

Abstract
Henoch–Schönlein purpura (HSP) is a small vessel vasculitis characterized by increased serum IgA and IgA-dominant immune complex deposition in lesions. The involvement of IgA implies a probable role for TGF-β, a major factor in IgA production, in the pathogenesis of HSP. Among IgA antibodies, serum IgA anti-cardiolipin antibodies (aCL) have been found in many diseases, including vasculitis. In addition to the clinical presentations and laboratory parameters, we further investigated the roles of IgA aCL and TGF-β in childhood HSP. Twenty-six Chinese children with the diagnosis of HSP were enrolled. Blood samples from these patients were collected at both acute and convalescent stages. Intracellular staining of lymphocytes was performed to enumerate type 1 (interferon-gamma-secreting), type 2 (IL-4-secreting), and type 3 (TGF-β-secreting) helper T cells. Serum levels of TGF-β were detected by ELISA. Serum IgA aCL of 21 of 26 patients at the acute stage, 11 of them at the convalescent stage, were measured by ELISA. The data showed that IgA aCL serum levels were significantly elevated in patients compared with healthy controls (P < 0·001), and those patients at the convalescent stage (P < 0·001). In addition, TGF-β-secreting T cells were significantly elevated during the acute stage, and decreased at the convalescent stage. Although more studies are needed, the high prevalence of IgA aCL and increased TGF-β-secreting T cells in children with acute HSP revealed some points which should permit a better understanding of the pathogenesis of HSP.
Keywords: TGF-β, type 3 helper T cells, IgA anti-cardiolipin antibodies, childhood, Henoch–Schönlein purpura
Introduction
Henoch–Schönlein purpura (HSP), one of the most common types of vasculitis in childhood, is characterized by palpable non-thrombocytopenic purpura over the lower extremities and buttocks, arthritis or arthralgia, abdominal pain with or without gastrointestinal haemorrhage, and less commonly glomerulonephritis [1]. Histologically, it reveals the change of leukocytoclastic vasculitis, and has been well regarded as a specific clinicopathological entity by the vascular deposition of IgA-dominant immune complexes and elevated serum IgA levels [2,3]. The prognosis is generally good and depends on the extent of renal involvement. Treatment is usually supportive; non-steroid anti-inflammatory drugs (NSAID) are almost all that is needed and steroids are usually reserved for severe cases [4–6].
Antibodies directed against negatively charged phospholipids such as cardiolipin are a heterogeneous group of immunoglobulins of the IgG, IgM, IgA classes [7]. Among them, there is a growing interest in the IgA isotype of anti-cardiolipin antibodies (aCL). Although some studies have reported that these antibodies were rarely detected and of limited clinical significance [8,9], it is generally accepted that IgA aCL, as well as IgG aCL and IgM aCL, are significantly associated with thrombocytopenia, thrombotic complications, and recurrent fetal loss in patients with systemic lupus erythematosus (SLE) and/or anti-phospholipid syndrome (APS) [10–12]. In addition, IgA aCL has also occasionally been found alone in a variety of diseases including Guillain–Barré syndrome [13], pulmonary fibrosis in systemic sclerosis [14], infections [15,16], and leukocytoclastic vasculitis [17,18]. Many cells, including helper T cells, secrete TGF-β, which has been found to enhance class switching of IgA [19,20].
To further clarify the roles of IgA aCL and TGF-β in childhood HSP, this study was designed not only to observe the clinical aspects of HSP, but also to compare these immunological parameters at different disease status.
PATIENTS and METHODS
Patients and controls
A total of 26 previously healthy Chinese children with acute HSP were enrolled in this study. There were 13 males and 13 females with an age range of 3–17 years. The diagnosis of HSP was made according to the American College of Rheumatology 1990 criteria [21] during the period from October 1996 to April 1999 at National Taiwan University Hospital. Informed consent and Institutional approval were obtained for the study. Blood samples were collected at the time of diagnosis (acute stage) and then 6–8 months later (convalescent stage). The clinical presentations were recorded. Twenty healthy children with comparable sex and age distributions were recruited as normal controls. In the assay of IgA aCL, 10 patients with active juvenile rheumatoid arthritis (JRA) (four pauciarticular type, four polyarticular, and two systemic type, none with thrombocytopenia, thrombotic complications, or associated vasculitis) were also enrolled as another control group.
Serological studies
All serum samples were stored at −70°C before the assay. The serum levels of C3, C4, IgG, IgA, IgM and anti-streptolysin O (ASLO) titre were assayed with laser nephelometry (Behring Diagnostics GmbH, Marburg, Germany). Serum TGF-β levels were measured with sandwich ELISAs. In vitro activation by transient acidification of serum samples prior to assay was performed, since TGF-β was secreted in a biologically inactive form.
IgA, IgG, and IgM anti-cardiolipin antibodies
Because of the limitations regarding blood sampling from some patients, only 21 of 26 patients at the acute stage and 11 of these 21 patients at the convalescent stage were included for the detection of serum levels of aCL. The IgG, IgM, and IgA aCL of 21 patients, IgA aCL of 11 of them at the convalescent stage, and IgA aCL of both control groups were measured by commercially standardized ELISAs (ORGenTec ELISA kit; Germany). Briefly, the microplate was coated with highly purified cardiolipin and saturated with human β2-glycoprotein I (β2-GPI). Polyclonal rabbit anti-human IgG, IgM, and IgA antibodies labelled with horseradish peroxidase were used as conjugate solutions to recognize aCL of the three isotypes. This assay system is calibrated against the internationally recognized reference sera from E. N. Harris (Louisville, KY) [22]. The plate was finally read in a spectrophotometer at 450 nm. The cut-off values for IgG, IgM, and IgA aCL were set at 10 GPL U/ml, 7 MPL U/ml, and 10 APL U/ml, respectively.
Lymphocyte phenotyping and intracellular staining
Phenotyping of lymphocytes was performed by a lysed whole-blood method. T cells (CD4, CD8, CD45RA−, TCR-αβ, TCR-γδ), B cell subsets, and natural killer (NK) cells were analysed using matched combinations of murine MoAbs conjugated with FITC or PE. For intracellular staining of cytokines, peripheral blood mononuclear cells (PBMC) were isolated and stimulated with phorbol 12-myristate 13-acetate (PMA; 20 ng/ml), ionomycin and monensin (4 nm) for 12 h. They were then washed in PBS, fixed with 4% paraformaldehyde, permeabilized with saponin, and stained with specificity-matched combinations of fluorescence-labelled antibodies (CD4 Cy-chrome plus interferon-gamma (IFN-γ)–FITC for Th1, IL-4–PE for Th2, and TGF-β1–FITC for Th3). Fluorescein-stained cells were detected with a FACScan flow cytometer, and the results were analysed with SimulSET software (Becton Dickinson Immunocytometry Systems, San Jose, CA).
Statistical analysis
The values of IgA aCL were presented as median with ranges, and the Mann–Whitney U-test was used to compare the IgA aCL levels between patients and controls, and at different stages. The prevalence of three isotypes of aCL of HSP was compared by McNemar's test. The relationship between serum total IgA and IgA aCL was analysed by correlation coefficient test. Other laboratory data were expressed as means ±s.e.m. Comparisons of these data between different disease stages of patients were performed with the non-parametric Wilcoxon signed ranks test. A two-tailed P value of <0·05 was considered statistically significant.
Results
Clinical presentations and courses
Most of the children (23/26) in this series had onset of HSP during either the autumn or winter seasons. Table 1 summarizes the clinical characteristics of the 26 patients in this study. All of them had typical characteristics of HSP with palpable purpura over the lower extremities. Abdominal pain with or without gastrointestinal bleeding occurred in 19 of 26 patients (73%). Arthritis or arthralgia was noted in 17 of 26 patients (65%). The ankle was the most common joint involved, followed by the knee joint. Five patients had leg oedema, and one had oedema of all four limbs. A history of upper respiratory tract infection (URI) was noted in 16 of 26 patients (61%). Only one patient developed glomerulonephritis, with presentations including heavy proteinuria and haematuria. None of the patients exhibited any features of APS, except for one patient with thrombocytopenia (110 000/mm3). The outcomes were generally good. All patients were prescribed NSAIDs. Short-term steroid was used in 19 patients and azathioprine (Imuran) in three patients because of either nephritis, GI bleeding, or severe abdominal pain despite NSAIDs. Twenty-two patients (including the 11 patients detected for IgA aCL at a mean follow-up duration of 7 months) were symptom-free during follow up (6–8 months); four patients (15%) had recurrent symptoms, mostly as leg purpura and abdominal pain.
Table 1.
Clinical characteristics and treatment of patients with Henoch–Schönlein purpura
| No. of patients (%) | |
|---|---|
| Symptoms and signs | |
| Purpura over lower extremities and/or buttocks | 26 (100) |
| Abdominal pain with/without SOB | 19 (73) |
| Arthritis/arthralgia | 17 (65) |
| Extremities oedema | 6 (23) |
| Glomerulonephritis | 1 (4) |
| Previous URI | 16 (61) |
| Treatment | |
| NSAID | 26 (100) |
| Steroid | 19 (73) |
| Azathioprine | 3 (12) |
SOB, Stool occult blood; URI, upper respiratory tract infection; NSAID, non-steroidal anti-inflammatory drug.
Haemogram and lymphocyte subsets
The leucocyte, neutrophil percentage, platelet count and C-reactive protein (CRP) were significantly higher at the acute stage than at the convalescent stage (Table 2). Serum C3, C4, and IgA levels were also significantly higher at the acute stage than at the convalescent stage. By contrast, IgG and IgM levels were within normal limits and did not differ significantly between disease stages. ASLO was increased in 19 of the 26 patients (Table 2).
Table 2.
Laboratory data of 26 patients at both acute and convalescent stage and that of 20 healthy controls
| Acute | Convalescence | Control | |
|---|---|---|---|
| Haematological and serological parameter | |||
| Leucocytes (mm3) | 11 001·9 ± 635·8* | 6790·0 ± 374·8 | ND |
| Neutrophils (%) | 63·4 ± 2·6* | 46·6 ± 3·0 | ND |
| Lymphocytes (%) | 33·8 ± 3·5 | 45·0 ± 2·9 | ND |
| Platelet count ( × 103/mm3) | 384·9 ± 16·6* | 311·4 ± 12·1 | ND |
| Elevation of CRP (no./total)† | 11/23* | 0/25 | ND |
| C3 (mg/dl) | 115·6 ± 3·1* | 100·8 ± 4·4 | 116·1 ± 3·7 |
| C4 (mg/dl) | 33·6 ± 2·5* | 25·1 ± 2·5 | 23·7 ± 1·5 |
| IgG (mg/dl) | 1133·2 ± 62·1 | 1072·2 ± 53·6 | 1071·9 ± 68·2 |
| IgA (mg/dl) | 269·4 ± 18·4* | 210·4 ± 21·9 | 170·8 ± 15·9 |
| IgM (mg/dl) | 181·8 ± 14·9 | 156·7 ± 12·8 | 181·1 ± 13·7 |
| TGF-β (ng/dl) | 32·0 ± 4·6 | 41·9 ± 5·1 | 30·4 ± 4·6 |
| Elevation of ASLO (no./total)‡ | 19/26* | 11/26 | 0/20 |
| Phenotype analysis of lymphocyte | |||
| T cell | 70·3 ± 1·8 | 71·9 ± 1·3 | 65·0 ± 1·6 |
| B cell | 17·3 ± 1·5 | 14·9 ± 0·8 | 15·2 ± 1·2 |
| CD4+ T cell | 35·8 ± 1·8 | 35·7 ± 1·6 | 32·0 ± 2·5 |
| CD8+ B cell | 27·6 ± 1·5 | 14·8 ± 1·1 | 12·5 ± 0·9 |
| CD45RA−CD4+ T cell | 15·2 ± 1·8 | 14·8 ± 1·1 | 12·5 ± 0·9 |
| CD45RA+CD4+ T cell | 20·7 ± 2·0 | 20·8 ± 1·5 | 19·5 ± 2·4 |
| αβ-T cell | 57·5 ± 2·1 | 59·3 ± 1·3 | ND |
| γδ-T cell | 7·5 ± 0·8 | 8·4 ± 0·7 | ND |
| NK cell | 10·1 ± 1·5 | 11·0 ± 1·5 | 13·1 ± 1·8 |
| CD5+ B cell | 8·2 ± 1·0 | 6·6 ± 0·5 | 8·9 ± 1·0 |
Values are presented as mean ±s.e.m.
ND, Not done.
P < 0·05 for differences between acute and convalescent stage.
Normal value of CRP: < 0·09.
Anti-streptolysin O (ASLO) level >58 U/ml was viewed as elevated.
T lymphocytes at both the acute and convalescent stages were increased significantly compared with the controls; CD5+ B cells decreased at the convalescent stage, although the difference was not statistically significant. Serum TGF-β levels at the acute stage were lower than those at the convalescent stage, but it did not show statistical significance when comparison was conducted between the different stages and the controls (Table 2).
Serum levels of IgA, IgG, and IgM anti-cardiolipin antibodies
Seventeen (81%) of 21 patients with HSP had a positive test result for IgA aCL. The median level was 17·4 APL U/ml with a range from 7·6 APL U/ml to 125·7 APL U/ml. IgA aCL was detected in only one (11·2 APL U/ml) of the 20 healthy controls (median 0·9 APL U/ml, range 0–11·2 APL U/ml), and none of the JRA controls (median 0·8 APL U/ml, range 0–7·5 APL U/ml). The difference in IgA aCL levels between patients and controls was statistically significant (P < 0·001) (Fig. 1). The IgA aCL levels of all 11 of the follow-up patients decreased significantly during the convalescent stage (P < 0·001) (Fig. 2). Four patients (4/21) and two JRA controls were positive for IgG aCL, and three of these four patients also had detectable levels of IgA aCL. The IgM aCL was not detected in any of the patients and controls. Statistically, the prevalence rate of IgA aCL was higher than that of IgG (P < 0·05) or IgM isotype (P < 0·05). No correlation was found between IgA aCL and total serum IgA antibodies (P = 0·596).
Fig. 1.
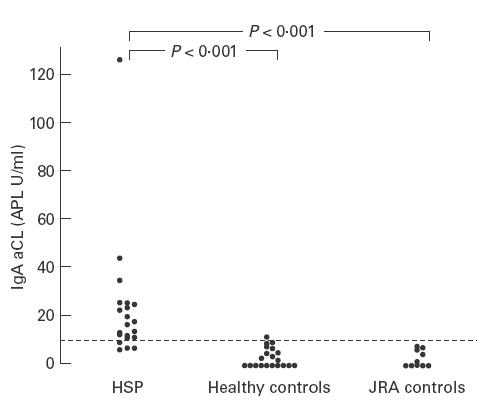
Distribution of the IgA anti-cardiolipin antibody (aCL) levels of patients with acute Henoch–Schönlein purpura (HSP) (n = 21), of healthy controls (n = 20), and of juvenile rheumatoid arthritis (JRA) controls (n = 10). The difference of serum levels of IgA aCL between patients and both control groups was statistically significant.
Fig. 2.
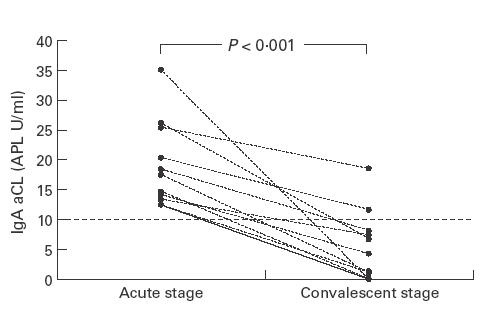
The IgA anti-cardiolipin antibody (aCL) levels of 11 patients with Henoch–Schönlein purpura (HSP) during the acute stage and the convalescent stage. The serum levels of IgA aCL decreased significantly during the convalescent stage.
Levels of cytokine-secreting T cells
Intracellular staining of stimulated PBMC with anti-cytokine antibodies demonstrated the presence of a particular subset of TGF-β-secreting CD4+ T cells in all patients during the acute stage. These TGF-β-secreting T cells were not found in normal control and JRA control groups (0%). A significant elevation in TGF-β-secreting (Th3) cells during the acute stage was demonstrated in all of the patients; this declined to nearly undetectable levels when disease activity subsided (Fig. 3). By contrast, there was no difference in the percentage of either Th1 or Th2 during the disease process of HSP (Fig. 4).
Fig. 3.
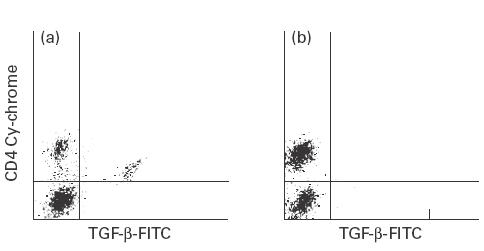
Photography of flow cytometry demonstrating the distribution of TGF-β-secreting helper T cells (Th3) during the acute (a) and the convalescent (b) stage. TGF-β-secreting helper T cells can only be detected in patients at the acute stage, not in those at the convalescent stage.
Fig. 4.
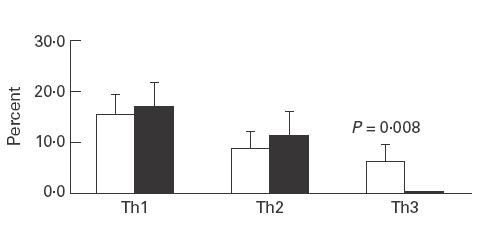
The percentage of IFN-γ-secreting (Th1), IL-4-secreting (Th2), and TGF-β-secreting (Th3) cells at both stages. The population of Th3 cells is significantly higher at the acute stage (□) than at the convalescent stage (▪) (6·2% versus 0·4%; P = 0·008).
Discussion
In the current study, elevation of leucocytes, neutrophil percentage, platelet counts and CRP at the onset of HSP reflected the possible nature of acute inflammatory process; increased serum C3, C4 and IgA levels implied involvement of immunological elements in the pathogenesis of HSP. There have been inconsistent results regarding the serum complement concentrations in patients with HSP—both classic and alternative pathways of complement activation were suggested [23–26]. The previous reports and our current study had the same finding of elevated serum IgA in this group of patients [27–30]. Since IgA activates the complement system via the alternative pathway [31], the involvement of the alternative pathway in HSP is more rational. No consensus has been reached on the distribution of lymphocyte subsets in HSP [32–35]. In our study, the population of total T cells increased significantly irrespective of disease status. Furthermore, CD5+ B cells, committed to the production of polyreactive autoantibodies and involved in the pathogenesis of autoimmune diseases [36], depressed at the convalescent stage of disease.
In 1994, Burden et al. reported detection of IgA aCL in a 51-year-old patient with HSP [17]. Two years later, in a prospective study, Burden found a high prevalence (60%) of IgA aCL in patients with cutaneous leukocytoclastic vasculitis. However, in their subsequent study, only one of 10 patients with childhood HSP had positive IgA aCL [18]. In 1998, Tajima et al. reported that IgA aCL was an independent risk factor for vasculitis in patients with various collagen diseases [37]. In the present study, 21 of 26 Chinese children without any collagen diseases who were suffering from acute onset HSP had a higher prevalence of IgA aCL than healthy and JRA controls. Similar to the results of previous studies [17,18,37], the other two isotypes of aCL were rarely detected in patients of this series. All four of the patients with positive IgG aCL in this series had low levels (11·3 GPL U/ml, 13·3 GPL U/ml, 13·6 GPL U/ml, 11·1 GPL U/ml). This result suggests that aCL restricted to IgA isotype may play an important role in the pathogenesis of HSP.
The presence of IgA aCL has been associated with thrombotic events, thrombocytopenia, fetal loss in patients with autoimmune diseases [10–12], and also independently with infections [15,16], neurological diseases [13,16], and vasculitis [17,18,31]. Recent studies have identified two different types of aCL [38]: one requires a cofactor for binding to cardiolipin (cofactor dependent), and the other one does not (cofactor independent). The cofactor, which was identified as β2-GPI, a 50-kD β2-globulin, enhances the reaction between cardiolipin and aCL [39]. In the present study, the test kits coated with cardiolipin and highly purified human β2-GPI were specific only for autoantibodies (IgG, IgM, and IgA) directed against cardiolipin and the complex of cardiolipin and β2-GPI. No cross-reactivity was observed to anti-DNA antibodies and antibodies occurring in syphilis. Therefore, regardless of whether the aCL was cofactor-dependent or cofactor-independent, it could be measured by this method.
An increased serum IgA concentration is a characteristic laboratory finding during the acute stage of HSP [1]. Some studies have reported that autoantibodies, such as IgA rheumatoid factor [40] and IgA anti-neutrophil cytoplasmic antibodies (ANCA) [41], were associated with HSP. Although no significant correlation was noted between total serum IgA and IgA aCL in the present study, the finding of the high prevalence of IgA aCL in patients with acute HSP suggested that IgA aCL is an important component of circulating IgA antibodies.
A variety of cells have been well documented to secret TGF-β. It has been suggested that a subset of CD4+ T cells called type 3 T helper (Th3) cells in the gut-associated lymphoid tissue (GALT) exert active cellular suppression via the secretion of TGF-β [42–45]. TGF-β, in addition to its potent immunosuppressive and anti-inflammatory effects, acts as a switch factor for IgA production [19,20]. Our study showed no significant correlation between serum TGF-β and disease activity. However, it clearly demonstrated the activation of TGF-β-secreting T cells (Th3) at the acute stage of HSP. The serum level of TGF-β is influenced by interactions of other cytokines, and the measurement by ELISA is unable to reflect correctly the microenvironment in the pathogenesis of disease; intracellular staining of cytokines may be a better tool to evaluate the frequency of TGF-β-secreting cells in HSP [46].
Although HSP is a common childhood disease, its pathogenesis remains undetermined. Previous epidemiological studies have found striking seasonal variations in HSP, with most cases occurring in the winter [47,48]. HSP has also been associated with a history of preceding infections, especially URI [48]. These results raise the possibility of infection as a direct cause or a potential trigger of this disease. Several microorganisms have been reported to be associated with the development of HSP, including β-haemolytic streptococcus [49], Mycoplasma pneumoniae [50], varicella [51], and mumps virus [52]. However, no definite pathogen could be isolated from the majority of patients with HSP. The association of infections and autoantibody production had been studied. Vaarala et al. found raised IgA aCL levels in patients with mumps infection [16]. Recently, Wilson et al. reported a high prevalence of IgA aCL in 42 Jamaicans with T cell lymphotrophic virus I (HTLV-I)-associated tropical spastic paraparesis (TSP) [15]. In the present study, 23 patients (90%) developed HSP during the autumn or the winter. More than half of patients in this series had preceding URI. The levels of IgA aCL decreased in parallel with the improvement of clinical symptoms. Taken together with increased Th3 cells, these findings may suggest that one of the associated microorganisms with specific antigenic structures which mimic cardiolipin enters the respiratory or GI tract and stimulates the T cells residing in the mucosa. Inflammatory responses are elicited with the participation of various immunocompetent cells and their humoral counterparts. The secreted TGF-β thus drives clonal IgA isotype switch in B cells, and thereby contributes to the formation of IgA antibodies. These antibodies to the structures may cross-react with host cardiolipin to form IgA immune complexes which damage the vessels. It is also possible that these pathogens could disrupt the structure of the vessels, resulting in the release of the hidden antigens, which induce IgA aCL production, and cause subsequent vascular inflammation.
In conclusion, the results of our study demonstrate a significant association between IgA isotype of aCL, Th3 cells, and childhood HSP during the acute stage. The detection of these antibodies and cells may also be valuable in the following of disease activity. In this study however, only two patients received skin lesion biopsies, which could not only provide the pathological evidence of leukocytoclastic vasculitis, but also demonstrate the deposition of IgA-containing complexes by fluorescence microscopy [1]. Recent studies have revealed that infection-related IgA aCL were cofactor-independent. The test kits used in the present study could not differentiate between these two subclasses of IgA aCL [22,38]. Although more work needs to be done, the data herein did reveal some immunological clues for a better understanding of the pathogenic mechanisms of HSP.
Acknowledgments
This study was supported by a grant from The C-L. Chen Paediatric Research Scholarship Foundation.
References
- 1.Cassidy JT, Petty RE. Textbook of pediatric rheumatology. 3. Philadelphia: W.B. Saunders Co.; 1995. pp. 384–8. [Google Scholar]
- 2.Knight JF. The rheumatic poison: a survey of some published investigations of the immunopathogenesis of Henoch–Schönlein purpura. Pediatr Nephrol. 1990;4:533–41. doi: 10.1007/BF00869841. [DOI] [PubMed] [Google Scholar]
- 3.Faille-Kuyber EH, Kater L, Kooiker CJ, Dorhout Mees EJ. IgA-deposits in cutaneous blood-vessel walls and mesangium in Henoch–Schönlein syndrome. Lancet. 1973;1:892–3. doi: 10.1016/s0140-6736(73)91471-2. [DOI] [PubMed] [Google Scholar]
- 4.Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature system of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 5.Jennette JC, Falk RJ. Small-vessel vasculitis. N Eng J Med. 1997;337:1512–23. doi: 10.1056/NEJM199711203372106. [DOI] [PubMed] [Google Scholar]
- 6.Sneller MC, Fauci AS. Pathogenesis of vasculitis syndromes. Adv Rheumatol. 1997;81:221–42. doi: 10.1016/s0025-7125(05)70512-5. [DOI] [PubMed] [Google Scholar]
- 7.Harris EN, Gharavi AE, Hughes GRV. Antiphospholipid antibodies. Clin Rheum Dis. 1985;11:591–609. [PubMed] [Google Scholar]
- 8.Albert SO, Josep OR, Francesc MF, et al. IgA anticardiolipin antibodies—relation with other antiphospholipid antibodies and clinical significance. Thromb Haemost. 1998;79:282–5. [PubMed] [Google Scholar]
- 9.Merkel PA, Chang Y, Pierangeli SS, et al. The prevalence and clinical associations of anticardiolipin antibodies in a large inception cohort of patients with connective tissue diseases. Am J Med. 1996;101:576–83. doi: 10.1016/s0002-9343(96)00335-x. [DOI] [PubMed] [Google Scholar]
- 10.Pierangeli SS. Induction of thrombosis in a mouse model by IgG, IgM, and IgA immunoglobulins from patients with the antiphospholipid. Thromb Haemost. 1995;74:1361–7. [PubMed] [Google Scholar]
- 11.Wilson WA, Faghiriz Z, Taheri F, Gharavi AE. Significance of IgA antiphospholipid antibodies. Lupus. 1998;7(Suppl. 2):110–3. doi: 10.1177/096120339800700225. [DOI] [PubMed] [Google Scholar]
- 12.Monila JF, Sergio GU, Molina J, et al. Variability of anticardiolipin antibody isotype distribution in 3 geographic populations of patients with systemic lupus erythematosus. J Rheumatol. 1997;24:291–6. [PubMed] [Google Scholar]
- 13.Frampton G, Winer JB, Cameron JS, et al. Severe Guillain–Barré syndrome: an association with IgA anti-cardiolipin antibody in a series of 92 patients. J Neuroimmunol. 1988;19:133–9. doi: 10.1016/0165-5728(88)90042-2. [DOI] [PubMed] [Google Scholar]
- 14.Gollier D, Enzenauer R, Santos M, et al. IgA anticardiolipin antibody associated with interstitial lung disease in systemic sclerosis. Arthritis Rheum. 1992;35(Suppl. 9):346. [Google Scholar]
- 15.Wilson WA, Morgan Ost C, Barton EN, et al. IgA antiphospholipid antibodies in HTLV-I associated tropical spastic paraparesis. Lupus. 1995;4:138–41. doi: 10.1177/096120339500400210. [DOI] [PubMed] [Google Scholar]
- 16.Vaarala O, Palosuo T, Kleemola M, Aho K. Anticardiolipin response in acute infections. Clin Immunol Immunopathol. 1986;41:8–15. doi: 10.1016/0090-1229(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 17.Burden AD, Gibson IW, Rodger RSC, Tillman DM. IgA anticardiolipin antibodies associated with Henoch–Schönlein purpura. J Am Acad Dermatol. 1994;31:857–60. doi: 10.1016/s0190-9622(94)70246-2. [DOI] [PubMed] [Google Scholar]
- 18.Burden AD, Tillman DM, Foley P, Holme E. IgA class anticardiolipin antibodies in cutaneous leukocytoclastic vasculitis. J Am Acad Dermatol. 1996;35:411–5. doi: 10.1016/s0190-9622(96)90606-4. [DOI] [PubMed] [Google Scholar]
- 19.Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–44. doi: 10.1084/jem.170.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Valsselaer P, Punnonen J, De Vries JE. Transforming growth factor beta. Immunology. 1998;9:215–20. [Google Scholar]
- 21.Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch–Schönlein purpura. Arthritis Rheum. 1990;33:1114–21. doi: 10.1002/art.1780330809. [DOI] [PubMed] [Google Scholar]
- 22.Harris EN, Gharavi AE, Patel SP, Hughes GVR. Evaluation of the anti-cardiolipin antibody test: report of an international workshop held 4 April 1986. Clin Exp Immunol. 1987;68:215–22. [PMC free article] [PubMed] [Google Scholar]
- 23.Levy M, Broyer M, Arsan A, Levy-Bentolila D, Habib R. Anaphylactoid purpura nephritis in childhood: natural history and immunopathology. Adv Nephrol. 1976;6:183–228. [PubMed] [Google Scholar]
- 24.Schulman ST, Barratt TM, Soothill JF. Immunoconglutinin and complement studies in congenital nephrotic syndrome and Henoch–Schönlein purpura in children. Arch Dis Child. 1971;46:838–41. doi: 10.1136/adc.46.250.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Fuentes M, Martin A, Chantler C, Williams DG. Serum complement components in Henoch–Schönlein purpura. Arch Dis Child. 1978;53:417–9. doi: 10.1136/adc.53.5.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen S, Taainig E, Soderstrom T, Pilgaard B, Tranebjaerg L, Christensen T, Nielsen HE. Immunoglobulin and complement studies in children with Schönlein–Henoch syndrome and other vasculitic diseases. Acta Paediatr Scand. 1991;80:1037–43. doi: 10.1111/j.1651-2227.1991.tb11780.x. [DOI] [PubMed] [Google Scholar]
- 27.Casanueva B, Rodrigus-Valverde V, Merion J, Arias M, Garcia-Fuentes M. Increased IgA producing cells in the blood of patients with Henoch–Schönlein purpura. Arthritis Rheum. 1983;26:854–60. doi: 10.1002/art.1780260706. [DOI] [PubMed] [Google Scholar]
- 28.Trygstad CW, Stiehm ER. Elevated serum IgA globulin in anaphylactoid purpura. Pediatrics. 1971;47:1023–8. [PubMed] [Google Scholar]
- 29.Kondo N, Kasahara K, Shinoda S, Orii T. Accelerated expression of secreted a-chain gene in anaphylactoid purpura. J Clin Immunol. 1992;12:193–6. doi: 10.1007/BF00918088. [DOI] [PubMed] [Google Scholar]
- 30.Beale MG, Nash GS, Bertovich MJ, MacDermott RP. Evidence of enhanced immunoglobulin synthesis and defective immune regulation in Henoch–Schönlein purpura. Contr Nephrol. 1983;35:46–60. doi: 10.1159/000407452. [DOI] [PubMed] [Google Scholar]
- 31.Heimstra PS, Biewenga J, Gorter A. Activation of complement by human serum IgA, secretory IgA and IgA1 fragment. Mol Immunol. 1988;26:527–33. doi: 10.1016/0161-5890(88)90074-0. [DOI] [PubMed] [Google Scholar]
- 32.Lin SC, Tsai MJ, Huang MT, Wu KH, Wang LH, Chiang BL. Immunological studies of children with anaphylactoid purpura. Acta Paed Sin. 1998;39:247–52. [PubMed] [Google Scholar]
- 33.Musumeci S, Sciotto A, Rosalba A, Volti SL, Mollica F. Lymphocyte subpopulations in anaphylactoid purpura. Arch Dis Child. 1980;55:541–3. doi: 10.1136/adc.55.7.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun XC, Chen MY, Cheng AS, Xu DL. Immunologic changes in children with Henoch–Schönlein purpura in the acute phase. Chin Med J. 1989;102:533–6. [PubMed] [Google Scholar]
- 35.Senger DPS, Acharya CD, Wolfish NM. HLA specificities, lymphocytes subsets, and mitogenic response in Henoch–Schönlein purpura nephritis. Int J Pediatr Nephrol. 1984;5:197–200. [PubMed] [Google Scholar]
- 36.Casali P, Notkins AL. CD5+ B lymphocytes, polyreactive antibodies and the human B-cell repertoire. Immunol Today. 1989;10:364–8. doi: 10.1016/0167-5699(89)90268-5. [DOI] [PubMed] [Google Scholar]
- 37.Tajima C, Suzuki Y, Mizushima Y, Ichikawa Y. Clinical significance of immunoglobulin A antiphospholipid antibodies: possible association with skin manifestations and small vessel vasculitis. J Rheumatol. 1998;25:1730–6. [PubMed] [Google Scholar]
- 38.Ordi J, Selva A, Monegal F, et al. Anticardiolipin antibodies and dependence of a serum cofactor. A mechanism of thrombosis. J Rheumatol. 1993;20:1321–4. [PubMed] [Google Scholar]
- 39.Koike T. Anticardiolipin antibodies and β2-glycoprotein I. Clin Immunol Immunopathol. 1994;72:187–92. doi: 10.1006/clin.1994.1128. [DOI] [PubMed] [Google Scholar]
- 40.Saulsburg FT. Heavy and light chain composition of serum IgA and IgA rheumatoid factor in Henoch–Schönlein purpura. Arthritis Rheum. 1992;35:1377–80. doi: 10.1002/art.1780351121. [DOI] [PubMed] [Google Scholar]
- 41.Ronda N, Esnault VL, Layward L, et al. Antineutrophil cytoplasm antibodies (ANCA) of IgA isotype in adult Henoch–Schönlein purpura. Clin Exp Immunol. 1994;95:49–55. doi: 10.1111/j.1365-2249.1994.tb06013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–43. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 43.Fontana A, Constam DB, Frei K, Malipiero U, Pfister HW. Modulation of the immune response by transforming growth factor beta. Int Arch Allergy Immunol. 1992;99:1–7. doi: 10.1159/000236328. [DOI] [PubMed] [Google Scholar]
- 44.Letterio JJ, Roberts AB. TGF-β: a critical modulator of immune cell function. Clin Immunol Immunopathol. 1997;84:244–50. doi: 10.1006/clin.1997.4409. [DOI] [PubMed] [Google Scholar]
- 45.Strober W, Kelsall B, Fuss I, Marth T, Ludviksson B, Ehrhardt R, Neurath M. Reciprocal IFN-γ and TGF-β responses regulate the occurrence of mucosal inflammation. Immunol Today. 1997;18:61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- 46.Jung T, Schauer U, Heusser C, Neumann C, Reiger C. Detection of intracellular cytokines by flow cytometry. J Immunol Methods. 1993;159:197–207. doi: 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- 47.Farely TA, Gillespei S, Rasoulpour M, et al. Epidemiology of a cluster of Henoch–Schönlein purpura. Am J Dis Child. 1989;143:798–883. doi: 10.1001/archpedi.1989.02150190048019. [DOI] [PubMed] [Google Scholar]
- 48.Atkinson SR, Barker DJ. Seasonal distribution of Henoch–Schönlein purpura. Br J Prev Soc Med. 1976;30:22–25. doi: 10.1136/jech.30.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.al-Sheyyab M, el-Shanti H, Ajlouni S, et al. Henoch–Schönlein purpura; clinical experience and contemplations on a streptococcal association. J Trop Paediatr. 1996;42:200–3. doi: 10.1093/tropej/42.4.200. [DOI] [PubMed] [Google Scholar]
- 50.Steare SE, Wiselka MJ, Kurinczuk JJ, Nicholson KG. Mycoplasma pneumoniae infection associated with Henoch–Schönlein purpura. J Infection. 1988;16:305–7. doi: 10.1016/s0163-4453(88)97796-1. [DOI] [PubMed] [Google Scholar]
- 51.Pedersen FK, Petersen EA. Varicella followed by glomerulonephritis. Treatment with corticosteroid and azathioprine resulting in recurrence of varicella. Acta Paediatr Scand. 1975;64:886–90. doi: 10.1111/j.1651-2227.1975.tb03943.x. [DOI] [PubMed] [Google Scholar]
- 52.Oner A, Demircin G, Tinaztepe K, Akinci A, Tezic T. Henoch–Schönlein purpura nephritis associated with subacute thyroiditis. Turkish J Pediatr. 1996;38:131–553. [PubMed] [Google Scholar]