be killed by mouse natural killer (NK) cells. To minimize this possibility we used NOD/LtSz-scid/scid mice. This strain of mice is specifically bred not only to produce no T or B cells, but also to have no NK activity (although the animals retain non-functional NK cells). In addition, we found no evidence of free/unbound 111In in the plasma of the animals (data not shown). The second more likely possibility, that huPBL were sequestered in the mouse RES, was confirmed by analysing radioactivity levels in murine organs where liver and spleen showed the highest radioactive counts (data not shown).
The low proportion of huPBL migrating to the synovial grafts could be construed as a major limitation of this model. On the other hand it can be argued that these cells represent an important component of circulating huPBL, i.e. those which are programmed to traffic specifically to the synovium. Although this concept remains controversial, a large body of evidence supports the idea that different subsets of T cells localize specifically to different tissues. Such evidence includes the observation that the majority of lymphocytes entering lymph nodes (LN) express the naive/resting phenotype (CD45RA) [33], while in inflamed tissue the cellular infiltrate is composed mainly of CD4 memory/activated (CD45RO) T cells. Within this latter population, different subsets of memory/effectors cells have been proposed to migrate preferentially to various inflamed tissues such as the gut, the joint and the skin [33–35]. The control of lymphocyte ‘homing’ is not completely understood, but there is compelling evidence to indicate that a series of integrated adhesion and signalling events is involved. Such events include the distinctive interaction of lymphocyte-associated adhesion molecules, called ‘homing receptors’ with their MVE tissue-specific counter-receptors, called ‘addressins’, and of specific chemokines (CK) with their correspondent CK-receptor (CK-R) preferentially expressed by different lymphocyte populations. A recent example of this has been published by Campbell et al. [36], who demonstrated that CCR4, the CK-R for thymus and activation-regulated chemokine (TARC), is preferentially found on skin but not on the intestinal homing subset as defined by the expression of CLA and α4β7 molecules, respectively. This preferential migration can be explained on the basis of the selective production of TARC by skin but not intestinal MVE. This paper therefore provides further support for the role of the combinatorial use of adhesion and signalling molecules in the preferential recruitment of a small circulating lymphocyte subset to a specific tissue.
Leaving aside the obvious artificial conditions of the model we have presented, the synovial transplants may function as natural targets for synovial homing PBL which bind to synovial specific ‘addressins’ presented by the graft MVE. This would be made possible by the fact that the transplants maintain the natural microenvironment for the retention of the tissue-specific MVE phenotype. We believe that this model represents an ideal medium through which organ-specific MVE ligands and their cognate homing receptors expressed by specific subsets of circulating lymphocytes can be identified.
Acknowledgments
We would like to acknowledge Dr David Walsh for valuable discussions on angiogenesis and anastomosis, Dr Martyn Robinson for the generous gift of antibodies, and Dr Francesca Sinigaglia for critically reviewing the paper. We also would like to thank the Wellcome Trust and the Arthritis Research Campaign for supporting this research.
REFERENCES
- 1.Pitzalis C. Role of adhesion mechanisms in the pathogenesis of chronic synovitis. (The Michael Mason Prize Essay 1996) B J Rheumatol. 1996;35:1198–215. doi: 10.1093/rheumatology/35.12.1198. [DOI] [PubMed] [Google Scholar]
- 2.Haskard DO. Cell adhesion molecules in rheumatoid arthritis. Curr Opin Rheumatol. 1995;7:229–34. doi: 10.1097/00002281-199505000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Koch AE. Angiogenesis: implications for rheumatoid arthritis. Arthritis Rheum. 1998;41:951–62. doi: 10.1002/1529-0131(199806)41:6<951::AID-ART2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 4.Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–90. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 5.Pitzalis C, Kingsley G, Lanchbury JS, Murphy J, Panayi GS. Expression of HLA-DR, DQ and DP antigens and interleukin-2 receptor on synovial fluid T lymphocyte subsets in rheumatoid arthritis: evidence for ‘frustrated’ activation. J Rheum. 1987;14:662–6. [PubMed] [Google Scholar]
- 6.Pitzalis C, Kingsley G, Murphy J, Panayi G. Abnormal distribution of the helper-inducer and suppressor-inducer T-lymphocyte subsets in the rheumatoid joint. Clin Immunol Immunopathol. 1987;45:252–8. doi: 10.1016/0090-1229(87)90040-7. [DOI] [PubMed] [Google Scholar]
- 7.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–6. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 9.Adams DH, Shaw S. Leucocyte–endothelial interactions and regulation of leucocyte migration. Lancet. 1994;343:831–6. doi: 10.1016/s0140-6736(94)92029-x. [DOI] [PubMed] [Google Scholar]
- 10.Salmi M, Jalkanen S. How do lymphocytes know where to go: current concepts and enigmas of lymphocyte homing. Adv Immunol. 1997;64:139–218. doi: 10.1016/s0065-2776(08)60889-5. [DOI] [PubMed] [Google Scholar]
- 11.Gimbrone Ma, Jr, Bevilacqua MP, Cybulsky MI. Endothelial-dependent mechanisms of leukocyte adhesion in inflammation and atherosclerosis. Ann NY Acad Sci. 1990;598:77–85. doi: 10.1111/j.1749-6632.1990.tb42279.x. [DOI] [PubMed] [Google Scholar]
- 12.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med. 1993;177:1277–86. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann B, Machleidt T, Lifka A, et al. Crucial role of 55-kilodalton TNF receptor in TNF-induced adhesion molecule expression and leukocyte organ infiltration. J Immunol. 1996;156:1587–93. [PubMed] [Google Scholar]
- 14.Probert L, Akassoglou K, Alexopoulou L, et al. Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice. J Leuk Biol. 1996;59:518–25. doi: 10.1002/jlb.59.4.518. [DOI] [PubMed] [Google Scholar]
- 15.Williams RO. Combination therapy in mice: what can we learn that may be useful for understanding rheumatoid arthritis. Springer Semin Immunopathol. 1998;20:165–80. doi: 10.1007/BF00832005. [DOI] [PubMed] [Google Scholar]
- 16.Feldmann M, Brennan FM, Elliott MJ, Williams RO, Maini RN. TNF alpha is an effective therapeutic target for rheumatoid arthritis. Ann NY Acad Sci. 1995;766:272–8. doi: 10.1111/j.1749-6632.1995.tb26675.x. [DOI] [PubMed] [Google Scholar]
- 17.Feldmann M, Brennan FM, Williams RO, Elliott MJ, Maini RN. Cytokine expression and networks in rheumatoid arthritis: rationale for anti-TNF alpha antibody therapy and its mechanism of action. J Inflamm. 1995;47:90–96. [PubMed] [Google Scholar]
- 18.Eigler A, Sinha B, Hartmann G, Endres S. Taming TNF: strategies to restrain this proinflammatory cytokine. Immunol Today. 1997;18:487–92. doi: 10.1016/s0167-5699(97)01118-3. [DOI] [PubMed] [Google Scholar]
- 19.Feldmann M, Elliott MJ, Woody JN, Maini RN. Anti-tumor necrosis factor-alpha therapy of rheumatoid arthritis. Adv Immunol. 1997;64:283–350. doi: 10.1016/s0065-2776(08)60891-3. [DOI] [PubMed] [Google Scholar]
- 20.Moreland LW. Inhibitors of tumor necrosis factor for rheumatoid arthritis. J Rheum. 1999;26(Suppl. 57):7–15. [PubMed] [Google Scholar]
- 21.Breedveld F. New tumor necrosis factor-alpha biologic therapies for rheumatoid arthritis. Eur Cytokine Network. 1998;9:233–8. [PubMed] [Google Scholar]
- 22.Tak PP, Taylor PC, Breedveld FC, et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthrtis Rheum. 1996;39:1077–81. doi: 10.1002/art.1780390702. [DOI] [PubMed] [Google Scholar]
- 23.Rendt KE, Barry TS, Jones DM, Richter CB, McCachren SS, Haynes BF. Engraftment of human synovium into severe combined immune deficient mice. Migration of human peripheral blood T cells to engrafted human synovium and to mouse lymph nodes. J Immunol. 1993;151:7324–36. [PubMed] [Google Scholar]
- 24.Jorgensen C, Couret I, Canovas F, et al. Mononuclear cell retention in rheumatoid synovial tissue engrafted in severe combined immunodeficient (SCID) mice is up-regulated by tumour necrosis factor-alpha (TNF-alpha) and mediated through intercellular adhesion molecule-1 (ICAM-1) Clin Exp Immunol. 1996;106:20–25. [PubMed] [Google Scholar]
- 25.Geiler T, Kriegsmann J, Keyszer GM, Gay RE, Gay S. A new model for rheumatoid arthritis generated by engraftment of rheumatoid synovial tissue and normal human cartilage into scid mice. Arthritis Rheum. 1994;37:1664–71. doi: 10.1002/art.1780371116. [DOI] [PubMed] [Google Scholar]
- 26.Pitzalis C, Cauli A, Pipitone N, et al. Cutaneous lymphocyte antigen-positive T lymphocytes preferentially migrate to the skin but not to the joint in psoriatic arthritis. Arthritis Rheum. 1996;39:137–45. doi: 10.1002/art.1780390118. [DOI] [PubMed] [Google Scholar]
- 27.Pitzalis C, Kingsley G, Haskard D, Panayi G. The preferential accumulation of helper-inducer T lymphocytes in inflammatory lesions: evidence for regulation by selective endothelial and homotypic adhesion. Eur J Immunol. 1988;18:1397–404. doi: 10.1002/eji.1830180915. [DOI] [PubMed] [Google Scholar]
- 28.Bevilacqua MP, Stengelin S, Gimbrone Ma, Jr, Seed B. Endothelial leukocyte adhesion molecule 1: an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–5. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- 29.Bevilacqua MP, Nelson RM. Selectins. J Clin Invest. 1993;91:379–87. doi: 10.1172/JCI116210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maini RN, Elliott M, Brennan FM, Williams RO, Feldmann M. TNF blockade in rheumatoid arthritis: implications for therapy and pathogenesis. APMIS. 1997;105:257–63. doi: 10.1111/j.1699-0463.1997.tb00567.x. [DOI] [PubMed] [Google Scholar]
- 31.Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998;41:1258–65. doi: 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Ko Y, Totzke G, Gouni-Berthold I, Sachinidis A, Vetter H. Cytokine-inducible growth factor gene expression in human umbilical endothelial cells. Mol Cellular Probes. 1999;13:203–11. doi: 10.1006/mcpr.1999.0236. [DOI] [PubMed] [Google Scholar]
- 33.Mackay CR. Homing of naive, memory and effector lymphocytes. Curr Opin Immunol. 1993;5:423–7. doi: 10.1016/0952-7915(93)90063-x. [DOI] [PubMed] [Google Scholar]
- 34.Mackay CR, Marston WL, Dudler L, Spertini O, Tedder TF, Hein WR. Tissue-specific migration pathways by phenotypically distinct subpopulations of memory T cells. Eur J Immunol. 1992;22:887–95. doi: 10.1002/eji.1830220402. [DOI] [PubMed] [Google Scholar]
- 35.Picker LJ, Michie SA, Rott LS, Butcher EC. A unique phenotype of skin-associated lymphocytes in humans. Preferential expression of the HECA-452 epitope by benign and malignant T cells at cutaneous sites. Am J Pathol. 1990;136:1053–68. [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell JJ, Haraldsen G, Pan J, et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 1999;400:776–80. doi: 10.1038/23495. [DOI] [PubMed] [Google Scholar]