

Abstract
Glycosylation in room temperature ionic liquid is demonstrated using unprotected and unactivated donors. Modest yields of simple benzyl glycosides and disaccharides of glucose, mannose and N-acetylgalactosamine were obtained in 1-ethyl-3-methylimidazolium benzoate with Amberlite IR-120 (H+) resin or p-toluenesulfonic acid as promoters.
Keywords: glycosylation, room temperature ionic liquids, unprotected donor, unactivated donor
Glycosylation occurs between a donor and an acceptor in the presence of a promoter, which activates the donor. Glycosylation reactions in the synthesis of complex carbohydrates can prove to involve challenging chemistry. There are various factors that need to be considered when carrying out the synthesis of glycosides including the presence and choice of a leaving group at the anomeric position of the donor, the manipulation of protecting groups in both donor and acceptor, the architecture of donor and acceptor, the solvent system and the choice of promoter.[1-3] Since the initial introduction of Fischer glycosylation,[4] many innovative glycosylation strategies have been developed that have improved overall yield, chemoselectivity, regioselectivity and stereoselectivity.[5] Most of these glycosylation reactions require temporary protection of both acceptor and donor. There is a plethora of literature describing the introduction of protecting groups on both donor and acceptor, the activation of donor, glycosylation reactions, recovery of oligosaccharide products, and the deprotection of oligosaccharide products. These strategies involve extensive manipulation of protecting groups and often a large number of synthetic steps.
Glycosylation using unprotected and unactivated donors is often preferable as it can reduce the number of steps, enhance reactivity, allow for different stereochemistry and increase prospects for further process modifications.[6,7] The chemical synthesis of unprotected carbohydrates possesses a number of challenges, including their poor solubility in most conventional organic solvents. Only very polar organic solvents, such as formamide, dimethylformamide, dimethyl sulfoxide, and pyridine, easily dissolve significant amounts of sugars.[8] Furthermore, these are reactive, nonvolatile solvents, difficult to dry and thus, they pose problems for both carrying out glycosylation reactions and the recovery of products. It is important to investigate new solvent systems that dissolve carbohydrates, support glycosylation reactions of unprotected sugars, and facilitate product recovery.
Room temperature ionic liquids (RTILs) are becoming increasingly used as solvents for a wide variety of chemical reactions,[9] including reactions used in carbohydrate synthesis.[10-12] RTILs also display desirable solvent properties such as low vapor pressure, low volatility, thermal stability, large liquidus range and tunable physical properties and are capable of dissolving both polar and nonpolar solutes. These ionic solvents have the potential of replacing conventional volatile organic solvents in carbohydrate chemistry. Our previous studies showed that dialkylimidazolium benzoate RTILs are useful solvent/catalysts for the peracetylation and perbenzoylation of simple and sulfated saccharides with good to excellent yields using short reaction times.[13] A number of reactions involving carbohydrates have utilized RTILs, resulting in new methodologies and enhanced yields. [10, 14]
In this work, we report the glycosidation of various simple, unprotected monosaccharides in RTILs to give benzyl glycosides, disaccharides and oligosaccharides. RTILs facilitate the use of unactivated and unprotected donors in these reactions, resulting in a simple synthetic strategy involving a single glycosylation step. Stereoselectivity is observed in these glycosylation reactions including α-selective glycosylation using N-acetyl-D-galactosamine (GalNAc) donor. Finally, the synthesis of galactose oligomers was also possible using these novel solvents.
D-Glucose (Glc), D-mannose (Man), D-galactose (Gal), N-acetyl-D-galactosamine (GalNAc) were used as unactivated donors in the RTIL, 1-ethyl-3-methylimidazolium benzoate ([emIm][ba]), with Amberlite IR-120 (H+) resin or p-toluenesulfonic acid (TsOH) as promoters to form an array of O-glycosylated products. The RTIL [emIm][ba] was shown in our previous studies to be a useful solvent and catalyst for the protection of carbohydrates.[13]
Our initial studies utilized benzyl alcohol as a glycosyl acceptor. Monosaccharide donors were used as α, β-anomeric mixtures, as determined from their 1H-NMR spectra in D2O. Benzyl glycosidation of Glc, Man, and GalNAc afforded fair to good isolated yields of exclusively α-glycosides (Table 1). RTILs are relatively viscous liquids, [emIm][ba] with a viscosity of 425 cP at 25 °C, making both stirring and product recovery difficult.[13]
Table 1.
Benzyl α-glycosidation of unprotected and unactivated monosaccharides in[emIm][ba] a.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Glycosyl donor | Product | Acceptor (eq) | Time (h) | Temp. (°C) | Yield (%)b |
 |
||||||
| 1 | Glc | 20 | 16 | 30 | 20 | |
| 6 | 50 | 64 | ||||
 |
||||||
| 2 | Man | 20 | 16 | 30 | 33 | |
| 6 | 50 | 61 | ||||
 |
||||||
| 3 | GalNAc | 10 | 6 | 50 | 27 | |
Carried out under argon,
Isolated yields.
Isolated yields of glycosylation products could be improved by increasing the reaction temperature and/or the reaction time. Further increase in reaction time or reaction temperature failed to improve the isolated product yields. Like other sugars (Glc and Man), GalNAc afforded only the α-glycoside, which is in keeping with the α-selectivity reported previously for the reaction of GalNAc with benzyl alcohol in the presence of HCl at 70-80 °C.[15] While the previous success of synthesis of benzyl α-glycoside of GalNAc by Fischer glycosidation suggests that ionic liquid is unnecessary, the reaction of GalNAc with benzyl alcohol at 30-50 °C in the presence of Amberlite (H+) fails to take place in the absence of ionic liquid. The added ionic liquid made these reactions possible under relatively mild conditions preventing the formation of undesired side products. More detailed mechanistic studies are underway to understand the precise role of ionic liquid in these reactions.
Our efforts next turned towards the synthesis of disaccharides from unprotected donors and partially protected acceptors using the same strategy. Glycosyl acceptors were synthesized using standard methods from methyl α-D-glucoside. 2,3,4-Tri-O-allyl- or 2,3,4-tri-O-benzyl-protected methyl α-D-glucoside leave the primary 6-hydroxyl group unprotected and available for reaction. Attempts to perform glycosylation reactions of these acceptors with unprotected Glc, Man and GalNAc donors under conditions successfully used in the benzyl glycosidation afforded modest yields of the α-linked disaccharides. The α-configuration was inferred from the chemical shifts and coupling constants of the anomeric proton signals. The reduced yields obtained using sugar-based acceptors, in place of benzyl alcohol acceptor, is believed to be associated with the increased viscosity of the reaction mixture, decreasing the mobility of the reaction components.
The mobility of the reactants could be slightly enhanced by replacing the insoluble promoter (Amberite IR-120 (H+) resin) by the soluble promoter, TsOH, and increasing the reaction temperature. The incorporation of smaller allyl protecting groups into the acceptor also improved the reaction yields. This glycosylation strategy forms disaccharides in fair yields, with excellent stereoselectivity (Table 2). While TLC showed each reaction to be nearly complete with a single major product formed, the isolated yields were modest. We ascribe this modest yield to the difficulty in recovering the products from benzoate-based ionic liquids.[13]
Table 2.
Synthesis of disaccharides using unactivated and unprotected donors in [emIm][ba] a.
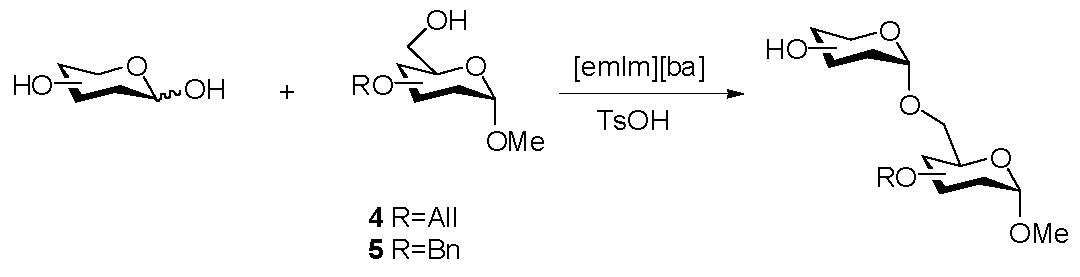 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product | Donor/Acceptor ratio | Time (h) | Temp. (°C) | Yield (%)b |
 |
|||||||
| 1/10 | 6 | 50 | 35 | ||||
| 1 | Glc | 4 | 1/1 | 4 | 50 | 25 | |
| 1/10 | 4 | 80 | 36 | ||||
 |
|||||||
| 2 | Man | 4 | 1/10 | 6 | 50 | 41 | |
| 1/2 | 6 | 50 | 22 | ||||
 |
|||||||
| 3 | Man | 5 | 1/2 | 10 | 50 | 20 | |
 |
|||||||
| 4 | GalNAc | 4 | 1/10 | 6 | 50 | 20 | |
Carried out under argon,
Isolated yields.
Finally, the synthesis of disaccharides has been extended to oligosaccharide synthesis by utilizing an isopropylidene-protected acceptor (Scheme 1). While strategies using both unprotected donor and unprotected acceptor were unsuccessful, we found that the glycosyl acceptor 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (10) could be used for the synthesis of α-1→6-linked galactose oligosaccharides. This reaction makes use of the instability of the isopropylidene protecting group under acidic conditions to afford di-, tri-, tetra-, penta-saccharide products within 4 h at 50 °C, as demonstrated by MS analysis (Figure 1a). Fractionation of the reaction mixture afforded the four disaccharides shown in Scheme 1 (See Figure 1b for MS). Deprotection of disaccharide products by treatment with Amberlite IR-120 (H+) resin in H2O at 80 °C for 4 h affords the exclusively α-linked disaccharide 12 (41%) (Table 3). Treatment of the entire reaction mixture with Amberlite IR-120 (H+) resin afforded a mixture of unprotected α-1→6-linked Gal oligosaccharides 11 (Figure 1c).
Scheme 1.
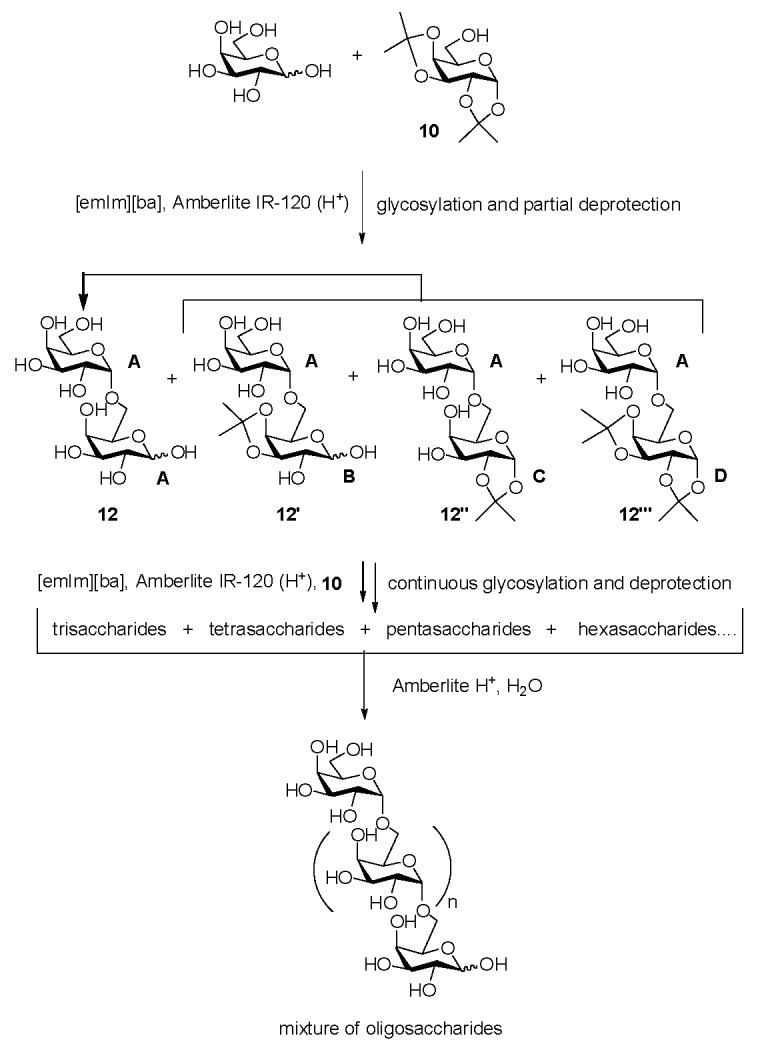
Synthesis of galactose oligosaccharides in [emIm][ba].
Figure 1.
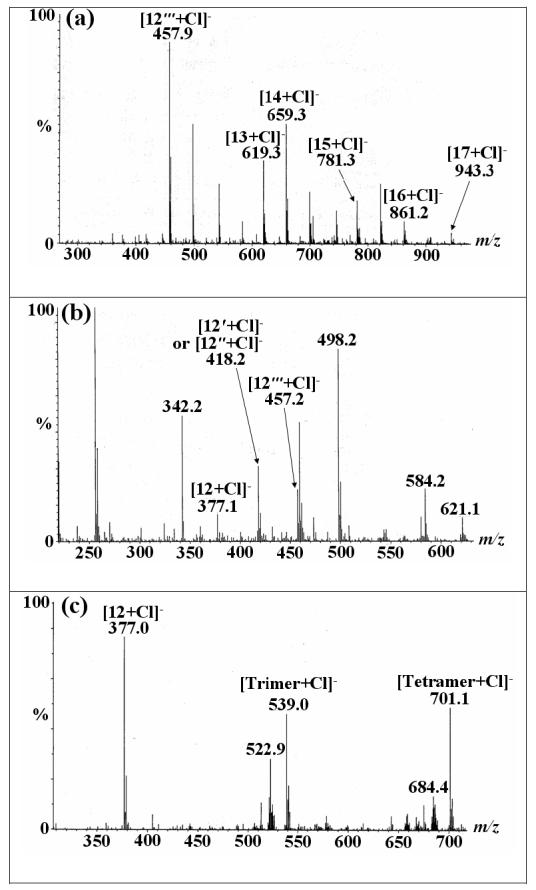
Negative-ion mode ESI/MS spectra of oligomerization reaction.
(a) The oligosaccharide mixture of unprotected and partially protected galactopyranosyl residues comprise compounds 12‴ (AD, see Scheme 1 for definition of A, B and D), 13 (AAD), 14 (ABD), 15 (AAAD), 16 (ABBD), 17 (AAAAD) that are observed as their chloride adducts. (b) Fractionation of the reaction mixture (c) Oligosaccharide mixture after deprotection: dimer (12), trimer (AAA) and tetramer (AAAA) are observed as their chloride adducts.
Table 3.
One-pot synthesis of oligosaccharides mixture a.


Carried out under argon,
Isolated yields,
Yield is estimated from average product size.
In summary, benzyl glycosides were synthesized from unactivated donors in good yield in a single synthetic step. Glycosylation of partially protected monosaccharide acceptors gave the expected disaccharides with α-stereoselectivity and without protection or covalent activation of the glycosyl donor. Additional studies including acceptors having multiple unprotected hydroxyl groups and a wider array of unactivated donors are currently under way.
1. Experimental
General methods
Room temperature ionic liquid [emim][ba] was synthesized according to reported procedure[13] and all chemicals were purchased from Fisher, Sigma, or Aldrich. 1H NMR and 13C NMR spectra were acquired at 25°C, in CDCl3 or D2O (Varian). Chemical shifts (δ) were indicated in ppm and coupling constants (J) in Hz. HRMS and LRMS were recorded on Micromass Autospec high resolution mass spectrometer and Agilent 1100 series LC/MSD trap, respectively. Thin-layer chromatography (TLC) was carried out using E. Merck plates of silica gel 60 with fluorescent indicator and the spots were visualized by dipping the plates into 5% sulfuric acid in ethanol followed by heating. Flash chromatography was performed using silica gel (230-400 mesh, Natland International Corporation).
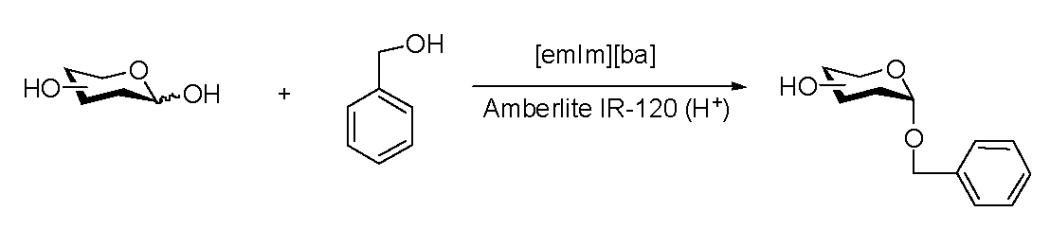
Benzyl α-D-glucopyranoside (1)
Amberlite IR-120 (H+) (300mg) was added to a solution of D-glucose (47 mg, 0.26 mmol) and anhydrous benzyl alcohol (562.3 mg, 5.2 mmol) in [emim][ba] (312 mg, 1.34 mmol) at room temperature or 50°C and the reaction mixture was stirred in an argon atmosphere until the reaction was complete as demonstrated by TLC. After adding methanol (5ml), the resin was removed by filtration and the mixture was concentrated by rotary evaporation. Flash chromatography of the residue [dichloromethane→ dichloromethane-methanol (96:4)] gave pure 1 (45 mg, 64 % at 50°C). 1H NMR (500 MHz, D2O), δ: 7.47-7.39 (5H, ArH), 5.01 (1H, d, J = 4 Hz, H-1), 4.77 (1H, d, J = 11.5 Hz, CH2Bn), 4.61 (1H, d, J = 11.5 Hz, CH2Bn), 3.91 (1H, dd, J1 = 2 Hz, J2 = 12.5 Hz, H-6a), 3.78 - 3.65 (3H, m, H-6b, H-5, H-3), 3.55 (1H, dd, J1 = 4 Hz, J2 = 10 Hz, H-2), 3.47 - 3.37 (1H, m, H-4);13C NMR (300 MHz, D2O), δ: 137.29, 128.96, 128.76, 128.54, 97.90, 76.19, 73.35, 72.18, 71.55, 69.91, 60.64; HRMS m/z calcd for C H + 13 18O6Na [M + Na] 293.1025, found 293.1028.
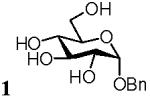
Benzyl α-D-mannopyranoside (2)
Glycoside 2 was obtained by the same protocol as described for 1. Flash chromatography afforded compound 2 (43 mg, 61 % at 50°C). 1H NMR (500 MHz, D2O), δ: 7.44 - 7.39 (5H, ArH), 4.96 (1H, J = 2 Hz, H-1), 4.76 (1H, d, J = 11 Hz, CH2Bn), 4.57 (1H, d, J = 11 Hz, CH2Bn), 3.94 (1H, dd, J1 = 2 Hz, J 2 = 3.5 Hz H-2), 3.86 (1H, dd, J 1 = 1.5 Hz, J 2 = 12 Hz, H-5), 3.80 - 3.74 (2H, m, H-6a, H-3), 3.70 - 3.64 (2H, m, H-4, H-6b); 13C NMR (300 MHz, D2O), δ: 136.84, 128.96, 128.76, 128.62, 99.49, 73.08, 70.73, 70.20, 69.60, 66.88, 61.01; HRMS m/z calcd for C13H18O6Na [M+Na]+ 293.1017, found 293.1028.
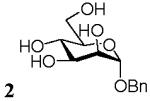
Benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (3)
Amberlite IR-120 (H+) (300mg) was added to a solution of D-GalNAc (57.5mg, 0.26mmol) and anhydrous benzyl alcohol (281.2 mg, 2.6 mmol) in [emim][ba] (312mg, 1.34mmol) at room temperature or 50°C and the mixture was stirred under argon as described for 1. The mixture was then purified by flash chromatography on silica gel to afford 3 (22 mg, 27 %). 1H NMR (500 MHz, D2O), δ: 7.47-7.40 (5H, ArH), 6.19 (1H, broad signal, NHAc), 4.97 (1H, d, J = 4 Hz, H-1), 4.71 (1H, d, J = 12 Hz, CH2Bn), 4.51 (1H, d, J = 12 Hz, CH2Bn), 4.11 (1H, dd, J 1 = 4 Hz, J 2 = 11 Hz, H-2), 4.03 (1H, m, H-5), 3.99 (1H, d, J = 3 Hz, H-4), 3.92 (1H, dd, J 1 = 3 Hz, J 2 = 11 Hz, H-3), 3.75 (1H, m, H-6a), 3.73 (1H, m, H-6b), 1.96 (3H, s, CH3Ac); 13C NMR (300 MHz, D2O), δ: 172.22, 136.54, 129.18, 128.43, 128.41, 99.18, 72.77, 69.90, 69.29, 66.58, 60.70, 50.11, 22.02; HRMS calcd for C15H21NO6Na [M+Na]+ 334.1266, found 334.1267.
Methyl 2,3,4-tri-O-allyl-6-O-(α-D-glucopyranosyl)-α-D-glucopyranoside (6)
A mixture of D-glucose (17.48 mg, 0.097 mmol), acceptor 4 (305mg, 0.97mmol), and TsOH (9 mg, 0.047 mmol) in [emim][ba] (300mg, 1.29mmol) was stirred under argon for 6 h at 50°C. Purification by flash chromatography on silica [dichloromethane→dichloromethane-methanol (98:2)→(95:5)] then gave the disaccharide 6 (16 mg, 35%). 1H NMR (500 MHz, CDCl3), δ: 6.02 - 5.85 (3H, m, 3 × CHAll), 5.32-5.15 (6H, m, 3 × CH=CH2 All), 4.93 (1H, d, J = 3 Hz, H-1′), 4.76 (1H, d, J = 3 Hz, H-1), 4.40 - 4.08 (8H, m, H-5′, H-4′, 3 × CH2-CHAll), 3.90-3.71 (5H, m, H-6a, H-6b, H-4, H-3, H-3′), 3.63 (1H, m, H-6b′), 3.45 - 3.32 (4H, m, H-5, H-2, H-6a′, H-2′), 3.41 (3H, s, OCH3); 13C NMR (300 MHz, CDCl3), δ: 134.74, 134.51, 118.20, 117.34, 99.09, 97.68, 79.15, 75.00, 73.58, 72.90, 72.72, 71.92, 70.81, 70.63, 67.97, 65.85, 62.48, 61.95, 60.34, 55.38; HRMS m/z calcd for C22H36O11Na [M+Na]+ 499.2164, found 499.2155.
Methyl 2,3,4-tri-O-allyl-6-O-(α-D-mannopyranosyl)-α-D-glucopyranoside (7)
Disaccharide 7 was obtained by the same protocol as described for 6. 1H NMR (500 MHz, CDCl3), δ: 6.01 - 5.84 (3H, m, 3 × CHAll), 5.34-5.14 (6H, m, 3 × CH2 All), 4.92 (1H, br.s, H-1′), 4.74 (1H, d, J = 3.5 Hz, H-1), 4.41 - 4.05 (6H, m, 3 × CH2 All), 3.99-3.92 (3H, m, H-6b′, H-4′, H-2′), 3.89 - 3.82 (2H, m, H-6a, H-3′), 3.78 - 3.63 (4H, m, H-5, H-3, H-6a′, H-6b,), 3.60 (1H, m, J = 10 Hz, H-5′), 3.42 - 3.36 (1H, m, H-2), 3.39 (3H, s, OCH3), 3.30 (1H, t, H-4); 13C NMR (300 MHz, CDCl3), δ: 135.37, 134.98, 117.88, 116.92, 100. 35, 98.24, 81.75, 79.78, 75.72, 74.50, 74.13, 73.88, 72.76, 72.12, 71.97, 70.52, 69.88, 66.45, 64.78, 55.34; HRMS m/z calcd for C22H36O11Na [M+Na]+ 499.2156, found 499.2155.
Methyl 2,3,4-tri-O-benzyl-6-O-(α-D-mannopyranosyl)-α-D-glucopyranoside (8)
A mixture of D-mannose (7.5 mg, 0.042 mmol), acceptor 5 (38.8 mg, 0.0836 mmol), and TsOH (9 mg, 0.047 mmol) in [emim][ba] (100mg, 0.43 mmol) was stirred under argon for 10 h at 50°C. Purification by flash chromatography on silica [dichloromethane→dichloromethane-methanol (98:2)→(95:5)] then gave the disaccharide 8 (5.4 mg, 20%) 1H NMR (500 MHz, CDCl3), δ: 7.34 - 7.21 (15H, 3 × ArH), 4.97 (1H, d, J = 11 Hz, CH2Bn), 4.90 (1H, d, J = 11 Hz, CH2Bn), 4.82 (1H, br.s, H-1′), 4.78 (1H, d, J = 11 Hz, CH2Bn), 4.74 (1H, d, J = 12 Hz, CH2Bn), 4.63 (1H, d, J = 12 Hz, CH2Bn), 4.56 (1H, d, J = 3.5 Hz, H-1), 4.52 (1H, d, J = 11 Hz, CH2Bn), 3.99 - 3.90 (3H, m, H-3, H-5′, H-2′), 3.82 - 3.69. (4H, m, H-6a, H-5, H-6a′, H-6b′), 3.57 - 3.50 (3H, m, H-6b, H-2, H-3′), 3.46 - 3.42 (2H, m, H-4, H-4′), 3.32 (3H, s, OCH3); 13C NMR (300 MHz, CDCl3), δ: 138.43, 138.39, 138.24, 128.68, 128.63, 128.26, 127.91, 127.72, 100.38, 98.11, 82.25, 80.65, 77.45, 77.39, 77.02, 76.96, 75.04, 74.34, 73.55, 73.25, 70.94, 68.57, 60.61, 55.38; HRMS calcd for C34H42O11Na [M+Na]+ 649.2629, found 649.2625.
Methyl 2,3,4-tri-O-allyl-6-O-(2-acetamido-2-deoxy-α-D-galactopyranosyl)-α-D-glucopyranoside (9)
Disaccharide 9 was obtained by the same protocol as described for 6. 1H NMR (500 MHz, CDCl3), δ: 5.99 - 5.90 (3H, m, 3 CH All), 5.34-5.19 (6H, m, 3 × CH2 All), 4.84 (1H, d, J = 3.5 Hz, H-1′), 4.77 (1H, d, J = 3.5, H-1), 4.36 - 4.12 (6H, m, 3 × CH2 All), 3.92-3.76 (6H, m, H-6a, H-6b, H-3, H-6b′, H-5′, H-3′), 3.67 - 3.56 (3H, m, H-5, H-6a′, H-4′), 3.51 (1H, m, H-2), 3.43 (3H, s, OCH3), 3.37 - 3.30 (2H, m, H-4, H-2′), 1.68 (3H, s, CH 13 3CO); C NMR (300 MHz, CDCl3), δ: 171.27, 135.13, 134.70, 133.31, 117.64, 117.07, 116.56, 106.62, 98.14, 86.69, 81.29, 79.45, 77.52, 77.25, 74.21, 73.79, 72.58, 70.55, 69.59, 65.41, 64.36, 61.42, 55.20, 52.12, 23.05; LRMS calcd for C24H39NO11Na [M+Na]+ 540.2, found 540.2.
6-O-(α-D-Galactopyranosyl)-D-galactopyranose (12)
A mixture of D-galactose (152.82 mg, 0.848 mmol), acceptor 10 (1260mg, 4.84 mmol), and Amberlite IR-120 (H+) (300mg) in [emim][ba] (1200mg, 5.155 mmol) was stirred under argon at 50 °C. After 4 h, H2O was added and the mixture was filtered to remove Amberlite IR-120 (H+) and benzoic acid liberated from [emim][ba]. The filtrate was treated with Amberlite IR-120 (H+) (1500mg) in water at 80 °C for 4 h to remove the protecting groups to provide unprotected oligosaccharide 12. Purification by flash chromatography [ethyl acetate - methanol - H2O (85:10:5)→(70:20:10)→(55:30:15)] then gave the oligosaccharide 12. 1H NMR (500 MHz, D2O), δ: α-anomer, 5.24 (1H, d, J = 3.5 Hz, H-1), 5.02 (1H, d, J = 1.5 Hz, H-1′), 4.22 (1H, dd, J1 = 3.5, J2 = 8 Hz, H-5′), 4.09-4.04 (2H, m, H-6a, H-2′), 3.97 - 3.90 (3H, m, H-6b, H-5, H-3′), 3.88 - 3.76 (3H, m, H-4, H-2, H-4′), 3.74 - 3.67 (2H, m, H-3, H-6b′) 3.65 - 3.61 (1H, m, H-6a′); β-anomer, 5.02 (1H, d, J = 1.5 Hz, H-1′), 4.56 (1H, d, J = 7.75 Hz, H-1), 4.22 (1H, dd, J1 = 3.5, J2 = 8 Hz, H-5′), 4.09 - 4.04 (2H, m, H-6a, H-2′), 3.97 - 3.90 (2H, m, H-5, H-3′), 3.88 - 3.76 (3H, m, H-6b, H-4, H-4′), 3.74 - 3.67 (1H, m, H-6b′), 3.65 - 3.61 (2H, m, H-6a′, H-3), 3.47 (1H, dd, J = 7.75, J2 = 10 Hz, H-2); 13 1 C NMR (300 MHz, D2O), δ: 107.95, 96.63, 92.51, 83.05, 81.19, 76.88, 73.88, 72.89, 71.99, 70.99, 69.08, 67.6, 62.87; LRMS m/z, calcd for C H + 12 22O11Na [M+Na] 365.0, found 365.0.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Green LG, Ley SV. Carbohydr. Chem. Biol. 2000;1:427–448. [Google Scholar]
- 2.Nitz M, Bundle DR. Glycoscience. 2001;2:1497–1542. [Google Scholar]
- 3.Ernst B, Hart GW, Sinaÿ P. Carbohydrates in Chemistry and Biology. Wiley, Weinheim; Germany: 2000. [Google Scholar]
- 4.Fischer E. Ber. Dtsch. Chem. Ges. 1893;26:2400–2412. [Google Scholar]
- 5.Balzer D, Luders H. Nonionic Surfactants - Alkyl polyglucosides. Marcel Dekker; New York: 2000. [Google Scholar]
- 6.Hanessian S. Preparative Carbohydrate Chemistry. Marcel Dekker; New York: 1997. [Google Scholar]
- 7.Hanessian S, Lou BL. Chem. Rev. 2000;100:4443–4463. doi: 10.1021/cr9903454. [DOI] [PubMed] [Google Scholar]
- 8.Foley KM, Griffin CC, Amaya E. US. Pat. 1990 App. 898124. [Google Scholar]
- 9.Sheldon R. Chem. Commun. 2001;23:2399–2407. doi: 10.1039/b107270f. [DOI] [PubMed] [Google Scholar]
- 10.Murugesan S, Linhardt RJ. Curr. Org. Synth. 2005;2:437–451. [Google Scholar]
- 11.Rencurosi A, Lay L, Russo G, Caneva E, Poletti L. Carbohydr. Res. 2006;341:903–908. doi: 10.1016/j.carres.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 12.Rencurosi A, Lay L, Russo G, Caneva E, Poletti L. J. Org. Chem. 2005;70:7765–7768. doi: 10.1021/jo050704x. [DOI] [PubMed] [Google Scholar]
- 13.Murugesan S, Karst N, Islam T, Wiencek JM, Linhardt RJ. Synlett. 2003;9:1283–1286. [Google Scholar]
- 14.Poletti L, Rencurosi A, Lay L, Russo G. Synlett. 2003;15:2297–2300. [Google Scholar]
- 15.Sharma M, Potti GG, Simmons OD, Korytnyk W. Carbohydr. Res. 1987;162:41–51. doi: 10.1016/0008-6215(87)80199-4. [DOI] [PubMed] [Google Scholar]