

Abstract
Overexpression of the cytotoxic T cell (CT) GalNAc transferase (Galgt2) in the skeletal muscles of transgenic mdx mice inhibits the development of muscular dystrophy. The profound effect of Galgt2 on muscular dystrophy in transgenic mice, where overexpression is begins from embryonic stages, is complicated by its additional effects on muscle growth and neuromuscular structure. Here, we use Adeno-associated virus (AAV) to show that overexpression of Galgt2 in skeletal myofibers in the early postnatal period is equally effective in inhibiting muscular dystrophy, but that it does so without altering muscle growth or neuromuscular structure. Unlike embryonic overexpression, postnatal overexpression of Galgt2 did not reproducibly increase the expression of utrophin, synaptic laminins, or dystrophin-associated glycoproteins along infected myofibers. Moreover, Galgt2 overexpression inhibited muscular dystrophy to the same extent in utrophin-deficient mdx muscles as it did in utrophin-expressing mdx muscles. Thus, Galgt2 is a molecular target for therapy in DMD that can be utilized in a manner that separates its clinical benefit from its effects on development, and its clinical benefit is distinct from that achieved by utrophin.
Keywords: dystroglycan, utrophin, neuromuscular junction, glycosylation, muscle
1. Introduction
Duchenne muscular dystrophy (DMD) is an X-linked disorder caused by loss of dystrophin protein expression that leads to the wasting of skeletal muscles [1, 2]. mdx mice are a commonly used model for testing therapeutic approaches to DMD; mdx animals do not express dystrophin protein in most of their skeletal myofibers, and mdx myofibers exhibit several important pathological hallmarks of DMD[3, 4]. In addition, loss of dystrophin protein expression, both in DMD and mdx muscles, correlates with loss of membrane expression of proteins in the dystrophin-glycoprotein complex, including dystroglycans and sarcoglycans, along the myofiber membrane[5]. At the neuromuscular junction, the site where skeletal myofibers are innervated by the motor nerve terminal, there exists a complex of synaptic proteins that likely subserves the same function that the dystrophin-glycoprotein complex does along the rest of the myofiber membrane[6]. The expression of these neuromuscular proteins, however, is not affected in DMD or mdx muscles[5]. One of these neuromuscular proteins is utrophin, a closely related homologue of dystrophin[7]. Another neuromuscular component is the cytotoxic T cell (CT) carbohydrate[8].
The CT carbohydrate is synthesized by the cytotoxic T cell (CT) GalNAc transferase, or Galg t2[9], an enzyme that is also concentrated at the neuromuscular synapse in skeletal muscle[10]. Transgenic overexpression of either utrophin [11-13] or Galgt2 [14] from embryonic time points onward in skeletal muscle can increase the expression of dystrophin-associated glycoproteins, including dystroglycan and sarcoglycans, along the myofiber membrane of mdx animals [11, 12]. Overexpression of either utrophin[11-13] or Galgt2[14] also inhibits the development of muscular dystrophy in the skeletal muscles of transgenic mdx mice. In addition to its effect on muscular dystrophy, embryonic overexpression of Galgt2 has a profound effect on neuromuscular development and skeletal muscle growth[10]. These phenotypes complicate the interpretation of how Galgt2 inhibits muscular dystrophy and suggest features of its expression that one would not desire in a therapy. Here, we use adeno-associated virus (AAV) to introduce the Galgt2 transgene (AAV-Galgt2) at later developmental time points and show that the beneficial effects of Galgt2 in inhibiting muscular dystrophy can be divorced from its unwanted developmental effects on muscle growth and neuromuscular structure. Surprisingly, while embryonic Galgt2 transgene expression stimulates the overexpression of utrophin, synaptic laminins, and dystrophin-associated glycoproteins along transgenic myofibers[10, 14], we show here that this does not occur with postnatal Galgt2 overexpression and that postnatal inhibition of muscular dystrophy by Galgt2 overexpression still occurs in mdx mice lacking utrophin. Thus, the therapeutic effects of Galgt2 overexpression are likely to occur via a novel molecular mechanism.
2. Materials and Methods
2.1 Materials
N-acetylgalactosamine (GalNAc) and N-acetylglucosamine (GlcNAc), were obtained from Calbiochem (San Diego, CA). Agarose bound lectins (Wisteria floribunda agglutinin, WFA, and Wheat Germ agglutinin, WGA) were purchased from EY laboratories (San Mateo, CA). AAV2-Galgt2 was made and purified by the Vector Development lab at UC San Diego. AAV1-Galgt2 was made and purified by Virapure (San Diego, CA). AAV8-like Galgt2 (rh.74-Galgt2) was made by the Viral Vector Core at Columbus Children’s Research Institute. Monoclonal antibodies to dystrophin, utrophin, β dystroglycan, α sarcoglycan, and β sarcoglycan, γ sarcoglycan, and δ sarcoglycan were obtained from Nova Castra (Newcastle Upon Tyne, UK). Antibodies to actin were obtained from Sigma (St. Louis, MO). Antibodies to α dystroglycan (VIA4-1 and IIH6) were obtained from Upstate Biotech nology (Lake Placid, NY). Antibodies β dystroglycan, CT carbohydrate (CT1 and CT2), and the CT GalNAc transferase were produced in our laboratory. Polyclonal antibodies to integrin α7B, utrophin, dystrophin, α sarcoglycan, and caveolin 3 were a generous gift from Ling Guo (UC San Diego) and Eva Engvall (Burnham Institute, La Jolla). Rhodamine-α-bungarotoxin was purchased from Molecular Probes (Eugene, OR). Secondary antibodies conjugated to horseradish peroxidase, fluorescein isothiocyanate, and Cy2 were purchased from Jackson Immunochemicals (Seattle, WA).
2.2 Transgenic mice
Transgenic mice that express the CT GalNAc transferase (Galgt2) specifically in skeletal muscles (via the skeletal α actin promoter[15]) were described by us previously[10, 16], as were Galgt2 transgenic mdx mice[14]. Additional mdx mice were bred in our colony from animals purchased from The Jackson laboratory (Bar Harbor, ME). Mdx mice lacking utrophin were bred from animals given to us by Jill Rafael-Fortney (Ohio State).
2.3 Histology
Muscles were dissected and snap-frozen in liquid nitrogen-cooled isopentane and cut at 8-10μm on a cryostat. Sections were either stained with hematoxylin and eosin or immunostained with various antibodies as previously described[10, 14, 17]. Quantitation of central nuclei, myofiber diameters, and neuromuscular diameters were all also done as previously described[10, 14, 17].
2.4 Infection of skeletal muscles with Adeno-associated virus
AAV vectors were produced, purified, and titered using the triple transfection method[18]. The tibialis anterior or gastrocnemius muscle on the left side of mdx or wild type mice of varying ages (see Table 1) were injected with between 5×109 vector genomes (vg) to 1 × 1011 vg of AAV2-Galgt2 or AAV1-Galgt2. Utrophin-deficient mdx mice (mdxutrn-/-) and control animals (mdxutrn+/-) were injected at 4 days to 1 week of age as above, only the left gastrocnemius and left quadriceps muscles were each injected with 1-2×1010vg of AAV1-Galgt2 or AAV8-like-Galgt2 (rh.74-Galgt2). Gastrocn emius and quadriceps muscles were injected in a volume of 50μl of sterile PBS, while tibialis anterior muscles were injected in a 25μl volume. All contralateral muscles (on the right side) were injected with a similar volume of PBS alone. After 4-12 weeks, mice were sacrificed and muscles dissected and snap-frozen in liquid nitrogen-cooled isopentane.
Table 1.
Summary of AAV-Galgt2 intramuscular injection experiments in mdx mice.
| Serotype | Titer | Age | Percent central myofiber nuclei | |||
|---|---|---|---|---|---|---|
| Injected | Analyzed | Muscle | CT overexpressing | Non-overexpressing | ||
| 1. AAV1 | 1×109 | 1wk | 6wks | TA | 6 | 87 |
| 2. AAV1 | 1×109 | 1wk | 6wks | TA | 4 | 80 |
| 3. AAV1 | 1×109 | 1wk | 6wks | TA | 3 | 87 |
| 4. AAV2 | 1×1010 | 2wks | 6wks | TA | 2 | 70 |
| 5. AAV2 | 1×1010 | 2wks | 6wks | TA | 3 | 61 |
| 6. AAV2 | 1×1010 | 2wks | 6wks | TA | 3 | 69 |
| 7. AAV2 | 1×1010 | 2wks | 6wks | TA | 4 | 76 |
| 8. AAV2 | 1×1010 | 2wks | 6wks | TA | 3 | 63 |
| 9. AAV2 | 1×1010 | 2wks | 6wks | G | 5 | 66 |
| 10. AAV1 | 1×1010 | 2wks | 6wks | TA | 1 | 75 |
| 11. AAV1 | 1×1010 | 2wks | 6wks | TA | 6 | 87 |
| 12. AAV1 | 1×1010 | 2wks | 6wks | TA | 6 | 82 |
| 13. AAV1 | 1×1010 | 2wks | 6wks | G | 1 | 44 |
| 14. AAV1 | 1×1010 | 2wks | 10wks | TA | 6 | 68 |
| 15. AAV1 | 1×109 | 2wks | 10wks | TA | 7 | 56 |
| 16. AAV1 | 1×1010 | 2wks | 10wks | G | 4 | 83 |
| 17. AAV1 | 1×1010 | 2wks | 14wks | TA | 14 | 92 |
| 18. AAV1 | 1×1011 | 2wks | 14wks | G | 8 | 82 |
| 19. AAV1 | 1×1010 | 3wks | 15wks | G | 25 | 76 |
| 20. AAV1 | 1×1010 | 4wks | 16wks | TA | 33 | 75 |
| 21. AAV2 | 1×1010 | 6mos | 7mos | TA | 70 | 75 |
| 22. AAV2 | 1×1010 | 15mos | 16mos | TA | 81 | 88 |
| 23. AAV2 | 1×1010 | 15mos | 16mos | G | 89 | 87 |
| Average of AAV1 or 2 | 1-2 wks | 6wks | 4±2*** | 72±12 | ||
| Average of AAV1 | 2wks | 10-14wks | 8±4*** | 76±14 | ||
Adeno-associated virus (AAV) serotype 1 or 2 containing the CT GalNAc transferase (Galgt2) transgene was delivered by intramuscular injection in the titers indicated. After 1-3 months, muscles were analyzed for CT carbohydrate overexpression and for the presence of centrally located myofiber nuclei, an indicator of dystrophic pathology. In mdx mice, dystrophic pathology begins to develop after 3 weeks of age. If transgene was delivered at 1-2 weeks of age, a highly significant reduction in pathology was observed in myofibers expressing transgene. Expression was evident up to three months post-injection. TA, Tibialis anterior; G, Gastrocnemius; Errors are standard deviations
P<0.001 for a 2-tailed Student’s t test.
2.5 Immunoblotting and Lectin Precipitation
Immunoblotting and lectin precipitations were done as previously described[17, 19].
2.6 Statistics
Determinations of significance were done using a two-tailed Student’s t-test.
3. Results
3.1 Length of inhibition of muscular dystrophy by Galgt2
In our original study, we demonstrated that transgenic expression of the CT GalNAc transferase (Gal gt2) in mdx mouse muscles inhibited the development of muscular dystrophy for up to 6 months of age[14]. We have now followed significant numbers of these mice up to 18 months of age, with some to 24 months. Several additional findings are relevant with respect to these older animals. First, all seven mdx/CT muscles studied maintained their inhibition of muscle pathology up to 18 months of age (Fig. 1A). Here we show the percentage of centrally located myofiber nuclei because it is one the most robust pathology measures in mdx muscles. As normal myofibers mature, nuclei migrate out to the periphery of the myofiber, such that fewer than 5% remain in a central position[20-23]. However, if dystrophin-lacking muscles are damaged and undergo regeneration in the adult mouse, nuclei within the regenerated myofiber remain in a central position for most of the life of the animal. Thus, the presence of increased central nuclei is a marker of mdx muscles that have undergone a cycle of degeneration and regeneration. Even at 18 months, mdx/CT myofibers maintained central nuclei at levels that were significantly lower than their mdx littermates in all muscle examined (P<0.001 for all) (Fig. 1A). The increase in central nuclei in several mdx/CT muscles above wild type levels may be due to diminished transgene expression at this age; CT (non-mdx) animals also had increased levels of myofibers with central nuclei at this age (Fig. 1). Like younger animals, skeletal muscles in 18 month-old CT and mdx/CT animals were significantly smaller than mdx or wild type littermates (Fig. 1B). All skeletal muscles examined had a reduction in average myofiber diameter that was significant and exceeded 50%. Similarly, neuromuscular junctions in non-mdx CT transgenic animals were irregular as compared to wild type, suggesting that the aberrant neuromuscular structure previously reported in younger animals remained even at 18 months of age (Fig. 1C). These experiments demonstrate that transgenic Galgt2 overexpression inhibits muscular dystrophy in mdx mice for almost the entire lifespan of the animal, but that it also equivalently maintains the dramatic suppression of muscle growth and altered neuromuscular structure.
Figure 1.

Overexpression of CT carbohydrate inhibits muscle pathology up to 18 months of age in Galgt2 transgenic mdx mice.
(A) Central nuclei were quantitated in skeletal muscles taken from wild type (Wt), Galgt2 transgenic (CT), mdx, and Galgt2-transgenic mdx (mdx/CT) mice at the indicated ages. Skeletal muscles analyzed (from left to right) were gastrocnemius, trapezius, diaphragm, triceps, quadriceps, tibialis anterior, and gluteus maximus. CT muscles began to develop some central nuclei at 18 months of age, as did mdx/CT muscles, however, the percentage of myofibers with central nuclei in mdx/CT animals was still significantly lower than mdx littermates for all muscles (P<0.001 for all) at 12 and 18 months of age. Errors are SEM for n=250 fibers per muscle for 3-4 animals per condition. (B) Hematoxylin and eosin staining of muscles at 18 months of age showed that CT and mdx/CT myofibers were significantly smaller than wild type (gastrocnemius is shown) and relatively free from muscle damage. Bar is 50μm. (C) Rhodamine-α-bungarotoxin of Wt and CT muscles in cross-section (larger panels) or in whole mount (smaller panels) shows aberrant neuromuscular junctions in CT muscles as compared to wild type at 18 months of age as well. Bar is 25μm (larger panels) or 12.5μm (smaller panels).
3.2 Inhibition of muscular dystrophy by AAV-Galgt2
The inhibition of muscle growth in Galgt2 transgenic mdx animals is a significant complicating factor in understanding the therapeutic aspects of this transgene. Myofiber diameters in transgenic muscles differ by only 5% from non-transgenic littermates at birth[10] but are reduced by over 50% by 6 weeks of age (and thereafter)[10]. The reduction of muscle growth correlates with an increase of an order of magnitude in intramuscular satellite cells[10], cells that significantly contribute to postnatal muscle growth in the first two postnatal weeks. Therefore, the effects of Galgt2 on development are likely to occur prior to the second postnatal week. Similarly, neuromuscular development is largely complete by 2 weeks of age[24]. In mdx mice, by contrast, muscular dystrophy is not present for the first three weeks of life[4]. There is a window, therefore, in 2-3 week old mdx animals when the contribution of satellite cells to myofiber growth has largely occurred but when muscular dystrophy is not yet present. We chose to introduce the Galgt2 transgene during this period to determine if its effect on inhibiting muscular dystrophy could be divorced from its effects on myofiber growth and neuromuscular development. Because single stranded AAV vectors can take up to a week after infection to begin to express transgene[25], we chose to infect mdx mice at 1-2 weeks of age.
To overexpress Galgt2 transgene postnatally, we infected skeletal muscles with AAV vectors containing the Galgt2 cDNA driven by the cytomegalovirus (CMV) promoter. Two different serotypes, AAV1 and AAV2, were used. mdx or wild type mice were infected with AAV1-Galgt2 or AAV2-Galgt2 in tibialis anterior or gastrocnemius muscle of the left leg, while an equivalent volume of PBS was injected into the same muscle on the contralateral side of the animal. Muscles were analyzed for expression of the CT carbohydrate 4, 8, or 12 weeks after infection (Fig. 2). In some experiments, CT carbohydrate was visualized with Dolichos biflorus agglutinin (DBA) (Fig 2A). DBA is a GalNAc-binding lectin that can recognize the terminal βGalNAc linkage synthesized by the CT carbohydrate[26, 27]. Because DBA normally only stains the neuromuscular junction[28], its e xtrasynaptic expression was easily visualized in infected cells. Use of an antibody to the CT carbohydrate antigen, CT2 (which recognizes GalNAcβ1,4[NeuGcα2,3]Galβ1,4GlcNAcβ-)[29], was also used to follow carbohydrate overexpression (Fig. 2B). AAV-Galgt2 infected muscles showed sustained expression of the CT carbohydrate for up to 3 months post-injection in mdx myofibers (Table 1), with AAV1-Galgt2 being superior to AAV2-Galgt2 in causing more myofibers to overexpress CT carbohydrate on a per vg basis (not shown). In most cases, AAV1-Galgt2 caused CT overexpression in 20-50% of myofibers within the tibialis, with 80% of myofibers overexpressing CT carbohydrate in several instances. In muscles with high levels of AAV-Galgt2 infection, Galgt2 gene expression (measured by RT-PCR) was equivalent (elevated by 40-fold) to levels of overexpression observed in Galgt2 transgenic mdx muscle (both compared to mdx controls (not shown)).
Figure 2.


Postnatal overexpression of CT carbohydrate in mdx skeletal muscle inhibits the development of muscle pathology but not muscle growth.
(A) AAV2-Galgt2 was used to overexpress CT carbohydrate in mdx muscle (tibialis anterior, infection at 2 weeks, staining at 6 weeks). Muscle was stained with DBA to identify CT carbohydrate overexpressing myofibers (left) and with hematoxylin and eosin on the next section in a serial series to determine myofibers with central nuclei. Myofibers overexpressing CT carbohydrate were protected from developing central nuclei. (B) Gastrocnemius muscle in an mdx mouse was infected with AAV1-Galgt2 at 2 weeks of age and analyzed for CT carbohydrate overexpression (green) and for central nuclei (red) at 6 weeks of age. Myofibers overexpressing CT carbohydrate were protected from muscle damage and did not contain central nuclei. Bar is 50μm in A and B.
Myofibers infected at 1-2 weeks of age with AAV-Galgt2 were inhibited from developing muscular dystrophy (Fig. 2 and Table 1). The degree of muscular dystrophy was quantified by comparing the percentage of myofibers with central nuclei in AAV-Galgt2-infected myofibers (CT overexpressing) to the percentage of myofibers with central nuclei in non-overexpressing cells. At the time of infection (1-2 weeks), the percentage of myofibers in mdx mice with central nuclei is not significantly different from wild type animals, however, by 6 weeks of age, most mdx muscles have increased levels of central nuclei that are highly significant compared to wild type [14](Table 1). In all mdx muscles infected with AAV-Galgt2, myofibers overexpressing the CT carbohydrate had significantly fewer myofibers with central nuclei (P<0.001), and these levels did not significantly differ from wild type muscle (Table 1). Thus, postnatal overexpression at this time was as effective as the previously demonstrated clinical benefit shown using embryonic Galgt2 overexpression [14]. AAV-Galgt2-infected myofibers were protected from developing muscular dystrophy for up to 3 months after infection using this early postnatal overexpression paradigm (Table 1).
We also analyzed infection of mouse muscles at later developmental time points, including 3-6 weeks of age, 6 months, and 15 months (Table 1). As expected, the percentage of CT overexpressing myofibers with central nuclei increased at these later time points, as more myofibers would have had central nuclei present prior to infection. These data, importantly, show that regenerated myofibers with central nuclei were not prohibited from being infected with AAV-Galgt2 or from overexpressing the CT carbohydrate.
3.3 Lack of Galgt2 effect on muscle growth and neuromuscular structure with postnatal expression
We next determined whether postnatal infection of mdx or wild type muscles with AAV-Galgt2 altered muscle growth. This was done by comparing the myofiber diameter in CT-overexpressing myofibers that had been infected at 2 weeks of age (and analyzed at 6 weeks of age) with non-overexpressing myofibers in the same animals. As expected, there was no significant decrease in myofiber diameter as a result of Galgt2 overexpression in these experiments (Table 2). The slight increase in average myofiber diameter in mdx myofibers expressing the CT carbohydrate was likely due to the presence of more small regenerating myofibers in the non-overexpressing (dystrophic) pool. These smaller regenerating myofibers, however, were not increased in total number as a result of AAV-Galgt2 administration. Indeed, the number of smaller regenerating myofibers was higher in mock-infected mdx muscles (157±22%, n=6) than in AAV-Galgt2 infected muscles. We presume that this was due to the fact that CT-overexpression in AAV-Galgt2-infected muscles protected myofibers from damage, and that the reduced dystrophy in such muscles would result in the reduced presence of small regenerating myofibers.
Table 2.
Lack of change in muscle or neuromuscular size as a result of postnatal AAVGalgt2 infection of myofibers at 2 weeks of age in mdx and wild type mice after 1 month of expression.
| Strain |
Myofiber Diameter (μm) |
NMJ Diameter (μm) |
||
|---|---|---|---|---|
| CT carbohydrate overexpression: | - | + | - | + |
| Wild type (gastroc) | 36±6 | 37±6 | 7±1 | 7±1 |
| Wild type (tibialis) | 25±2 | 22±2 | 7±1 | 7±2 |
| Mdx (gastroc) | 30±5 | 37±3 | ||
| Mdx (tibialis) | 25±6 | 35±4 | ||
P>0.05 in all cases
We also determined the average cross-sectional length of neuromuscular junctions in myofibers expressing CT carbohydrate as compared with those not infected with AAV-Galgt2 (Table 2). Quantification of neuromuscular length was done in wild type animals because of the presence of small regenerating myofibers complicated the analysis of mdx animals. Neuromuscular length in CT-overexpressing myofibers was indistinguishable from that in non-overexpressing myofibers for both the tibialis and gastrocnemius (p=0.55 for both, Table 2). Neuromuscular junctions in mdx myofibers overexpressing CT carbohydrate also appeared normal when compared to neuromuscular synapses on non-expressing myofibers (Fig. 3). In non-infected myofibers, CT carbohydrate was still present at the neuromuscular junction (Fig. 3), as we have previously shown[8]. Therefore, the inhibition of muscular dystrophy by the CT carbohydrate can be divorced from its effects both on muscle growth and on neuromuscular structure if overexpression occurs after the second postnatal.
Figure 3.

Neuromuscular junctions in mdx myofibers appear normal after postnatal overexpression of CT carbohydrate.
Mdx muscles were infected with AAV1-Galgt2 at 2 weeks of age and analyzed at 6 weeks. Muscles were co-stained for CT carbohydrate overexpression (using the CT2 monoclonal antibody, right) and with rhodamine-α-bungarotoxin (left) to label nicotinic acetylcholine receptors at the neuromuscular junction. Neuromuscular junctions appear normal in mdx muscles where CT carbohydrate is overexpressed. Bar is 40μm (top panels) and 10μm (bottom panels).
3.4 AAV-Galgt2 inhibits muscular dystrophy via a utrophin-independent mechanism
Previous experiments have showed that Galgt2 overexpression in transgenic mdx mice caused the overexpression of utrophin and the upregulation of dystroglycan and sarcoglycans along the myofiber membrane[11, 12]. Because α dystroglycan was the primary glycoprotein glycosylated by Galgt2 in transgenic skeletal muscles in these experiments[10, 14], the upregulation of utrophin and associated membrane glycoproteins suggested that utrophin was central to the inhibition of muscular dystrophy observed. Such a model would be consistent with studies by Davies and colleagues showing overexpression of utrophin can inhibit muscular dystrophy and improve mechanical muscle strength in mdx animals by re-establishing the physical continuity of the actin cytoskeleton to the extracellular matrix via the dystroglycan-sarcoglycan complex [11-13, 30]. To test whether postnatal overexpression of Galgt2 caused similar effects, we serially stained AAV-Galgt2 infected mdx muscles for CT carbohydrate and either utrophin, dystrophin (which should not be present), laminins (α2, α4, α5), dystroglycan (α,β), sarcoglycans (α-δ), integrin α7B, and caveolin 3 (Fig. 4 and not shown). Surprisingly, none of the molecules overexpressed in Galgt2 transgenic mdx mouse muscles were overexpressed using this postnatal overexpression paradigm, despite the fact that postnatal Galgt2 overexpression inhibited the development of muscle pathology just as well as embryonic overexpression had previously (Table 1 and [14]). Dystroglycans, sarcoglycans, and utrophin were expressed primarily at neuromuscular junctions in mdx muscles, much as previously described by Campbell and colleagues[5], but none were overexpressed along myofibers that overexpressed CT carbohydrate. Caveolin 3 was expressed along all myofibers regardless of CT overexpression. Integrin α7B was overexpresed in some smaller, presumably regenerating myofibers, but again this did not correlate with CT overexpression. The fact that utrophin, dystroglycan, and sarcoglycans were not overexpressed in AAV-Galgt2 infected mdx muscles suggests that its ability to inhibit muscular dystrophy is not due to the re-establishment of a functional linkage of the actin cytoskeleton by these molecules.
Figure 4.

Expression of utrophin, synaptic laminins, and dystrophin-associated glycoproteins is not increased with postnatal overexpression of the CT carbohydrate in mdx skeletal muscles.
Mdx muscles were infected with AAV1-Galgt2 at 1-2 weeks of age and analyzed at 6 weeks. Serial sections were stained with antibodies to the CT carbohydrate (CT2) or with antibodies to the indicated proteins (panels below CT2 panels). Overexpression of the CT carbohydrate in postnatal mdx skeletal muscle did not increase expression of utrophin, dystroglycan, sarcoglycans, laminin α4, laminin α5, or integrin α7B along CT-overexpressing myofibers. Bar is 50μm.
We confirmed the lack of increased protein expression for utrophin, dystroglycan, and sarcoglycans by comparing immunoblots of whole muscle lysates taken from a Galgt2-infected mdx muscles (where over half of the myofibers overexpressed CT carbohydrate) and contralateral (mock-infected) controls. No increase in total protein was observed for any of these molecules in Galgt2-infected muscle. Similarly, little or no increase in CT glycosylation of α dystroglycan was observed with postnatal Galgt2 overexpression using previously published lectin pull-down experiments (Fig. 5). While there was some increase in α dystroglycan precipitated by GalNAc-binding lectin (WFA agarose) in AAV-Galgt2-infected muscles, it paled in comparison to the amount precipitated from Galgt2 transgenic (CT) mdx muscles (Fig. 5). Moreover, postnatal overexpression did not correlate with increased CT carbohydrate on α dystroglycan, while embryonic overexpression did increase CT glycosylation of α dystroglycan (Fig. 5). The amount of α dystroglycan precipitated by WGA agarose, a GlcNAc/sialic acid-binding lectin known to precipitate α dystroglycan[31, 32], showed equivalent amounts of α dystroglycan were present in Galgt2-infected and mock-infected muscle lysates (Fig. 5). Thus, postnatal Galgt2 overexpression in mdx muscle does not correlate with increased glycosylation of α dystroglycan with the CT carbohydrate.
Figure 5.
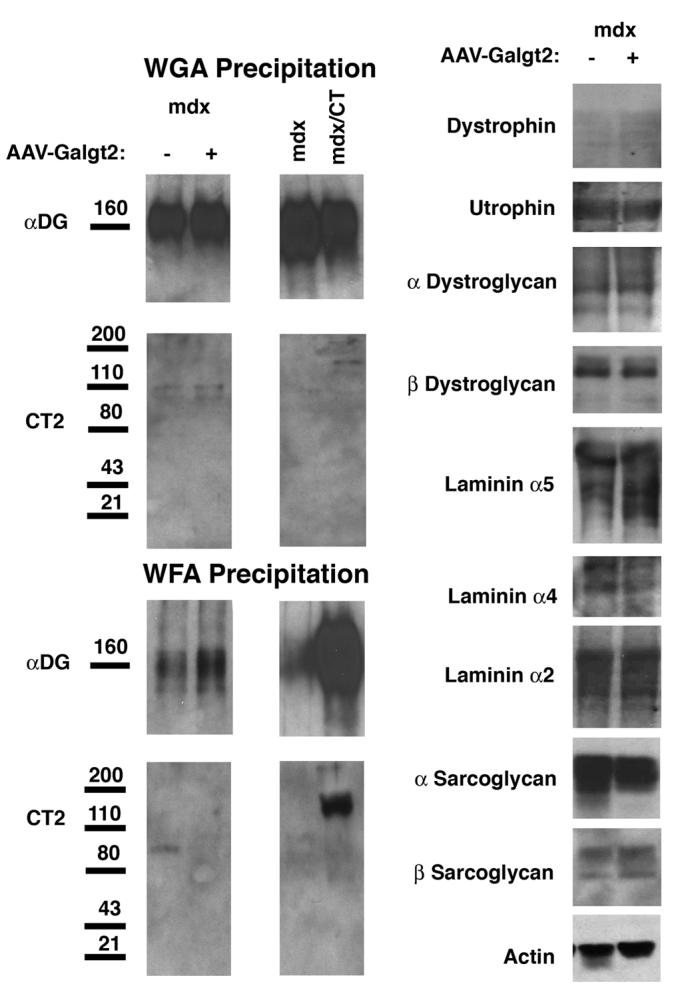
Postnatal overexpression of Galgt2 does not increase CT glycosylation of α dystroglycan or expression of utrophin, synaptic laminins, or dystrophin-associated glycoproteins in mdx muscle.
Left, 150μg of NP-40 detergent protein lysate was precipitated with a GalNAc-binding lectin that can identify the CT carbohydrate (WFA agarose) or with a control lectin known to precipitate α dystroglycan (WGA agarose). Proteins were eluted with GalNAc (for WFA) or GlcNAc (for WGA) and analyzed by Western blot for α dystroglycan or CT carbohydrate (CT2). Postnatal overexpression of Galgt2 using AAV caused a slight increase in α dystroglycan precipitated with WFA, but this protein was not glycosylated with the CT carbohydrate. By contrast, WFA precipitated CT-glycosylated α dystroglycan from Galgt2 transgenic mdx muscle (mdx/CT). WGA precipitation showed equal amounts of α dystroglycan in each pair of samples. Right, comparison of whole cell muscle lysates showed postnatal overexpression of Galgt2 did not increase expression of utrophin, α dystroglycan, β dystroglycan, laminin α2, laminin α4, laminin α5, α sarcoglycan, or β sarcoglycan protein. Blots were stripped and reprobed for actin as a control for protein loading and transfer.
The fact that postnatal overexpression of Galgt2 in myofibers did not increase utrophin expression (Fig. 4) suggested that utrophin was not required for the resulting inhibition of muscular dystrophy. To prove this, we injected AAV1 or rh.74 (AAV8-like)-Galgt2 into skeletal muscles of mdx mice lacking utrophin (mdxutrn-/-; Fig. 6 and Table 3). Overexpression of Galgt2 in mdxutrn-/- muscle still inhibited muscular dystrophy; myofibers overexpressing CT carbohydrate in mdxutrn-/- muscles had central nuclei at levels that were equivalent to those found in mdx mice that did express utrophin (mdxutrn+/-, Table 3). In both cases, the percentage of myofibers having central nuclei did not exceed levels normally found in non-dystrophic mouse muscle[14, 23]. Average myofiber diameters were larger, on average, in Galgt2-overexpressing mdxutrn-/- myofibers than in non-expressing myofibers (36±5μm in CT-overexpressing vs. 26±3μm in non-expressing, P<0.05 for n=4 experiments), much as was seen in utrophin-expressing mdx muscles (Table 2). The more pronounced pathology and reduced myofiber size in mdxutrn-/- muscles led to a significant change between groups in this instance. Experiments were not extended beyond 6 weeks because of premature lethality in mdxutrn-/- animals[33, 34]. These experiments show that utrophin is not required for Galgt2-mediated inhibition of muscular dystrophy in mdx mice and suggest that Galgt2 works via a distinct therapeutic mechanism to inhibit DMD disease progression.
Figure 6.

Postnatal overexpression of Galgt2 inhibits muscular dystrophy in mdx mice lacking utrophin.
Mdx mice lacking utrophin (mdxutrn-/-) or expressing utrophin (mdxutrn+/-) were infected with AAV1-Galgt2 or AAV8-like (rh.74) Galgt2 at 4 days to 1 week of age and analyzed at 6 weeks of age. Infected muscles were stained with antibody to the CT carbohydrate (CT2) in the fluorescein channel (green) and with a non-specific antibody to mark central nuclei in the rhodamine channel (red). Mdx myofibers overexpressing the CT carbohydrate did not have central nuclei, regardless of utrophin expression. Bar is 50μm.
Table 3.
Summary of AAV-Galgt2 intramuscular injection experiments in utrophindeficient mdx mice.
| Genotype | Age | Percent central myofiber nuclei | |||
|---|---|---|---|---|---|
| Injected | Analyzed | Muscle | CT overexpressing | Non-overexpressing | |
| 1. mdxutrn+/- | 1wk | 6wks | G | 3 | 57 |
| 2. mdxutrn+/- | 1wk | 6wks | G | 1 | 64 |
| 3. mdxutrn+/- | 1wk | 6wks | Q | 1 | 73 |
| 4. mdxutrn+/- | 1wk | 6wks | G | 1 | 63 |
| 5. mdxutrn+/- | 1wk | 6wks | G | 0 | 62 |
| 6. mdxutrn+/- | 1wk | 6wks | Q | 4 | 62 |
| 7. mdxutrn+/- | 1wk | 6wks | G | 4 | 66 |
| 8. mdxutrn+/- | 1wk | 6wks | Q | 5 | 60 |
| 9. utrn-/- | 1wk | 6wks | G | 0 | 0 |
| 10. mdxutrn-/- | 1wk | 6wks | G | 4 | 35 |
| 11. mdxutrn-/- | 1wk | 6wks | Q | 3 | 57 |
| 12. mdxutrn-/- | 1wk | 6wks | G | 5 | 63 |
| 13. mdxutrn-/- | 4d | 6wks | G | 4 | 77 |
| 14. mdxutrn-/- | 4d | 6wks | Q | 5 | 64 |
| 15. mdxutrn-/- | 4d | 6wks | G | 3 | 52 |
| 16. mdxutrn-/- | 4d | 8wks | G | 0 | 65 |
| 17. mdxutrn-/- | 1wk | 8wks | G | 0 | 86 |
| 18. mdxutrn-/- | 1wk | 8wks | G | 0 | 75 |
| Average of mdxutrn+/- 1wk | 6wks | G,Q | 2±1*** | 63±2 | |
| Average of mdxutrn-/- 4d-1wk | 6-8wks | G,Q | 3±1*** | 63±5 | |
Adeno-associated virus (AAV) serotype 1 or 8-like (rh.74) containing the CT GalNAc transferase (Galgt2) transgene was delivered by intramuscular injection at 1-2×1010vg. After 1-2 months, muscles were analyzed for CT carbohydrate overexpression and for the presence of centrally located myofiber nuclei, an indicator of dystrophic pathology. In mdx mice, dystrophic pathology begins to develop after 3 weeks of age. If transgene was delivered at 4 days (d) to 1 week (wk) of age, a highly significant reduction in pathology was observed in myofibers expressing transgene, both in mdx mice that were heterozygous for utrophin (mdxutrn+/-) and in mice that were deficient in utrophin (mdxutrn-/-). G, Gastrocnemius; Q, Quadriceps. Errors are standard deviations
P<0.001 for a 2-tailed Student’s t test.
4.Discussion
These experiments demonstrate two important findings with regard to Galgt2 overexpression as a therapeutic approach to treating Duchenne muscular dystrophy (DMD). First, postnatal overexpression of Galgt2, unlike embryonic overexpression, inhibits muscular dystrophy in a way that does not alter muscle growth or neuromuscular structure. Our previous findings with Galgt2 transgenic mice[10, 14] showed clear effects on both of these parameters that would nullify their usefulness with regard to therapy in children with DMD. Here, we show that postnatal overexpression, which would be the period in which therapy would most likely be attempted, bypasses these unwanted developmental effects while maintaining the therapeutic benefit. Second, postnatal overexpression of Galgt2, unlike embryonic overexpression, does not increase levels of utrophin along myofibers, and Galgt2 inhibits mdx myofibers lacking utrophin from developing muscle pathology. These experiments demonstrate that Galgt2 acts independently of utrophin to inhibit muscular dystrophy in mdx mice.
Utrophin upregulation does occur in some mdx myofibers, particularly in regenerating myofibers in younger mdx animals[3, 35, 36]. Indeed, some of our early experiments with AAV2-Galgt2, which yielded far fewer infected myofibers on a vg basis than our later experiments with AAV1-Galgt2, showed that some utrophin upregulation did occur in some Galgt2 overexpressing myofibers. Subsequent analysis using all experiments shown in Table 1, however, showed this not to be the case in general and that upregulation of utrophin, when it did occur, bore no relationship to the inhibition of muscle pathology caused by Galgt2 overexpression. Moreover, the inhibition of muscle pathology by Galgt2 in mdxutrn-/- muscles clearly shows that it acts via a mechanism distinct from utrophin (Figs. 4 and 6).
That α dystroglycan is not increased after postnatal Galgt2 overexpression (Fig. 4) and is not increased in CT glycosylation (Fig. 5) suggest that dystroglycan is also not required Galgt2’s therapeutic mechanism. Indeed, no proteins appear to be highly glycosylated with CT carbohydrate as a result of postnatal Galgt2 overexpression in mdx muscle (Fig. 5). Recent work suggests that sialyl-N-acetyllactosaminyl-containing glycolipids can also be glycosylated by Galgt2 when it is overexpressed[37]. Thus, the postnatal therapeutic effects of Galgt2 could occur via glycolipid-mediated mechanisms. In addition, CT-glycosylated proteins where protein turnover has been stimulated or where protein secretion has occurred may not be evident in this type of assay. The lack of upregulation of integrin α7B, another molecule that can ameliorate aspects of muscular dystrophy in mdx utrn-/- muscles[38], also points to the novelty of the effect with Galgt2. Indeed, Galgt2 overexpression is the only therapy shown to inhibit the development of muscle pathology in mdx utrn-/- muscles, which again sets it apart from integrin α7B [38]. The demonstration that Galgt2 does not require utrophin to be therapeutic suggests it should be considered as a distinct target for therapeutic intervention in DMD.
Acknowledgements
This grant was supported by grants from the Muscular Dystrophy Association and the National Institutes of Health (grants AR050202 and (AR049722) to PTM. Matthew Glass and Kumar Chandrasekharan provided additional technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- [2].Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- [3].Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- [4].De la Porte S, Morin S, Koenig J. Characteristics of skeletal muscle in mdx mutant mice. Int Rev Cytol. 1999;191:99–148. doi: 10.1016/s0074-7696(08)60158-8. [DOI] [PubMed] [Google Scholar]
- [5].Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- [6].Martin PT. Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology. 2003;13:55R–66R. doi: 10.1093/glycob/cwg076. [DOI] [PubMed] [Google Scholar]
- [7].Ohlendieck K, Ervasti JM, Matsumura K, Kahl SD, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- [8].Martin PT, Scott LJ, Porter BE, Sanes JR. Distinct structures and functions of related pre- and postsynaptic carbohydrates at the mammalian neuromuscular junction. Mol Cell Neurosci. 1999;13:105–118. doi: 10.1006/mcne.1999.0737. [DOI] [PubMed] [Google Scholar]
- [9].Smith PL, Lowe JB. Molecular cloning of a murine N-acetylgalactosamine transferase cDNA that determines expression of the T lymphocyte-specific CT oligosaccharide differentiation antigen. J Biol Chem. 1994;269:15162–15171. [PubMed] [Google Scholar]
- [10].Xia B, Hoyte K, Kammesheidt A, Deerinck T, Ellisman M, Martin PT. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol. 2002;242:58–73. doi: 10.1006/dbio.2001.0530. [DOI] [PubMed] [Google Scholar]
- [11].Deconinck N, Tinsley J, De Backer F, Fisher R, Kahn D, Phelps S, Davies K, Gillis JM. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3:1216–1221. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- [12].Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- [13].Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- [14].Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Muscat GE, Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol Cell Biol. 1987;7:4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jayasinha V, Hoyte K, Xia B, Martin PT. Overexpression of the CT GalNAc transferase inhibits muscular dystrophy in a cleavage-resistant dystroglycan mutant mouse. Biochem Biophys Res Commun. 2003;302:831. doi: 10.1016/s0006-291x(03)00271-7. [DOI] [PubMed] [Google Scholar]
- [17].Hoyte K, Jayasinha V, Xia B, Martin PT. Transgenic overexpression of dystroglycan does not inhibit muscular dystrophy in mdx mice. Am J Pathol. 2004;164:711. doi: 10.1016/S0002-9440(10)63158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jayasinha V, Nguyen HH, Xia B, Kammesheidt A, Hoyte K, Martin PT. Inhibition of dystroglycan cleavage causes muscular dystrophy in transgenic mice. Neuromuscul Disord. 2003;13:365–375. doi: 10.1016/s0960-8966(03)00040-3. [DOI] [PubMed] [Google Scholar]
- [20].Allbrook DB, Han MF, Hellmuth AE. Population of muscle satellite cells in relation to age and mitotic activity. Pathology. 1971;3:223–243. doi: 10.3109/00313027109073739. [DOI] [PubMed] [Google Scholar]
- [21].Cardasis CA, Cooper GW. An analysis of nuclear numbers in individual muscle fibers during differentiation and growth: a satellite cell-muscle fiber growth unit. J Exp Zool. 1975;191:347–358. doi: 10.1002/jez.1401910305. [DOI] [PubMed] [Google Scholar]
- [22].Schmalbruch H, Hellhammer U. The number of nuclei in adult rat muscles with special reference to satellite cells. Anat Rec. 1977;189:169–175. doi: 10.1002/ar.1091890204. [DOI] [PubMed] [Google Scholar]
- [23].Bischoff R. In: Satellite cells in Myology, Basic and Clinical. Engel AG, Franzini-Armstrong C, editors. McGraw-Hill; New York: 1994. [Google Scholar]
- [24].Martin PT. Glycobiology of the neuromuscular junction. J Neurocytol. 2003;32:915–929. doi: 10.1023/B:NEUR.0000020632.41508.83. [DOI] [PubMed] [Google Scholar]
- [25].Herzog RW, Hagstrom JN, Kung SH, Tai SJ, Wilson JM, Fisher KJ, High KA. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc Natl Acad Sci U S A. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Martin PT, Sanes JR. Role for a synapse-specific carbohydrate in agrin-induced clustering of acetylcholine receptors. Neuron. 1995;14:743–754. doi: 10.1016/0896-6273(95)90218-x. [DOI] [PubMed] [Google Scholar]
- [27].Scott LJ, Bacou F, Sanes JR. A synapse-specific carbohydrate at the neuromuscular junction: association with both acetylcholinesterase and a glycolipid. J Neurosci. 1988;8:932–944. doi: 10.1523/JNEUROSCI.08-03-00932.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sanes JR, Cheney JM. Lectin binding reveals a synapse-specific carbohydrate in skeletal muscle. Nature. 1982;300:646–647. doi: 10.1038/300646a0. [DOI] [PubMed] [Google Scholar]
- [29].Hoyte K, Kang C, Martin PT. Definition of pre- and postsynaptic forms of the CT carbohydrate antigen at the neuromuscular junction: ubiquitous expression of the CT antigens and the CT GalNAc transferase in mouse tissues. Brain Res Mol Brain Res. 2002;109:146–160. doi: 10.1016/s0169-328x(02)00551-x. [DOI] [PubMed] [Google Scholar]
- [30].Hirst RC, McCullagh KJ, Davies KE. Utrophin upregulation in Duchenne muscular dystrophy. Acta Myol. 2005;24:209–216. [PubMed] [Google Scholar]
- [31].Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- [32].Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- [34].Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- [35].Yamane A, Akutsu S, Diekwisch TG, Matsuda R. Satellite cells and utrophin are not directly correlated with the degree of skeletal muscle damage in mdx mice. Am J Physiol Cell Physiol. 2005;289:C42–C48. doi: 10.1152/ajpcell.00577.2004. [DOI] [PubMed] [Google Scholar]
- [36].Pons F, Robert A, Fabbrizio E, Hugon G, Califano JC, Fehrentz JA, Martinez J, Mornet D. Utrophin localization in normal and dystrophin-deficient heart. Circulation. 1994;90:369–374. doi: 10.1161/01.cir.90.1.369. [DOI] [PubMed] [Google Scholar]
- [37].Kawamura YI, Kawashima R, Fukunaga R, Hirai K, Toyama-Sorimachi N, Tokuhara M, Shimizu T, Dohi T. Introduction of Sd(a) carbohydrate antigen in gastrointestinal cancer cells eliminates selectin ligands and inhibits metastasis. Cancer Res. 2005;65:6220–6227. doi: 10.1158/0008-5472.CAN-05-0639. [DOI] [PubMed] [Google Scholar]
- [38].Burkin DJ, Wallace GQ, Nicol KJ, Kaufman DJ, Kaufman SJ. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]