Abstract
The novel amino acid residue (2R,3R,4S)-4-amido-7-guanidino-2,3-dihydroxyheptanoic acid (AGDHE, 3), a constituent of the cyclic depsipeptides callipeltins A and D, and its (2S,3S,4S) diastereomer were synthesized from a protected l-ornithine derivative in 13 steps (15% overall yield), and its configurational assignment was reexamined by 1H NMR.
Callipeltin A (1, Figure 1), a cyclic depsidecapeptide isolated from the marine lithisda sponges Callipelta sp.1 and Latruncula sp.,2 was shown to possess antifungal and anti-HIV activity and cytotoxicity against selected human carcinoma cell lines,3 as well as powerful inhibition of the Na/Ca exchanger in guinea pig left atria.4 The structure of 1 was determined by Minale and co-workers;1 it contains a number of novel amino acids and a novel fatty acid, though the stereochemistry of the β-methoxytyrosine residue could not be determined. Current efforts in our laboratory are focused on the synthesis and study of 1 and related compounds.
Figure 1.

Comparison of the bioactivity of 1 with the related compound callipeltin B indicates that the side chain attached to the macrocycle, recently isolated as callipeltin D (2),2 is essential for anti-HIV activity.3 A key residue in the side chain is the novel amino acid (2R,3R,4S)-4-amido-7-guanidino-2,3-dihydroxyheptanoic acid (AGDHE, 3).5 The configuration of the vicinal diol was proposed based on 1H NMR coupling constants and molecular mechanics calculations, assuming a six-membered-ring hydrogen-bonded conformation of the β-hydroxyamide. This same assumption was used to assign the configuration of TMHEA ((2R,3R,4R)-3-hydroxy-2,4,6-trimethylheptanoic acid) in 1 and later was revised,6 calling into question the stereochemical assignment of AGDHE 3. The possible bioactivity of 3, as well as the need for a protected form of the residue for the synthesis of 1, has prompted us to develop a protocol for its construction and reexamine its configurational assignment.
The synthesis of 3 started from Nδ-benzyloxycarbonyl-Nα-tert-butyloxycarbonyl-l-ornithine (4, Scheme 1), a commercially available ornithine derivative. The original conception of the synthesis envisaged stereoselective addition of an α-hydroxyacetate synthon to an α-aminoaldehyde derived from a protected ornithine derivative. In practice, the carboxylic acid was converted to a mixed anhydride using isobutyl chloroformate and immediately reduced with NaBH4 to give alcohol 5 in 95% yield.7 Swern oxidation of 5 yielded the aldehyde 6, but this compound rapidly underwent cyclization to the cyclic aminal 7, which was unreactive and not a viable synthetic intermediate.8
Scheme 1a.

a Reagents and conditions: (a) (i) iPrOCOCl, NEt3, THF, −15 °C, (ii) NaBH4, H2O (95%, two steps); (b) (COCl)2, DMSO, NEt3, THF, −45 °C.
The undesired cyclization event was prevented by placement of an additional protecting group on the side-chain nitrogen of the ornithine residue (Scheme 2). Alcohol 5 was protected as the TBS ether 8, which was treated with di-tert-butyl dicarbonate and DMAP to effect regioselective addition of a Boc protecting group to the side chain nitrogen, giving intermediate 9 in 97% yield. TBAF deprotection of the TBS group provided alcohol 10 in essentially quantitative yield.
Scheme 2a.

a Reagents and conditions: (a) TBSCl, imidazole, DMF (97%); (b) Boc2O, DMAP, CH3CN (97%); (c) TBAF, THF (99%); (d) (i) (COCl)2, DMSO, CH2Cl2 −65 °C: NEt3, (ii) (CF3CH2O)2POCH2-CO2CH3, K2CO3, 18-C-6, PhCH3–CH3CN, −15 °C (92%).
Oxidation of 10 provided the desired aldehyde, which was not prone to the cyclization observed with 5. Although a number of literature procedures for stereospecific addition of α-alkoxyenolates and α-alkoxyenolate equivalents to aldehydes were examined,9-11 our eventual solution was to osmylate a Z-alkene derived from a Horner–Emmons olefination.12 Thus, the aldehyde was subjected without purification to Z-selective Horner–Emmons olefination conditions (Scheme 2).13 Trace amounts of E-alkene in the crude product mixture were removed by flash chromatography, yielding the desired Z-alkene 11 in 92% yield.14,15 Alkene 11 was subjected to a number of Sharpless asymmetric dihydroxylation16 conditions in an effort to obtain the desired diol 12a (Table 1). Reetz has previously examined the stereoselectivity of dihydroxylation of α-amino-E-alkenes and shown that S-configured amines (as in this case) underwent β-dihydroxylation,17 opposite the selectivity desired in our synthesis. It was expected that employment of Sharpless asymmetric dihydroxylation conditions might reverse the intrinsic selectivity of addition. In all cases, this chemistry failed to afford 12a as the major product. The best results (a 1:1 ratio) were obtained using catalytic osmium tetraoxide with NMO as the stoichiometric oxidant.
Table 1.
Survey of Ligands for the Asymmetric Dihydroxylation of 11
 |
| ligand | oxidant | solvent | 12a/12b |
|---|---|---|---|
| (DHQD)2PHAL | K3Fe(CN)6 | tBuOH/H2O | 1:5 |
| (DHQD)2PYR | K3Fe(CN)6 | tBuOH/H2O | <1:2 |
| (DHQD)2AQN | K3Fe(CN)6 | tBuOH/H2O | 1:5 |
| DHQD-IND | K3Fe(CN)6 | tBuOH/H2O | 2:3 |
| (DHQ)2PHAL | K3Fe(CN)6 | tBuOH/H2O | 1:1 |
| (DHQ)2PYR | K3Fe(CN)6 | tBuOH/H2O | 1:2 |
| (DHQD)2AQN | K3Fe(CN)6 | tBuOH/H2O | 1:2 |
| DHQD-IND | K3Fe(CN)6 | tBuOH/H2O | 1:5 |
| DHQD-IND | K3Fe(CN)6 | acetone/tBuOH/H2O | <1:2 |
| NMO | iPrOH | 1:1 | |
| NMO | acetone/tBuOH/H2O | 1:1 |
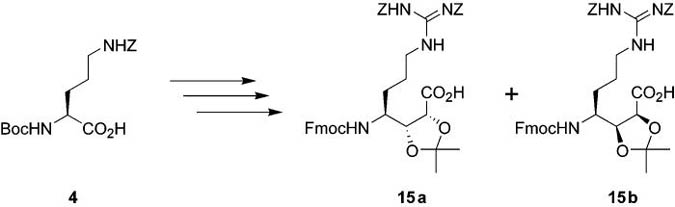
Alkene 11 was dihydroxylated (Scheme 3) using OsO4–NMO to afford a 1:1 mixture of the two diastereomeric diols 12a and 12b. After chromatographic separation of diastereomers, desired diol 12a was obtained in 49% isolated yield. Diol 12a was treated with TFA to remove both Boc protecting groups, followed by reprotection of the N-terminal amine with Fmoc in 82% yield over two steps. Protection of the diol was accomplished with 2,2-dimethoxypropane and catalytic CSA in DMF. The reaction was extremely sluggish at room temperature, but when run at 35 °C overnight yielded the desired acetonide 13a in 78% yield. Hydrogenolysis of the Z group provided the amine for the guanylation reaction. This transformation was accomplished by treatment of the acetate salt of the amine with Et3N and N,N′-di-Z-N″-trifluoromethanesulfonyl-guanidine,18 yielding the guanylated product 14a in 66% yield over two steps. Saponification of the methyl ester with 0.2 M aq LiOH in THF at 0 °C yielded 15a, a protected version of AGDHE suitable for incorporation into the solid-phase synthesis of callipeltin A. Compound 15a is somewhat unstable due to the presence of a carboxylic acid and the acid-labile acetonide protecting group in the same molecule, prompting the immediate use of 15a without purification in the synthesis of callipeltin A. Isomer 12b was subjected to analogous conditions providing 15b in almost the same yield as shown for its diastereomer 15a.
Scheme 3a.

a Reagents and conditions: (a) OsO4, NMO, MeSO2NH2, acetone–tBuOH–H2O (12a, 49%; 12b, 49%); (b) (i) 33% TFA–CH2Cl2, (ii) Fmoc–OSu, NEt3, CH2Cl2 (82%; 73%, two steps), (iii) CSA, (CH3)2C(OCH3)2, DMF, 35 °C (13a, 78%; 13b, 89%); (c) (i) H2, Pd/C, 5% AcOH–MeOH, (ii) N,N′-di-Z-N″-trifylguanidine, NEt3, CHCl3 (14a, 66%; 14b, 72%, two steps); (d) LiOH, THF, 0 °C (15a, 90%; 15b, 90%); (e) Dowex resin, 90% MeOH–H2O (16a and 16b in quantitative yield); (f) H2, Pd/C, 2.5% AcOH–MeOH (17a, 78%; 17b, 70%).
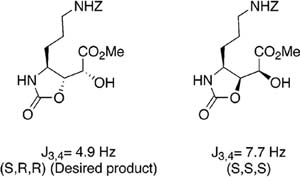
With both isomers 12a and 12b in hand, it was decided to examine the stereochemical assignment of Minale et al. by correlation of 1H NMR data. The methines of C-2, C-3, and C-4 were assigned the 2R,3R,4S configuration by Minale after determination of the vicinal coupling constants between the protons on these carbons (J2,3 = 9.1 Hz, J3,4 = 2.1 Hz) and comparison to the calculated value expected for J3,4 (2.0 Hz) in this configuration as determined computionally.1 The product of the dihydroxylation procedure, 12a, showed nearly identical coupling between H-3 and H-4 (2.0 Hz), suggesting that this intermediate has the same configuration at these stereogenic centers. The actual C-2, C-3 configuration of 12a was confirmed as (R,R) through spectroscopic analysis of oxazolidinone and lactam structures derived from 12a.19-21
More evidence for this configuration was found upon deprotection of the acetonides in both diastereomers 14a and 14b, giving diols 16a and 16b (Scheme 3). Comparison of the coupling constants again showed closer values for the 2R,3R,4S configuration rather than the 2S,3S,4S isomer (Table 2). Closer relation to the natural product was achieved when the benzyloxycarbonyl groups in 16a and 16b were successfully deprotected, yielding guanidines 17a and 17b. Examination of vicinal coupling constants in these isomers lent further support for the assignment of AGDHE as the (2S,3R,4R) diastereomer.
Table 2.
Comparison of Selected 1H NMR Data for Diols 16a,b, 17a,b, and Callipeltins A (1) and D (2)
| compd | H-2 d, ppm (J, Hz) | H-3d, ppm (J, Hz) |
|---|---|---|
| 1 | 3.99 d (9.1) | 3.65 dd (9.1, 2.2) |
| 2 | 4.02 d (7.1) | 3.76 dd (7.1, 2.7) |
| 16a | 3.87 d (8.4) | 3.58 dd (8.5, 2.0) |
| 16b | 4.11 d (3.0) | 3.71 s |
| 17a | 3.87 d (8.5) | 3.58 dd (8.4, 1.9) |
| 17b | 4.11 d (3.8) | 3.71 d (3.7) |
In summary, a protected derivative of (4S,3R,2R)-4-amido-7-guanidino-2,3-dihydroxyheptanoic acid (AGDHE, 15a) suitable for use in solid-phase synthesis was synthesized in 13 steps and 15% overall yield. In addition, the configurational assignment of this fragment was supported on the basis of a comparison of 1H NMR coupling constants with those of the natural products callipeltins A and D.
References
- 1.Zampella A, D'Auria V, Paloma L, Casapullo A, Minale L, Debitus C, Henin Y. J. Am. Chem. Soc. 1996;118:6202–6209. [Google Scholar]
- 2.Zampella A, Randazzo A, Borbone N, Luciani S, Trevisi L, Debitus C, D'Auria MV. Tetrahedron Lett. 2002;43:6163–6166. [Google Scholar]
- 3.D'Auria V, Zampella A, Paloma L, Minale L. Tetrahedron. 1996;52:9589–9596. [Google Scholar]
- 4.Trevisi L, Bova S, Carnelli G, Danieli-Betto D, Floreani M, Germinario E, D'Auria MV, Luciani S. Biochem. Biophys. Res. Commun. 2000;279:219–222. doi: 10.1006/bbrc.2000.3906. [DOI] [PubMed] [Google Scholar]
- 5.Chandrasekar S, Ramachandar T, Venkateswara Rao B. Tetrahedron: Asymmetry. 2001;12:2315–2321. [Google Scholar]; Synthesis of (2R,3R,4S)-4,7-diamino-2,3-dihydroxyheptanoic acid has been previously published.
- 6.Zampella A, D'Auria MV. Tetrahedron: Asymmetry. 2002;13:1237–1239. [Google Scholar]
- 7.Rodriguez M, Llinares M, Doulut S, Heitz A, Martinez J. Tetrahedron Lett. 1991;32(7):923–926. [Google Scholar]
- 8.Salituro GF, Agarwal N, Hofman T, Rich DH. J. Med. Chem. 1987;30:286–295. doi: 10.1021/jm00385a010. [DOI] [PubMed] [Google Scholar]
- 9.Barret AGM, Malecha JW. J. Org. Chem. 1991;56:5243–5245. [Google Scholar]
- 10.Brown HC, Narla G. J. Org. Chem. 1995;60:4686–4687. [Google Scholar]
- 11.Kobayashi S, Hayashi Y. J. Org. Chem. 1995;56:1098–1099. [Google Scholar]
- 12.All new compounds exhibited spectroscopic (IR, 1H, and 13C NMR) and analytical data in accord with the assigned structures.
- 13.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- 14.Pihko PM, Koskinen AMP. J. Org. Chem. 1998;63:92–98. doi: 10.1021/jo971167m. [DOI] [PubMed] [Google Scholar]
- 15.1H NMR indicated >60:1 ratio of Z/E alkenes.
- 16.Wang L, Sharpless KB. J. Am. Chem. Soc. 1992;114:7568–7570. [Google Scholar]
- 17.Reetz MT, Strack TJ, Mutulis F, Goddard R. Tetrahedron Lett. 1996;37:9293–9296. [Google Scholar]
- 18.Feichtinger K, Zapf Z, Sings HL, Goodman M. J. Org. Chem. 1998;63:3804–3805. [Google Scholar]
-
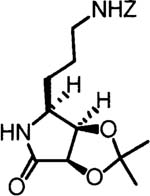
19.The configuration of the dihydroxylation products (12a and 12b) was determined by conversion of the aminodiol to an oxazolidinone and examination of the coupling constant between H-3 and H-4:
- 20.Futagawa S, Inui T, Shiba T. Bull Chem. Soc. Jpn. 1993;46:3308–3310. [Google Scholar]
-
21.Further confirmation of the (2S,3R,4R) stereochemical assignment was obtained from the observation of NOE enhancements in the lactam derived from 11a: