

Abstract
The aim of this study was to investigate the p53 status in two autoimmune diseases; juvenile chronic arthritis (JCA) and rheumatoid arthritis (RA). In a PCR-sequencing analysis of exons 4–9 of the p53 gene, no mutation was identified, except for the case of an RA synovectomy sample with two mutations of intron 7. p53 gene polymorphisms for codons 36, 47, and 213 were not detected. Codon 72 polymorphism showed an indication of an increased occurrence of the Pro/Pro allelotype in JCA. Expression of P53 protein was comparable for JCA and RA synovectomy samples. For all RA samples P53 protein was detectable, whereas one sample of a JCA patient failed to express P53 protein.
Keywords: juvenile chronic arthritis, rheumatoid arthritis, fibroblast-like synoviocytes, p53, tumour suppressor gene
Introduction
Juvenile chronic arthritis (JCA) and rheumatoid arthritis (RA), are both chronic inflammatory systemic diseases. They belong to the autoimmune diseases and are characterized by a pathological immune response that attacks synovial tissue, cartilage and bone cells resulting in an irrespective damage to joints, vasculature and back bone. JCA is the most common autoimmune rheumatic disease in childhood and presents different clinical subtypes. It is considered to be of a polygenic nature and its genetic background is still under investigation.
Besides several models describing pathogenesis [1,2], the invasive behaviour of transformed-appearing synovial fibroblasts encouraged several authors to see paralleles to tumourigenesis [3,4]. Synovial fibroblasts from RA patients can (i) grow independently of anchorage [4]; (ii) overexpress oncogenes as, e.g. H-ras, c-myc, c-fos [5,6]; (iii) grow in nude mice [7,8]; (iv) and express matrix degenerating enzymes such as cathepsins B and L and metalloproteinases [5,9]. Mice transgenic for c-fos are able to develop a destructive arthritis [10]. Additionally, the findings from inadequate apoptosis of synovial fibroblasts from RA patients [11] support this hypothesis.
Recently, alterations in the p53 status of RA patients have come into focus. The tumour suppressor p53 is a major regulator of apoptosis inducing pro-apoptotic genes as bax or Fas and suppressing antiapoptotic genes as bcl-2 [12,13]. Interestingly, both an overexpression of P53 protein and occurrence of p53 mutations were described for RA but not osteoarthritis patients [11,14–16]. However, Kullmann et al. [17] did not detect p53 mutations in synovial fibroblasts of RA-patients from Germany, but p53 mutations were found in clones from three additional RA synovial fibroblast cell lines from the USA. In short, the p53 mutational and expression status in inflammatory autoimmune diseases has not been comprehensively investigated to date. This is especially true for JCA; therefore we are studying it for the first time.
Materials and methods
Patients, tissues and cell lines
Between 1992 and 1998, synovial pannus tissues were taken from 16 patients with JCA (17 samples) and from 15 patients with RA (17 samples) these samples were collected from the Department of Paediatric Rheumatology and the Clinic of Orthopedics of the Martin Luther University Halle-Wittenberg, Germany. For RA patients, the time between diagnosis and operation was on average 6·25 years (range 1–22 years). Table 1 summarizes the clinical features of the patients.
Table 1.
Clinical features of JCA and RA patients
| Samples | Age at operation in years | Sex | RF | Diagnosis | Medication | Synovial pannus cells |
|---|---|---|---|---|---|---|
| JCA | ||||||
| G 12 | 13 | m | 0 | JCA (systemic) | CyA, Pred,SSZ, NSAID | fr |
| G 16 | 3 | m | 0 | JCA (systemic) | Pred, SSZ, NSAID | fr |
| G/Z 26 | 12 | m | 0 | JCA (systemic) | Pred, SSZ | fr/c |
| G 1 | 2 | f | 0 | JCA (poly) | MTX, NSAID | fr |
| G 10 | 9 | f | 0 | JCA (poly) | Pred, Gold | fr |
| G 11 | 12 | m | 0 | JCA (poly) | MTX, Pred, SSZ | fr |
| G 20 | 13 | f | 0 | JCA (poly) | Pred, NSAID | fr |
| G 22 | 7 | m | + | JCA (poly) | Pred, SSZ | fr |
| G 2 | 14 | m | 0 | JCA (pauci), iridocyclitis | Pred, SSZ | fr |
| G 7 | 20 | m | + | JCA(pauci) | Pred, SSZ, NSAID | fr |
| G 14 | 15 | f | (+) | JCA (pauci) | SSZ, NSAID | fr |
| G 23 | 11 | f | + | JCA (pauci) | Pred, SSZ, NSAID | fr |
| G 25 | 7 | m | 0 | JCA (pauci) | SSZ | fr |
| G 44 | 14 | m | + | JCA/psoriasis | Pred, SSZ | fr |
| G 48 | 10 | f | 0 | JCA/tendovaginitis | NSAID | c |
| G 24 | 6 | m | 0 | JCA/sarcoidosis | Leukeran, NSAID, Pred | fr |
| RA | ||||||
| G/Z 4 | 61 | f | 0 | RA + SLE | Pred, Azathioprin | fr/c |
| G/Z 5 | 70 | f | + | RA | Pred, Tauredon, NSAID | fr/c |
| G 9 | 71 | f | + | RA | MTX, Pred, NSAID | fr |
| G 13 | 40 | f | + | RA | Azathioprin | fr |
| G 15 | 41 | f | + | RA | SSZ | fr |
| G 17 | 61 | f | + | RA | Azathioprin, NSAID | fr |
| G 21 | 27 | f | 0 | RA | SSZ, Tauredon, NSAID | fr |
| G 28 | 58 | f | 0 | RA | Chloroquine | fr |
| G 31 | 34 | f | (+) | RA | MTX, Pred, NSAID | fr |
| G 32 | 62 | f | + | RA | Pred | fr |
| G 36 | 64 | f | 0 | RA | MTX, Pred | fr |
| G 37 | 63 | f | + | RA | MTX, Pred | fr |
| G 46 | 76 | f | + | RA | Predni, Azathioprin | fr |
| G 47 | 65 | f | (+) | RA | Predni, Endoxan | fr |
| G 49 | 53 | m | 0 | RA | MTX, Pred, NSAID | fr |
Poly, polyarticular onset; pauci, pauciarticular course; systemic, systemic course; SLE, systemic lupus erythematosus; RF, rheumatic factor; Pred, prednison; MTX, methotrexate; NSAID, nonsteroidal anti-inflammatory drugs; SSZ, sulphasalazine; fr, frozen tissue; c, culture of synovial fibroblast cells; +, positive; (+), weak positive; 0, negative.
Postsurgical specimens were placed on ice and were snap frozen. In four cases, cell lines of synovial fibroblasts were established from pannus, by dissociating the minced tissues enzymatically with HBSS containing 0·5 mg/ml, collagenase type II (Sigma, Deisenhofen, Germany), 0·15 mg/ml DNAse I (Boehringer Mannheim, Mannhein, Germany) and 5 mm Ca2+, as described earlier [18]. The cells were cultured in RPMI 1640 containing 10% foetal calf serum, antibiotics and glutamin. Cells were used at confluence at the third to fifth passage.
DNA preparation and PCR
Genomic DNA was isolated from 10 to 15 cryo-sections of 50 µm of synovial pannus tissue as previously described [19,20]. Exons 4–9 of the p53 gene were amplified in five separate PCR reactions. We used 20-mer oligonucleotide primers described by Mashiyama et al. [21], PCR conditions were as following: predenaturation at 92°C for 5 min and 35 cycles of denaturation at 92°C for 1 min, annealing for 30 s at 56°C (exons 8–9), 58°C (exon 5), 62°C (exons 4,6,7) and DNA synthesis at 72°C for 1 min, postsynthesis at 72°C for 5 min and storage of the PCR products at 4°C [19].
Sequencing and allelotyping
Using the same protocol and primers as for the PCRs in a cycle sequencing method (TSII sequenase kit/Pharmacia, Freiburg, Germany) amplification products were sequenced in both directions and subsequently analysed on an ABI 373 (Perkin Elmer, Weiterstadt, Germany).
p53-allelotyping for codon 72 was performed after PCR by an RFLP analysis as described by Klaes et al. [22]. Briefly, PCR products were digested with two restriction endonucleases having a restriction site either in the proline coding allele (BsaJI, New England Biolabs, Schwalbach, Germany) or in the arginine coding allele (BstUI, New England Biolabs). Digestion with BsaJI resulted for Arg/Arg allelotypes in four bands (138 bp, 75 bp, 49 bp and 34 bp), whereas in Pro/Pro allelotypes the 138 bp band was further separated in bands of 94 bp and 44 bp. Digestion with BstUI delivered for Pro/Pro allelotypes a 269-bp band that was further digested in Arg/Arg allelotypes to 167 bp and 127 bp bands.
Western blot analysis
Thirty µg of total protein was separated on a 12·5% polyacrylamide/SDS gel (Minigel system; Biometra, Göttingen, Germany). Afterwards, the proteins were transferred to a PVDF Immobilon membrane (Millipore; Eschborn, Germany) at 200 mA for 90 min (Miniblotter; Biometra). The membrane was blocked with 0·1% Tween 20 containing 3% BSA and incubated with anti-p53 or antiactin antibodies (DO-7; 1:500; Dianova, Hamburg, Germany or AL-40; 1:500; Sigma) for 1 h and with horseradish peroxidase-conjugated anti-mouse IgG antibody (1:1000; Dako, Gostrup, Denmark) at room temperature for 1 h. For protein detection the membrane was placed in ECL-substrate for 1 min (Amersham, Braunschweig, Germany) and exposed to Biomax film (Kodak, Braunschweig, Germany). As positive control for P53 protein expression, RD cells/ATCC CCL 136, carrying a p53 mutation and overexpressing P53 protein were used. Furthermore, for comparison of P53 expression, a human myeloid leukaemia cell line (HL-60; Serva, Heidelberg, Germany) and HUVEC cells were tested for P53 expression. First, for evaluation purposes, the band intensities were equalized to the positive control running on each blot and, second, the amount of P53 protein was related to the actin band which was determined in the same sample and expressed as the ratio p53 to actin signal intensity. The ratios were considered as week (≤ 0·3), moderate (> 0·3–0·9) and strong (> 0·9).
Results
Sequencing of p53 gene (exons 4–9)
The sequences of exons 4–9 of the p53 gene were determined from the 34 samples taken from 31 patients including 16 JCA-patients and 15 RA-patients. With the exception of one sample from a RA patient with two point mutations in intron 7, no p53 mutation could be detected. The two point mutations (nucleotides 14201 and 14121 according to the GenBank-Accension No. X54156, Table 2) do not concern splicing sites and may not influence synthesis of a correct P53 protein.
Table 2.
Sequencing of p53 gene and Western blot analysis of P53 protein
| Sequencing of p53 gene | |||||
|---|---|---|---|---|---|
| Samples | Diagnosis | Exon 4–9 | bp cod 72 | aa cod 72 | Western blot P53/actin |
| JCA | |||||
| G 12 | JCA (systemic) | wt p53 | CGC/CGC | Arg/Arg | 1·37 |
| G 16 | JCA (systemic) | wt p53 | CGC/CGC | Arg/Arg | 0·3 |
| G 26 | JCA (systemic) | wt p53 | CGC/CGC | Arg/Arg | 0 |
| Z 26 | JCA (systemic) | wt p53 | CGC/CGC | Arg/Arg | 0 |
| G 1 | JCA (poly) | wt p53 | CGC/CCC | Arg/Pro | 1·51 |
| G 10 | JCA (poly) | wt p53 | CGC/CCC | Arg/Pro | 1·45 |
| G 11 | JCA (poly) | wt p53 | CGC/CGC | Arg/Arg | 0·93 |
| G 20 | JCA (poly) | wt p53 | CGC/CCC | Arg/Pro | 1·01 |
| G 2 | JCA (pauci), iridocyclitis | wt p53 | CCC/CCC | Pro/Pro | 1·43 |
| G 7 | JCA(pauci) | wt p53 | CCC/CCC | Pro/Pro | 1·71 |
| G 14 | JCA (pauci) | wt p53 | CGC/CGC | Arg/Arg | 0·77 |
| G 22 | JCA (poly) | wt p53 | CGC/CCC | Arg/Pro | 0·51 |
| G 23 | JCA (pauci) | wt p53 | CGC/CCC | Arg/Pro | 1·88 |
| G 25 | JCA (pauci) | wt p53 | CGC/CGC | Arg/Arg | ND |
| G 44 | JCA/psoriasis | wt p53 | CCC/CCC | Pro/Pro | 0·08 |
| G 48 | JCA/tendovaginitis | wt p53 | CGC/CGC | Arg/Arg | ND |
| G 24 | JCA/sarcoidosis | wt p53 | CGC/CGC | Arg/Arg | 1·16 |
| RA | |||||
| G 4 | RA + SLE | wt p53 | CCC/CCC | Pro/Pro | 1·28 |
| Z 4 | RA + SLE | wt p53 | CCC/CCC | Pro/Pro | ND |
| G 5 | RA | wt p53 | CGC/CGC | Arg/Arg | 1·32 |
| Z 5 | RA+ | wt p53 | CGC/CGC | Arg/Arg | ND |
| G 9 | RA | wt p53 | CGC/CGC | Arg/Arg | 1·06 |
| G 13 | RA + SLE | wt p53 | CGC/CGC | Arg/Arg | ND |
| G 15 | RA | wt p53 | CGC/CGC | Arg/Arg | 1·07 |
| G 17 | RA | wt p53 | CGC/CCC | Arg/Pro | 0·54 |
| G 21 | RA | wt p53 | CGC/CCC | Arg/Pro | 1·65 |
| G 28 | RA | wt p53 | CGC/CCC | Arg/Pro | 1·04 |
| G 31 | RA | wt p53 | CGC/CCC | Arg/Pro | 1·00 |
| G 32 | RA | wt p53 | CGC/CCC | Arg/Pro | 1·48 |
| G 36 | RA | wt p53 | CGC/CGC | Arg/Arg | 1·51 |
| G 37 | RA | wt p53 | CGC/CGC | Arg/Arg | 0·24 |
| G 46 | RA | wt p53 | CGC/CCC | Arg/Pro | 0·50 |
| G 47 | RA | Intron 7* | CGC/CCC | Arg/Pro | 0·43 |
| G 49 | RA | wt p53 | CGC/CGC | Arg/Arg | 0·65 |
nt 14121 C to T, nt 14201 T to G.
Poly, polyarticular onset; pauci, pauciarticular course; systemic, systemic course; SLE, systemic lupus erythematosus; +, positive; (+), weak positive; 0, negative; bp, basepairs; aa, amino acids; Z-cell line samples; G‐frozen samples.
Analysis of polymorphisms in the p53 gene
Four polymorphisms for codon 36, codon 47, codon 72 (all three in exon 4) and codon 213 (in exon 6) of the p53 gene were investigated. Codons 36, 47 and 213 revealed no polymorphism in our study group. This result is not unexpected at reported frequencies of 98%, 95·3% and 96·8%, respectively [23]. The genotypes of codon 72 were determined by sequencing and additional RFLP analysis was subsequently performed (Fig. 1). The distribution of the three allelic types Pro/Pro, Pro/Arg and Arg/Arg was 12·9%, 38·7% and 48·4% and is comparable to the normal Caucasian controls from published research (10·2%, 44·2% and 45·6%, respectively) (Table 3). Pro alleles were identified in 32% of our JCA/RA patients and in the control group (Table 3), in accordance with results previously reported [24]. Considering the small number of JCA patients, a predominance of Pro/Pro (18·8%) is noticeable (Table 3), but requires further investigation.
Fig. 1.
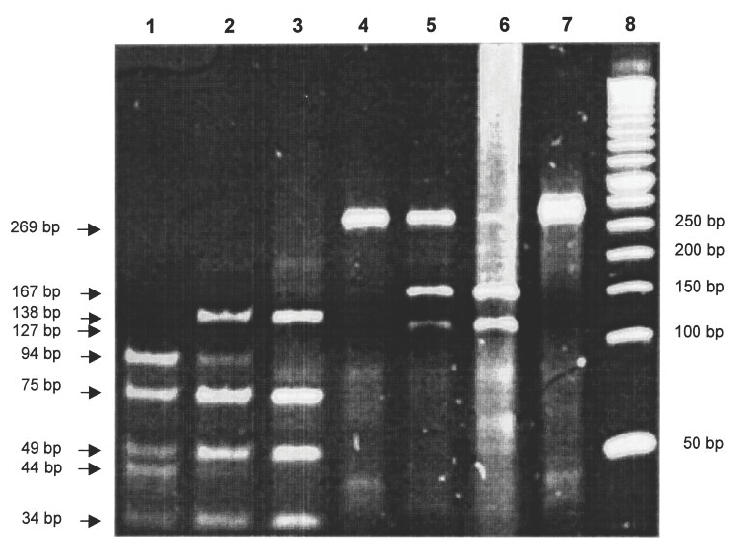
Determination of codon 72 polymorphisms by restriction endonuclease digests. PCR products of exon 4 were digested either with BsaJI (lane 1–3) or with BstUI (lane 4–6); undigested DNA (lane 7) and a 50-bp marker (lane 8). The samples were homozygously Pro/Pro (lane 1 and 4), heterozygously Arg/Pro (lane 2 and 5) and homozygously Arg/Arg (lane 3 and 6). In the heterozygous condition, note the somewhat weaker expression of bands (i.e. lane 2, bands at 94 bp and 44 bp; lane 5, band at 127 bp.
Table 3.
Frequency of codon 72 alleles of the p53 gene in JCA, RA and control Caucasians
Western blot analysis
P53 protein expression detected by Western hybridization did not differ on average between JCA (1·00) and RA synovectomy tissue (0·98). In both groups of diseases, cases with weak (0–0·3), moderate (> 0·3–0·9) or strong (> 0·9) P53 expression were found (Fig. 2, Table 2). On closer examination, samples of nine JCA and RA patients each showed a strong expression (> 0·9) of P53 protein. Two samples of JCA patients and four samples of RA-patients possessed a moderate P53 expression (> 0·3–0·9). For three samples of JCA patients and for one sample of an RA patient, a weak P53 expression (0–0·3) was detected. One sample from a patient with JCA was completely negative, whereas in RA synovectomy tissue, P53 protein was always detectable. In comparison to JCA and RA synovectomy tissue, a human myeloid leukaemia cell line (HL-60) and HUVEC cells showed values between 0 and 0·3 (data not shown).
Fig. 2.
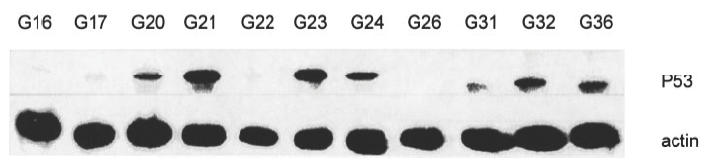
P53 protein expression in JCA and RA patient samples in a Western blot hybridization.
Discussion
In this study, we investigated the gene status and protein expression of tumour suppressor p53 in JCA and RA patients. We did not detect p53 mutations in synovial pannus in these patients, with the exception of two point mutations in intron 7, identified in a sample of an RA patient. Although, we can not exclude the fact that we may have missed mutations occurring in just a few cells, our result is in accordance with a recent study describing the failure to detect p53 mutations in RA synovial fibroblasts using three different techniques [17]. A reason for the detection of p53 mutations in synovial cells found in US RA patients but not found in German patients might be that the latter represent longstanding destructive diseases with chronic inflammation, oxydative stress and permanent genetic changes (including p53 mutations), resulting from synovectomies which were performed rather late in the USA [25]. To date, three groups could identify p53-mutations in RA tissue [14,15,26] with two mutations possessing a dominant negative effect suppressing wild-type p53 [27]. However, taken together, these few findings strengthen the opinion of Reme and colleagues [15], who stated that p53 abnormalities may not play a fundamental role in the aetiology of RA, which might also be true for JCA. Furthermore, in patients with another autoimmune disease, Sjogren‘s syndrome, there was no p53 mutation either, but in combination with non-Hodgkin’s lymphoma, it was detectable [28].
Besides p53 mutations, we analysed the status of five known p53 polymorphisms (four in exon 4 and one in exon 6). According to the expected frequencies, only for the codon 72 (exon 4) polymorphism could different haplotypes be identified. Taken together, the samples did not differ from the distribution in a normal Caucasian population. However, when considering only the JCA cases, a conspicuous occurrence of the Pro/Pro haplotype appeared. Based on the common features between autoimmune diseases and cancer, findings of codon 72 polymorphisms in cancer seem interesting. An early onset of lung cancer may correlate with occurrence of the Pro/Pro allele, suggesting that the Pro/Pro genotype predisposes individuals to an early outbreak of this disease [29]. For different cancers, the influence of the codon 72 polymorphism on prognosis may vary. On one hand, it can be associated with a higher risk for lung, breast, ovarian and colorectal cancer [24 29–32]; but this finding was not shared by other authors [23,33]. On the other hand, different allelotypes were found to have no effect on cervical cancer [22,34]. However, further investigations are necessary to find out if different allelotypes of the codon 72 polymorphism may increase susceptibility for autoimmune diseases.
Investigating P53 expression by Western hybridization, we agree with the findings of Firestein et al. [11] and Tak et al. [16], who all investigated RA synoviocytic cell lines and RA synovectomy samples and demonstrated P53 protein expression. In contrast to the suggestion of Tak and colleagues [16], in our samples of RA and JCA patients, P53 overexpression is not secondary to a p53 mutation. We observed that the expression level of P53 protein can widely differ between individual JCA and RA patients. Comparable differences in the expression level of P53 for RA samples were also found by Nickels et al. [35], who considered Western blot analysis to be the gold standard for demonstration of P53 expression in their study of inflammatory tissues. However, an increased P53 expression detected either immunohistochemically or by Western hybridization has been found by several authors, related to an increased presence of DNA strand breaks, a disturbance of apoptosis or a putative higher potential of aggressiveness of synovial fibroblasts [11,15,16,35]. In conclusion, we would like to emphasize that P53 protein expression is a characteristic feature of RA and JCA. Furthermore, for JCA patients, we found a conspicuous occurrence of the Pro/Pro haplotype for codon 72 of the p53 gene. This finding is still preliminary but could be remarkable, since this haplotype is suspected of possessing an increased cancer susceptibility.
Acknowledgments
The authors thank Mrs B. Wypior and Mrs M. Wolff for their excellent technical assistance and Mrs C. Burns-Klein for revising the manuscript. This study was supported by the Fritz Thyssen Stiftung Germany (AZ 926 98 003). Dr A. Meye was sponsored by grants from the ‘Marianne-und-Fritz-Walter-Fischer-Stiftung’ im Stifterverband der Deutschen Wissenschaft and the ‘Novartis Stiftung für therapeutische Forschung eV’ (Germany).
References
- 1.Feldman M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–10. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 2.Weynand CM, Goronzy JJ. The molecular basis of rheumatoid arthritis. J Mol Med. 1997;75:772–85. doi: 10.1007/s001090050167. [DOI] [PubMed] [Google Scholar]
- 3.Fassbender HG. Histomorphological basis of articular cartilage destruction in rheumatoid arthritis. Coll Relat Res. 1983;3:141–55. doi: 10.1016/s0174-173x(83)80040-5. [DOI] [PubMed] [Google Scholar]
- 4.Lafyatis R, Remmers EF, Roberts AB, et al. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogeneous platelet-derived growth factor and inhibition by transforming growth factor β and retinoids. J Clin Invest. 1989;83:1267–76. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trabandt A, Aicher WK, Gay RE, et al. Expression of the colagenolytic and ras-induced cystein proteinase cathepsin L and proliferation-associated oncogenes in synovial cells of MRL/1 mice and patients with rheumatoid arthritis. Matrix. 1990;10:349–61. doi: 10.1016/s0934-8832(11)80142-3. [DOI] [PubMed] [Google Scholar]
- 6.Mueller-Ladner U, Kriegsmann J, Gay RE, et al. Oncogenes in rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21:675–90. [PubMed] [Google Scholar]
- 7.Brinckerhoff CE, Harris ED. Survival of rheumatoid synovium implanted into nude mice. Am J Pathol. 1981;103:411–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller-Ladner U, Kriegsmann J, Franklin BN, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–15. [PMC free article] [PubMed] [Google Scholar]
- 9.Keyszer G, Lambiri I, Nagel R, et al. Circulating levels of matrix metalloproteinases MMP-3 and MMP-1, tissue inhibitor of metalloproteinases 1 (TIMP-1), and MMP-1/TIMP-1 complex in rheumatic disease. Correlation with clinical activity of rheumatoid arthritis versus other surrogate markers. J Rheumatol. 1999;26:251–8. [PubMed] [Google Scholar]
- 10.Shiozawa S, Tanaka Y, Fujita T, et al. Destructive arthritis without lymphocyte infiltration in H2-c-fos transgenic mice. J Immunol. 1992;148:3100–4. [PubMed] [Google Scholar]
- 11.Firestein GS, Nguyen K, Aupperle KR, et al. Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am J Pathol. 1996;149:2143–51. [PMC free article] [PubMed] [Google Scholar]
- 12.Owen-Schaub LB, Zhang W, Cusack JC, et al. Wild-type human p53 and a temperature sensitive mutant induce fas/APO1 expression. Mol Cell Biol. 1995;15:3032–40. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 14.Firestein GS, Echeverri F, Yeo M, et al. Somatic mutations in the p53 tumor suppressor gene. Proc Natl Acad Sci USA. 1997;94:10895–900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reme T, Travaglio A, Gueydon E, et al. Mutations of the p53 tumor suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998;111:353–8. doi: 10.1046/j.1365-2249.1998.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tak PP, Smeets TJ, Boyle DL, et al. p53 overexpression in synovial tissue from patients with early and longstanding rheumatoid arthritis compared with patients with reactive arthritis and osteoarthritis. Arthritis Rheum. 1999;42:948–53. doi: 10.1002/1529-0131(199905)42:5<948::AID-ANR13>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Kullmann F, Judex M, Neudecker I, et al. Analysis of the tumor suppressor gene in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 1999;42:1594–600. doi: 10.1002/1529-0131(199908)42:8<1594::AID-ANR5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Navarrete Santos A, Riemann D, Navarrete Santos A, et al. Treatment of fibroblast-like synoviocytes with IFN-γ results in the down-regulation of autotaxin mRNA. Biochem Biophys Res Comm. 1996;229:419–24. doi: 10.1006/bbrc.1996.1819. [DOI] [PubMed] [Google Scholar]
- 19.Taubert H, Würl P, Meye A, et al. Molecular and immunohistochemical status in liposarcoma and malignant fibrous histiocytoma. Cancer. 1995;76:1187–96. doi: 10.1002/1097-0142(19951001)76:7<1187::aid-cncr2820760714>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Würl P, Taubert H, Bache M, et al. Frequent occurence of p53 mutations in rhabdo- and leiomyosarcoma, but not in fibrosarcoma and malignant neural tumors. Int J Cancer. 1996;69:1996. doi: 10.1002/(SICI)1097-0215(19960822)69:4<317::AID-IJC14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Mashiyama S, Murakami Y, Yoshimoto T, et al. Detection of p53 gene mutations in human brain tumors by SSCP analysis of PCR products. Oncogene. 1991;6:1313–8. [PubMed] [Google Scholar]
- 22.Klaes R, Ridder R, Schaefer U, et al. No evidence of p53 allele-specific predisposition in human papillomavirus-associated cervical cancer. J Mol Med. 1999;77:299–302. doi: 10.1007/s001090050353. [DOI] [PubMed] [Google Scholar]
- 23.Weston A, Godbold JH. Polymorphisms of H-ras-1 and p53 in breast cancer and lung cancer: a meta-analysis. Environ Health Perspectives. 1997;105(Suppl. 4):919–6. doi: 10.1289/ehp.97105s4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawajiri K, Nakachi K, Imai K, et al. Germ line polymorphisms of p53 and CYP1A1 genes involved in human lung cancer. Carcinogenesis. 1993;14:1085–9. doi: 10.1093/carcin/14.6.1085. [DOI] [PubMed] [Google Scholar]
- 25.Tak PP, Zvaivler NJ, Green DR, Firestein GS. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82. doi: 10.1016/s0167-5699(99)01552-2. [DOI] [PubMed] [Google Scholar]
- 26.Inazuka M, Tahira T, Horiuchi T, et al. Analysis of p53 tumor suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology. 2000;39:262–6. doi: 10.1093/rheumatology/39.3.262. [DOI] [PubMed] [Google Scholar]
- 27.Han Z, Boyle DL, Shi Y, et al. Dominant negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999;42:1088–92. doi: 10.1002/1529-0131(199906)42:6<1088::AID-ANR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Tapinos NI, Polihronis M, Moutsopoulos HM. Lymphoma development in Sjogren syndrome: novel p53 mutations. Arthritis Rheum. 1999;42:1466–72. doi: 10.1002/1529-0131(199907)42:7<1466::AID-ANR21>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 29.Jin X, Wu X, Roth JA, et al. Higher lung cancer risk for younger African-Americans with the Pro/Pro p53 genotype. Carcinogenesis. 1995;16:2205–8. doi: 10.1093/carcin/16.9.2205. [DOI] [PubMed] [Google Scholar]
- 30.Själander A, Birgander R, Athin L, et al. P53 germ line haplotypes associated with increased risk for colorectal cancer. Carcinogenesis. 1995;16:1461–4. doi: 10.1093/carcin/16.7.1461. [DOI] [PubMed] [Google Scholar]
- 31.Själander A, Birgander R, Hallmans G, et al. P53 polymorphisms and haplotypes in breast cancer. Carcinogenesis. 1996;17:1313–6. doi: 10.1093/carcin/17.6.1313. [DOI] [PubMed] [Google Scholar]
- 32.Buller RE, Sood A, Fullenkamp C, et al. The influence of the codon 72 polymorphism on ovarian carcinogenesis and prognosis. Cancer Gene Ther. 1997;4:239–45. [PubMed] [Google Scholar]
- 33.Birgander R, Själander A, Rannung A, et al. P53 polymorphism and haplotypes in lung cancer. Carcinogenesis. 1995;16:2233–6. doi: 10.1093/carcin/16.9.2233. [DOI] [PubMed] [Google Scholar]
- 34.Rosenthal AN, Ryan A, Al-Jehani RM, et al. p53 codon 72 polymorphism and risk of cervical cancer in UK. Lancet. 1998;352:871–2. doi: 10.1016/S0140-6736(98)07357-7. [DOI] [PubMed] [Google Scholar]
- 35.Nickels A, Selter H, Pfreundschuh M, et al. Detection of p53 in inflammatory tissue and lymphocytes using immunohistology and flow cytometry: a critical comment. J Clin Pathol. 1997;50:654–60. doi: 10.1136/jcp.50.8.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de la Calle-Martin O, Fabregat V, Romero M, et al. Accii polymorphism of the p53 gene. Nucl Acids Res. 1990;18:4963. [PMC free article] [PubMed] [Google Scholar]
- 37.Ara S, Lee PSY, Hansen MF, et al. Codon 72 polymorphism of the TP53 gene. Nucleic Acids Res. 1990;18:4961. doi: 10.1093/nar/18.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olschwang S, Laurent-Puig P, Vassal A, et al. Characterization of a frequent polymorphism in the coding sequence of the TP53 gene in colonic cancer patients and a control population. Human Genet. 1991;86:369–70. doi: 10.1007/BF00201836. [DOI] [PubMed] [Google Scholar]
- 39.Weston A, Perrin LS, Forrester K, et al. Allelic frequency of a p53 polymorphism in human lung cancer. Cancer Epidemiol Biomarkers Prev. 1992;1:481–3. [PubMed] [Google Scholar]
- 40.Zhang W, Hu G, Deisseroth A. A polymorphism at codon 72 of the p53 gene in human acute myelogenous leukemia. Gene. 1992;117:271–5. doi: 10.1016/0378-1119(92)90738-b. [DOI] [PubMed] [Google Scholar]