

Abstract
Transforming growth factor beta (TGFβ) can modulate the activity of various MAP kinases. However, how this pathway may mediate TGFβ-induced malignant phenotypes remains elusive. We investigated the role of autocrine TGFβ signaling through MAP kinases in the regulation of cell survival in breast carcinoma MCF-7 cells and untransformed human mammary epithelial cells (HMECs). Our results show that abrogation of autocrine TGFβ signaling with the expression of a dominant negative type II TGFβ receptor (DNRII) or the treatment with a TGFβ type I receptor inhibitor significantly increased apoptosis in MCF-7 cell, but not in HMEC. The expression of DNRII markedly decreased activated/phosphorylated Erk, whereas increased activated/phosphorylated p38 in MCF-7 cells. In contrast, there was no or little change of phosphorylated Erk and p38 in HMECs after the expression of DNRII. Inhibition of Erk activity in MCF-7 control cell induced apoptosis whereas restoration of Erk activity in MCF-7 DNRII cell reduced apoptosis. Similarly, inhibition of p38 activity also inhibited apoptosis in MCF-7 DNRII cell. Thus, autocrine TGFβ signaling can enhance the survival of MCF-7 cells by maintaining the level of active Erk high and the level of active p38 low. Furthermore, the survival properties of TGFβ pathway appear related to transformation supporting the notion that it may be a potential target for cancer therapy.
Keywords: Autocrine TGF–beta, breast cancer, Apoptosis, MAP kinases
Introduction
Transforming growth factor beta (TGFβ) is a multifunctional polypeptide growth factor regulating a variety of biological activities including cell proliferation, cell migration, cell differentiation, immune cell behavior, and extracellular matrix remodeling [1]. TGFβ functions by binding to a heteromeric complex of transmembrane serine/threonine kinase receptors called type I (RI) and type II (RII) receptors [1]. Binding of TGFβ to RII recruits and transphosphorylates RI [2]. The activated RI phosphorylates intracellular Smad2 and Smad3, which then interact with Smad4 protein to form an oligomeric complex [3]. Once transported into nuclei, Smad2/Smad4 and Smad3/Smad4 complexes can act as transcriptional activator or repressor depending on cellular context [1].
TGFβ is also known to regulate the activation of Erk, p38, and JNK mitogen-activated protein kinase (MAPK) pathways in both Smad-dependent and Smad-independent manners [4]. Published studies have mostly focused on the effect of exogenous TGFβ-induced MAPK activation on gene transcription [5], cell death [6,7], and epithelial-mesenchymal transdifferentiation [8]. Few studies have addressed the regulation of MAPK pathways by autocrine TGFβ and the effect of autocrine TGFβ-modulated MAPK activities on cell survival.
TGFβ plays complex roles in carcinogenesis. It functions as a tumor suppressor in early epithelial carcinogenesis, but often becomes pro-oncogenic in late stages of tumor progression [9]. Early studies have shown that TGFβ is a potent inhibitor of cell cycle in various types of cells including epithelial cells [10]. Loss of TGFβ signaling components due to mutational inactivation of TGFβ receptors or Smads has been reported in gastrointestinal cancers as well as other types of carcinomas. For example, RII gene mutation occurs in both sporadic and hereditary colon cancer cells with microsatellite instability [11]. Inactivating mutations of Smad2 and Smad4 were reported in colorectal carcinomas and pancreatic carcinomas [12,13]. On the other hand, restoration of autocrine TGFβ signaling in colon carcinoma cells has been shown to inhibit their malignancy [14]. However, several studies showed that complete inactivation of TGFβ signaling through mutations of RII or Smad proteins is rare in many types of cancers including breast cancer [15,16]. In fact, blockade of TGFβ signaling with a dominant negative TGFβ receptor or Smad3 has been shown to inhibit malignant phenotypes in various mammary carcinogenesis models [17-20] suggesting that autocrine TGFβ signaling is required for breast cancer progression and may be a potential target for cancer therapy. We have previously shown that autocrine TGFβ contributes to the survival of human breast carcinoma MDA-MB-231 cells revealing a potential mechanism by which autocrine TGFβ maintains the malignancy of breast cancer cells [21].
In the current study, we used human breast cancer MCF-7 cells and telomerase-immortalized human mammary epithelial cells (HMEC) to investigate the effect of the modulation of MAP kinases by TGFβ on cell survival and to determine whether the survival-promoting activity of TGFβ is commonly operational in mammary epithelial cells. The proliferation of early passage MCF-7 cells is known to be inhibited by TGFβ. Interestingly, ectopic expression of a dominant negative TGFβ type II receptor (DNRII) in the cell showed no effect on the tumorigenicity and growth of the cell in vivo even though it abrogated the growth inhibitory activity of TGFβ [22]. This is in stark contrast with what has been observed in colon carcinoma cells that are growth-inhibited by TGFβ and became more tumorigenic when TGFβ signaling was blocked with ectopic expression of the DNRII [23]. We found that DNRII-expressing MCF-7 cells were significantly more apoptotic than the control cells in culture and that the increased apoptosis was mainly due to the alteration of MAP kinase signaling pathways. Thus, the loss of cell viability is likely to be the reason why the blockade of TGFβ-induced inhibition of cell proliferation did not lead to increased tumorigenicity in the MCF-7 cells. Interestingly, the survival promoting activity of TGFβ was not observed in untransformed HMECs suggesting that it may have evolved during mammary carcinogenesis.
Materials and methods
Cell Culture
Human breast cancer MCF-7 control cell line and dominant negative RII (DNRII) transfected cell line were kindly provided by Dr. Michael G. Brattain at Roswell Park Cancer Institute, Buffalo, N.Y. [22] and maintained in McCoy's 5A medium with 10% fetal bovine serum (FBS), pyruvate, vitamins, amino acids, and antibiotics. Human mammary epithelial cells (HMECs) were obtained from Cambrex (Maryland) and cultured in mammary epithelial cell growth medium (MEGM) plus MEGM Bullet kit from Cambrex. HMECs were immortalized with ectopic expression of human telomerase reverse transcriptase (hTERT) as reported [24]. Immortalized HMECs were infected with control pLPCX retrovirus vector (Clontech) or the pLPCX vector containing the DNRII cDNA that was used to generate MCF-7 DNRII cells. Working cultures were maintained at 37°C in a humidified incubator with 5% CO2.
Transient Transfection and Luciferase Assay
To determine the cell autocrine TGFβ activity, we measured a TGFβ-responsive promoter activity using p3TP-Lux [25] or pSBE4-Luc[26]. The promoter activity is reported by luciferase activity. MCF-7 cells and HMECs were plated in 12-well plates and cultured for one day. The p3TP-Lux or pSBE4-Luc plasmid (0.5 μg) and a β-galactosidase (β- gal) expression plasmid (0.1 μg) were transiently co-transfected into the cells using Fugene6 (Roche Molecular Biochemicals) according to the manufacturer's instruction. After 2 hr incubation, the cells were treated with or without TGFβ3 (2.0 ng/ml). The cells were lysed after an additional 24 hr incubation, and the activity of luciferase and β- galactosidase were assayed as described previously [27]. Luciferase activity was normalized to β-galactosidase activity and expressed as relative luciferase activity.
Cell Death Detection ELISA
DNA fragmentation during apoptosis was measured using the Cell Death Detection ELISA kit (Roche Molecular Biochemicals), which can quantify cytoplasmic histone-associated-DNA-fragments (mono- and oligonucleosomes) during apoptosis. The assay was carried out as previously described [21]. In some cases, cells were treated with the MEK inhibitor PD98059 or p38 MAP kinase inhibitor SB202190 (CalBiochem) prior to the ELISA as described in the figure legends.
Annexin V-FITC Staining
Apoptosis detection kit (Caltag Laboratories) including Annexin V-FITC and propidium iodide (PI) staining was used to identify cells at an early stage of apoptosis. The assay was performed as previously described [21].
Western Blot and Antibodies
Cell cultures were rinsed two times with ice-cold PBS and lysed in a cell lysis buffer (50mM Tris-HCl pH7.4, 150mM NaCl, 1%Nonidet P-40) containing protease and phosphatase inhibitors (Boreinger Mannheim EDTA protease inhibitors, 1mM NaVO3 and 1mM NaF). Equal amounts of protein were separated on SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Corp.). Blots were blocked in TBST [100mM Tris-HCl (PH 8.0), 150mM NaCl, 0.05% Tween-20] containing 5% non-fat powder milk. The membrane was then incubated with primary antibody for 1 hour at room temperature or overnight at 4°C. Antibodies used detected RII and DNRII (R&D Systems), phospho-Smad2 (Ser 465/467) (Upstate Biotechnology), total-Smad2/3 (BD Transduction Laboratories), PTEN (Oncogene), phospho-Erk, total-Erk, phospho-p38, total-p38 and phospho-JNK (Cell signaling Technology). After three washes with TBST, the membrane was incubated with HRP-linked secondary antibodies (1:3000 dilution, Santa Cruz) for 1 hr at room temperature and washed again. Bound Complexes were detected using chemiluminescence procedures according to the manufacturer's instruction (NEN Life Science Products).
MEK transfection and Annexin V-FITC Staining
MCF-7 control and DNRII cells were plated at 5×105 cells per 60-mm dish and cultured for one day. The cells were then co-transfected with a constitutively active MEK1 expression plasmid (3.0 μg) [28] and pDsRed1-N1 (0.3 μg) (Clontech). After 48 hr incubation, the cells were stained with Annexin-V-FITC and apoptotic cells that expressed the red fluorescent protein were analyzed with a flow cytometer.
Statistical analysis
Student t-tests were used to determine a significant difference between control and experimental data and one-way ANOVA was used followed with Newman-Keuls Multiple Comparison Test for the comparison of more than two means [29].
Results
Expression of DNRII attenuates TGFβ signaling in both MCF-7 and immortalized HMEC cells
To confirm the expression of DNRII protein after MCF-7 and HMEC cells were stably transfected with a DNRII expression construct, we performed Western blotting analysis to show the expression of DNRII (Fig. 1A). Since MCF-7 cells were transfected with a tetracycline-suppressible DNRII expression system, treatment with tetracycline almost completely blocked DNRII expression as shown in Fig. 1A. We previously showed that autocrine TGFβ loop in MCF-7 cells was operational and that DNRII expression significantly attenuated its activity [30]. Consistent with these observations, stable expression of the DNRII in the MCF-7 and HMEC cells reduced the basal levels of phosphorylated Smad2 and blocked TGFβ induction of Smad2 phosphorylation without affecting total Smad2 and 3 levels (Fig. 1B). The blockade of Smad phosphorylation also led to the inhibition of the transcriptional activity of Smad proteins as reported by the TGFβ-responsive promoter-luciferase construct, p3TP-Lux or pSBE4-Luc. The DNRII-transfected MCF-7 and HMEC cells had a significantly (P<0.05) lower basal as well as TGFβ-induced promoter activity than the control cells (Fig. 1C). Thus, the expression of DNRII antagonized both exogenous and endogenous/autocrine TGFβ activity in both MCF-7 and immortalized HMEC cells.
Fig. 1.
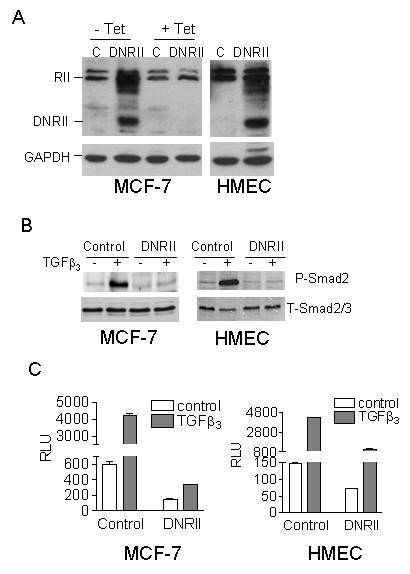
Blockade of TGFβ signaling in MCF-7 and HMEC cells by the expression of a TGFβ DNRII. (A) The expression of DNRII was detected with Western blotting using the lysate of control (C) and DNRII-transfected cells. Since the MCF-7 cells were transfected with a tetracycline (Tet)-suppressible DNRII expression system, they were treated with or without Tet at 1 μg/ml for 5 days before harvested for the Western blotting analysis. GAPDH was blotted to indicate equal sample loading. (B) Exponentially growing cultures of the control and DNRII-transfected MCF-7 and HMEC cells were treated with 2.0 ng/ml of TGFβ3 for 24 h. Cell lysates were collected afterward and Western Blotting was performed for phosphorylated Smad2 (P-Smad2) and total Smad2/3 (T-Smad2/3) as described in Materials and Methods. (C) Control and DNRII-transfected MCF-7 and HMEC cells were transiently co-transfected with a TGFβ-responsive promoter-luciferase construct (p3TP-Lux for MCF-7 cells or pSBE4-Luc for HMECs) and a β-gal expression construct. The transfected cells were treated with or without 2.0 ng/ml of TGFβ3. The activity of luciferase and β-gal in the cell lysates was measured 24h later. The β-gal-normalized luciferase activity (RLU) was plotted. The data represent the means ± SEM from triplicate transfections.
Abrogation of autocrine TGFβ signaling induces apoptosis in MCF-7 cells but not in immortalized HMEC cells
Previous studies showed that the expression of DNRII had no effect on the tumorigenicity of the MCF-7 cells in vivo [22]. The result was somewhat unexpected since the proliferation of the MCF-7 cells was inhibited by exogenous TGFβ [22] and ectopic expression of the wild-type RII in a late passage MCF-7 cell line with a low RII level reduced its tumorigenic potential in vivo [31]. To explain the apparent paradox, we examined the effect of DNRII expression on apoptosis in MCF-7 cells. As shown in Fig. 2A, DNRII expression induced apoptosis in MCF-7 cells by about 3-fold as detected with an apoptosis ELISA. Since the DNRII is expressed in a tetracycline-repressible expression system, we treated the control and DNRII cells with tetracycline to determine whether the inhibition of DNRII expression could restore cell viability. The treatment with tetracycline for 4 days caused a moderate, although not statistically significant, increase of apoptosis in both cells. In contrast, it significantly (P<0.05) inhibited the apoptosis in the DNRII cells to the level of the control cell indicating that the increased apoptosis in the DNRII cells was specifically due to the expression of DNRII (Fig. 2A). Interestingly, the expression of the DNRII in the immortalized HMECs did not induce apoptosis (Fig. 2A). Annexin V staining for apoptotic cells also produced similar results. The percentage of apoptotic MCF-7 control and DNRII cells was 3.50% and 12.04%, respectively (Fig. 2B). In contrast, the percentage of apoptotic HMEC control and DNRII cells was not very different, being 2.30% and 1.89% respectively (Fig. 2B). To ascertain that the apoptosis induced by the DNRII expression in MCF-7 was due to the attenuation of autocrine TGFβ signaling, we also investigated whether the apoptosis in MCF-7 cells could be induced with a TGFβ RI kinase inhibitor, which we have previously shown to specifically inhibit TGFβ signaling [32]. As shown in Fig. 2C, the treatment with RI kinase inhibitor produced a dose-dependent apoptosis in MCF-7 cells, but not in HMECs. These results indicate that blockade of autocrine TGFβ signaling can induce apoptosis in human breast cancer MCF-7 cells, but not in the untransformed human mammary epithelial cells.
Fig. 2.
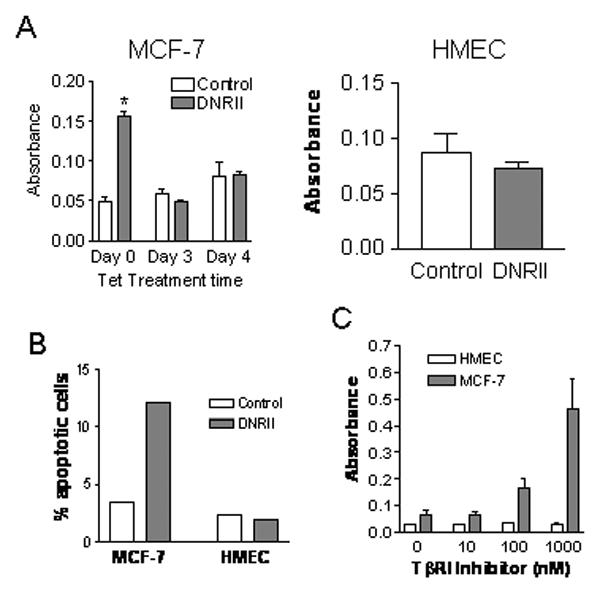
Effect of the blockade of autocrine TGFβ signaling on cell apoptosis. (A) MCF-7 control and DNRII cells were plated in 100-mm dishes at 500,000 cells per dish. Cells were treated with or without tetracycline (0.5 μg/ml), two days after plating, for the indicated time period. HMEC control or DNRII cells were plated at 250,000 cells per 60 mm dish and cultured for 7 days. Apoptosis was measured with Cell Death Detection ELISA using 30,000 cells from each well per manufacturer's instructions. The results plotted represent the means ± SEM from triplicate wells. The treatment with an asterisk “*” denotes a significant (P<0.05) difference from other treatments with one-way ANOVA followed with Newman-Keuls Multiple Comparison Test. (B) The control and DNRII cells of MCF-7 and HMEC were cultured in T-25 flasks and harvested at exponential growth phase. The cells were detached with a trypsin solution and stained with Annexin V-FITC and propidium iodide. The stained cells were analyzed with a flow cytometer. Percentage of apoptotic cells with positive staining by Annexin V-FITC and negative staining by PI is plotted. (C) Parental MCF-7 were plated at 0.5×106 cells per 60-mm dish in McCoy's 5A medium with 1% FBS. hTERT-immortalized HMEC cells were plated at 0.2×106 cells per 60-mm dish in the serum-free MEGM medium. The cells were treated with various concentrations of a TGFβ kinase inhibitor (Calbiochem) for 5 days after plating. The cells were then harvested for the apoptosis ELISA and the results plotted represent the means ± SEM from triplicate wells.
Alteration of MAP kinase pathways by the expression of DNRII in MCF-7 cells
TGFβ has been shown to activate various MAP kinase pathways including Erk, p38, and JNK pathways [33]. These pathways are known to regulate cell survival. In general, activation of Erk pathway is associated with cell survival, whereas activation of p38 and JNK can lead to cell death [34]. To elucidate the molecular mechanism of apoptosis induced by the blockade of autocrine TGFβ signaling, we examined the effect of DNRII expression on phosphorylation-associated activation of the three MAP kinases. As shown in Fig. 3A, the level of the phosphorylated Erk (p-Erk) was dramatically reduced whereas that of the phosphorylated p-38 (p-p38) was dramatically increased in MCF-7 DNRII cells in comparison to the control cells. This modulation of the phosphorylation of Erk and p38 was specifically due to the blockade of autocrine TGFβ signaling by the expression of DNRII because inhibition of DNRII expression with the treatment of tetracycline reversed the altered phosphorylation status of both Erk and p38. In contrast, the level of p-Erk was not changed and that of p-p38 was only moderated increased after stable expression of DNRII in HMECs (Fig. 3B). Although we observed a moderately lower levels of phosphorylated JNK1 and 2 in MCF-7 DNRII cells than in the control cells, the treatment with tetracycline did not increase the phosphorylation of JNK1 and 2 (Fig. 3A) suggesting that DNRII-induced apoptosis of MCF-7 cells was not mediated by JNK activation. PTEN is known to inhibit Akt phosphorylation and consequently promotes apoptosis in various cell systems. Its transcription was shown to be inhibited by TGFβ in human keratinocytes [35]. We have previously shown that autocrine TGFβ promotes the survival of human breast carcinoma MDA-MB-231 cells by suppressing PTEN expression [21]. Interestingly, blockade of autocrine TGFβ signaling did not affect PTEN in MCF-7 cells (Fig. 3A). Furthermore, the expression of DNRII showed little effect on the level of phosphorylated Akt. Thus, the apoptosis induced by the blockade of autocrine TGFβ signaling was apparently not mediated by PTEN/Akt pathway. We, therefore, focused our attention on Erk and p38 pathways.
Fig. 3.
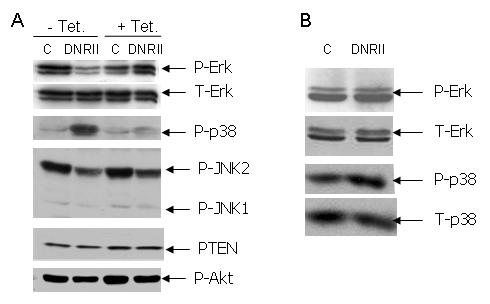
Effect of the blockade of autocrine TGFβ signaling by DNRII expression on the activation of MAP kinases and the level of PTEN. (A) MCF-7 control (C) and DNRII cells were treated with or without tetracycline at 1 μg/ml for 3-4 days and harvested at exponential growth phase for Western blotting as described in the Material and Methods. (B) Exponential growing HMEC control (C) and DNRII cells were harvested and Western blotting was performed as described in the Material and Methods. The phosphorylated MAP kinases are labeled with “p-“, whereas total MAP kinases are labeled with “T-“.
Erk activity is necessary for the survival of MCF-7 cells
To determine whether the reduction of p-Erk level was sufficient to induce apoptosis in MCF-7 cells, we treated the cells with PD98059 compound (Calbiochem) to specifically inhibit the activity of the Erk activator, MEK. As shown in Fig. 4A, the treatment with PD98059 blocked Erk phosphorylation. It also induced apoptosis in MCF-7 cells in a dose dependent manner (Fig. 4B). On the other hand, transient ectopic expression of a constitutively active MEK1 significantly increased the levels of p-Erk in MCF-7 control cells and restored the level of p-Erk in MCF-7 DNRII to that of the control cells (Fig. 5A). As a result, the percentage of apoptotic MCF-7 DNRII cells was significantly reduced after the transfection of the constitutively active MEK1 in comparison with the mock transfection (Fig. 5B). These data suggest that autocrine TGFβ signaling can enhance the survival of the MCF-7 cell by maintaining a high level of Erk signaling.
Fig. 4.
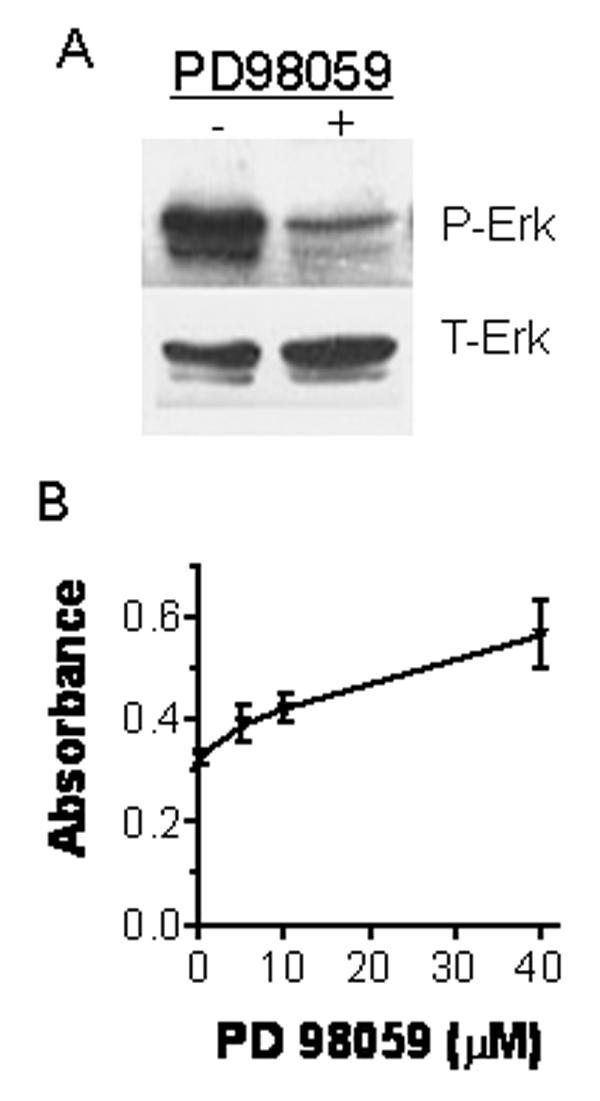
Induction of apoptosis in MCF-7 cells by the inhibition of Erk activity with PD98059 compound. (A) MCF-7 control cells were cultured in 60mm dishes until they were 80% confluent. The cells were treated without or with PD98059 at 40 μM for 24 hours. Cells were then harvested for Western blotting for phosphorylated Erk (p-Erk) and total Erk (T-Erk) as described in the Material and Methods. (B) MCF-7 control cells were cultured and treated with PD98059 at different concentrations in the same way as in panel A. Cell apoptosis was measured using Cell Death Detection ELISA. The results plotted represent the means ± SEM from triplicate measurements.
Fig. 5.
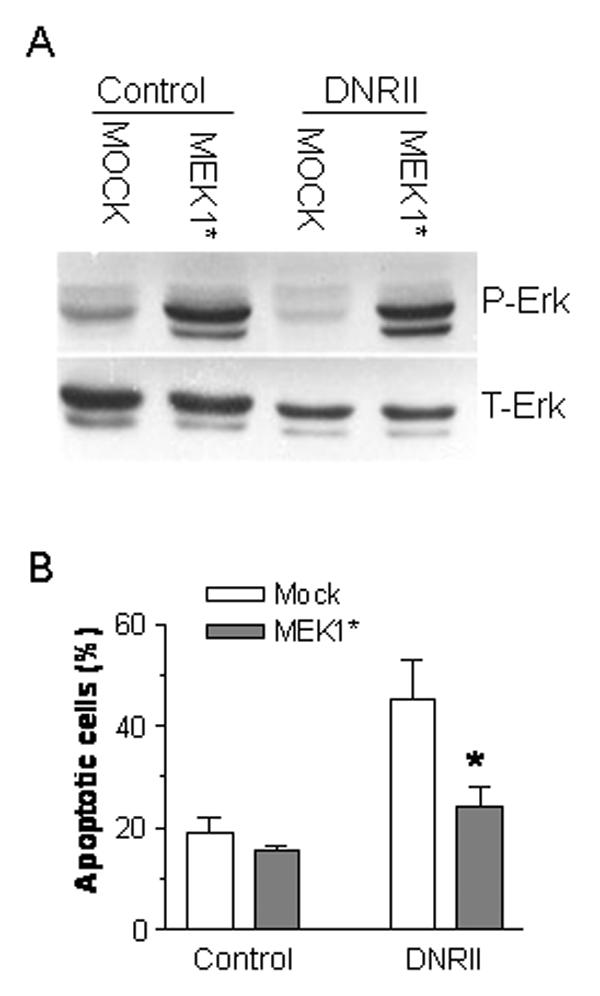
Inhibition of DNRII-induced apoptosis by the activation of Erk in MCF-7 cells. MCF-7 control and DNRII cells were plated at 500,000 cells per 60mm dish and cultured for 24 hr. The cells were co-transfected with a constitutively active MEK1 (MEK1*) expression plasmid and a red fluorescent protein expression vector pDsRed1-N1 (Clontech). After 48 hr, the cells were stained with Annexin V-FITC and the apoptotic cells stained with Annexin V-FITC and also expressing the red fluorescent protein were counted with a flow cytometer. The results plotted in Panel B represent the means ± SEM from three independent transfections. The cells recovered from flowcytometry were used for Western blotting for p-Erk and T-Erk as shown in Panel A.
Inhibition of p38 activity inhibits apoptosis in MCF-7 DNRII cells
Since the activation of p38 pathway is generally pro-apoptotic and the expression of DNRII in MCF-7 cells caused a dramatic increase of p-p38 level (Fig. 3A), we next examined whether inhibition of p38 activity could inhibit the apoptosis in MCF-7 DNRII cells. Treatment with a p38 inhibitor, SB202190 (Calbiochem), reduced the levels of p-p38 (Fig. 6A) and inhibited the apoptosis (Fig. 6B) in a dose-dependent manner in MCF-7 DNRII cells. Thus, autocrine TGFβ signaling appears to promote MCF-7 cell survival by increasing Erk activity as well as by decreasing p38 activity.
Fig. 6.
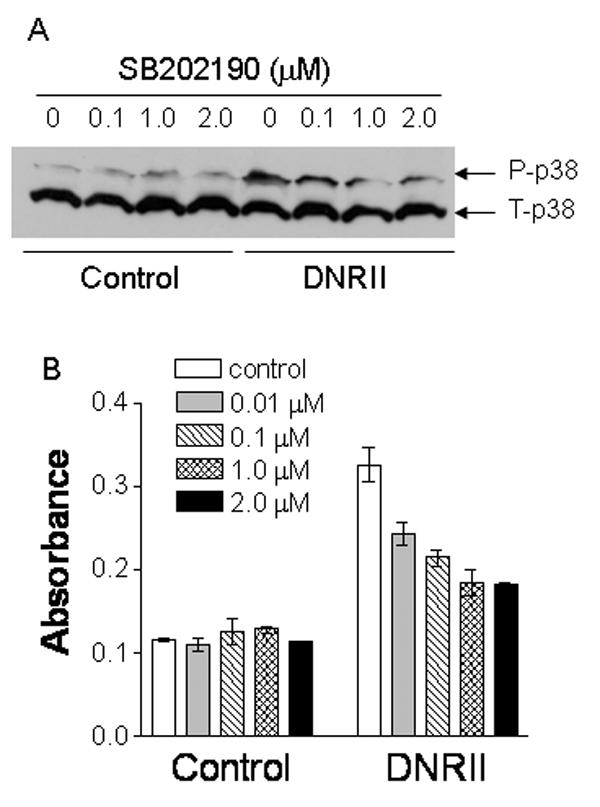
Inhibition of p38 activity by SB202190 suppresses the apoptosis in MCF-7 DNRII cells. (A) Exponential growing MCF-7 control and DNRII cells were treated with or without SB202190 at concentrations depicted for 24 hours. The cells were then harvested for Western blotting for phosphorylated and total p38 as described in the Material and Methods. (B) MCF-7 cells were cultured and treated in the same way as described in panel A. Cell apoptosis was measured using Cell Death Detection ELISA. The results plotted represent the average of two measurements with the error bars depicting the minimal and maximal values of the two measurements.
Discussion
Although loss of or reduced TGFβ growth inhibitory activity is common in many types of cancers including carcinomas, melanomas, lymphoid and myeloid malignancies, this is apparently due to the attenuation of TGFβ-mediated anti-mitogenic activity in many cases [9,36]. Genetic studies have shown that mutational inactivation of TGFβ signaling components is restricted to certain subsets of cancer. For example, Smad4 is frequently mutated or deleted in colon and pancreatic tumors, but not in other tumor types [37]. In breast cancer, mutations of TGFβ RII and Smad4 genes are uncommon [15,16]. Thus, TGFβ pathway appears to be necessary for tumorigenesis and tumor progression in various types of malignancy. Indeed, disruption of autocrine TGFβ signaling with the expression of a TGFβ DNRII in transformed mammary and colon epithelial cells significantly inhibited their invasive and metastatic potential [17,19]. Highly aggressive and metastatic U9 colon cancer cells were shown to use autocrine TGFβ1 to promote their growth and invasion [38].
While TGFβ in a tumor microenvironment is well known to act on various stromal cells to inhibit host immunosurveillance and promote angiogenesis and invasion, how TGFβ signaling in tumor cells to promote tumor progression is less well defined. One mechanism by which TGFβ may directly enhance the motility and invasiveness of tumor cells is through the induction of epithelial-to-mesenchymal transdifferentiation [39,40]. TGFβ is also known to stimulate the expression of various pro-oncogenic gene products. For example, TGFβ-induced parathyroid hormone-related protein and bone sialoprotein in mammary carcinoma cell line has been shown to promote their metastasis [41,42]. In the current study, we describe yet another mechanism by which autocrine TGFβ can enhance the malignant properties of tumor cells. Our study showed that abrogation of autocrine TGFβ signaling by the expression of a DNRII can induce the apoptosis in human breast cancer MCF-7 cells suggesting that TGFβ signaling might have evolved during the progression to support cell survival in this tumor model system.
MAPKs are serine-threonine protein kinases that are activated by diverse stimuli including cytokines, growth factors, neurotransmitters, hormones, cellular stress, and cell adherence. The three major MAPK family members are the extracellular signal-regulated kinases (Erks), p38, and c-Jun N-terminal kinases (JNKs) [43]. MAPKs have been shown to play a central role in mediating a diverse array of cellular signals. Activation of MAPK can exert anti- or pro-apoptotic effect that is important in the regulation of tumorigenesis and tumor progression. TGFβ has been shown to activate all three MAPK pathways in various cell types [33,44]. However, the activation of MAPKs was shown by the treatment with exogenous TGFβ during a relatively short period of time in the majority of published reports. Little is known on how steady-state autocrine TGFβ signaling or the loss of it affects the activity of the MAPKs and consequently cell survival. In our study, blockade of autocrine TGFβ signaling with the expression of the DNRII was found to down-regulate the level of active Erk, but up-regulate the level of active p38 in MCF-7 cells. Restoration of Erk activity by the ectopic expression of a constitutively active MEK1, an activator of Erk, in the MCF-7 DNRII cell significantly inhibited apoptosis, whereas inhibition of Erk activation by a MEK inhibitor induced apoptosis in a dose dependent manner in MCF-7 cells. On the other hand, inhibition of p38 activity by SB202190 also partially reversed the apoptosis in MCF-7 DNRII cells. Since MCF-7 mainly produces active TGFβ1 [27], our data indicate that the steady-state autocrine TGFβ1 signaling can enhance the survival of the MCF-7 cells by enhancing the signaling through the anti-apoptotic Erk pathway while suppressing the signaling through the proapoptotic p38 pathway. Since exogenous TGFβ can activate MAPKs via a Smad-independent pathway [44], it will be interesting to investigate whether the regulation of Erk and p38 activation by the autocrine TGFβ in the MCF-7 cell is also through a Smad-independent pathway in future studies.
In addition to the MAPK pathways, TGFβ has been shown to also stimulate the activity of PI3K/Akt pathway, which is required for the TGFβ-induced epithelial-tomesenchymal transdifferentiation [45]. Furthermore, treatment with exogenous TGFβ was shown to protect serum starvation-induced apoptosis through an Akt-dependent pathway [46]. We have previously reported that autocrine TGFβ can promote the survival of human breast cancer MDA-MB-231 cells by inhibiting the expression of PTEN, which antagonizes the activity of PI3K/Akt pathway [21]. Interestingly, this TGFβ-induced survival mechanism was not observed in MCF-7 cells since the abrogation of autocrine TGFβ signaling did not affect PTEN level (Fig. 3). Thus, while autocrine TGFβ signaling can promote the survival of various cancer cells, the molecular pathways that mediate its survival signal can differ among the model systems.
Autocrine TGFβ is known to suppress tumorigenesis and tumor progression in normal and early transformed epithelial cells by inhibiting cell division. Although exogenous TGFβ, usually at higher concentrations than what is required to inhibit cell division, has been shown to induce apoptosis in untransformed and transformed epithelial cells, little is known on how autocrine TGFβ may affect the survival of untransformed epithelial cells. Therefore, we investigated the effect of abrogation of autocrine TGFβ signaling on cell survival and MAPK activation in a telomerase-immortalized HMEC line. The results presented in this report show that HMECs possess an operational autocrine TGFβ pathway. However, blockade of the autocrine TGFβ signaling showed little effect on cell apoptosis, which is consistent with our observation that the abrogation of autocrine TGFβ signaling did not significantly alter MAPK activation in this model system. Thus, the survival promoting activity of autocrine TGFβ appears to be acquired during carcinogenesis. Currently, the molecular mechanisms by which TGFβ regulates MAPK activation are not well understood. Therefore, further studies are needed to elucidate the molecular pathways that are altered during carcinogenesis causing autocrine TGFβ to promote cell survival by modulating MAPK activation. Although our previous study has shown that TGFβ signaling in estrogen receptor negative breast cancer MDAMB-231 cells supported their survival [21], further studies are also needed to investigate whether the expression of estrogen receptor in MCF-7 cells, but not in HMECs, also contributes to the regulation of apoptosis by TGFβ. Nonetheless, our study suggests that TGFβ pathway may be targeted for the induction of apoptosis in breast cancer cells.
Supplementary Material
{kind=link}
Acknowledgements
This work was supported in part by NIH Grants CA75253 and CA79683 to L-Z. Sun. The authors thank Ms. Jennifer Nguyen for technical assistance in obtaining some of the results. The authors also thank Dr. Andrew Hinck (University of Texas Health Science Center at San Antonio) for the recombinant TGFβ3, Dr. Michael G. Brattain (Roswell Park Cancer Center, Buffalo, N.Y.) for the MCF-7 cell lines, Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center, New York, NY) for the p3TP-Lux plasmid, Dr. Bert Vogelstein (Johns Hopkins Oncology Center, Baltimore, Maryland) for the pSBE4-Luc plasmid, and Dr. Kun-Liang Guan (University of Michigan Medical School, Ann Arbor, MI) for the constitutively active MEK1 plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 2.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 3.Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu.Rev.Cell Dev.Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 4.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 5.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nature Cell Biology. 2001;3:708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- 7.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr., Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. Journal of Biological Chemistry. 2003;278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 8.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc.Natl.Acad.Sci.U.S.A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyons RM, Moses HL. Transforming growth factors and the regulation of cell proliferation. Eur.J.Biochem. 1990;187:467–473. doi: 10.1111/j.1432-1033.1990.tb15327.x. [DOI] [PubMed] [Google Scholar]
- 11.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain MG, Willson JKV. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 12.Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 13.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Sun L, Myeroff L, Wang X, Gentry LE, Yang J, Liang J, Zborowska E, Markowitz S, Willson JK, Brattain MG. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J.Biol.Chem. 1995;270:22044–22049. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- 15.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA, Jr., Meltzer PS, Hahn SA, Kern SE. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- 16.Tomita S, Deguchi S, Miyaguni T, Muto Y, Tamamoto T, Toda T. Analyses of microsatellite instability and the transforming growth factor-beta receptor type II gene mutation in sporadic breast cancer and their correlation with clinicopathological features. Breast Cancer Res.Treat. 1999;53:33–39. doi: 10.1023/a:1006167210269. [DOI] [PubMed] [Google Scholar]
- 17.Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr.Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 18.McEarchern JA, Kobie JJ, Mack V, Wu RS, Meade-Tollin L, Arteaga CL, Dumont N, Besselsen D, Seftor E, Hendrix MJ, Katsanis E, Akporiaye ET. Invasion and metastasis of a mammary tumor involves TGF-beta signaling. Int.J.Cancer. 2001;91:76–82. doi: 10.1002/1097-0215(20010101)91:1<76::aid-ijc1012>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 19.Tang BW, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. Journal of Clinical Investigation. 2003;112:1116–1124. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian F, DaCosta BS, Parks WT, Yoo S, Felici A, Tang B, Piek E, Wakefield LM, Roberts AB. Reduction in smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63:8284–8292. [PubMed] [Google Scholar]
- 21.Lei XF, Bandyopadhyay A, Le T, Sun LZ. Autocrine TGF beta supports growth and survival of human breast cancer MDA-MB-231 cells. Oncogene. 2002;21:7514–7523. doi: 10.1038/sj.onc.1205966. [DOI] [PubMed] [Google Scholar]
- 22.Ko Y, Koli KM, Banerji SS, Li W, Zborowska E, Willson JKV, Brattain MG, Arteaga CL. A kinase-defective transforming growth factor-β receptor type II is a dominant-negative regulator for human breast carcinoma MCF-7 cells. Int.J.Oncol. 1998;12:87–94. doi: 10.3892/ijo.12.1.87. [DOI] [PubMed] [Google Scholar]
- 23.Ye SC, Foster JM, Li WH, Liang JR, Zborowska E, Venkateswarlu S, Gong JG, Brattain MG, Willson JV. Contextual effects of transforming growth factor beta on the tumorigenicity of human colon carcinoma cells. Cancer Res. 1999;59:4725–4731. [PubMed] [Google Scholar]
- 24.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes & Development. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 26.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol.Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Wang XF, Sun LZ. Expression of transforming growth factor beta type III receptor restores autocrine TGF beta1 activity in human breast cancer MCF-7 cells. J.Biol.Chem. 1997;272:12862–12867. doi: 10.1074/jbc.272.19.12862. [DOI] [PubMed] [Google Scholar]
- 28.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 29.Zar JH. In: Biostatistical Analysis. Zar JH, editor. Prentice-Hall, Inc.; Englewood Cliffs, N.J.: 1984. [Google Scholar]
- 30.Yang H, Kyo S, Takatura M, Sun L. Autocrine transforming growth factor beta suppresses telomerase activity and transcription of human telomerase reverse transcriptase in human cancer cells. Cell Growth Differ. 2001;12:119–127. [PubMed] [Google Scholar]
- 31.Sun L, Wu G, Willson JK, Zborowska E, Yang J, Rajkarunanayake I, Wang J, Gentry LE, Wang XF, Brattain MG. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J.Biol.Chem. 1994;269:26449–26455. [PubMed] [Google Scholar]
- 32.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, Cornell JE, Pollock BH, Mundy GR, Sun LZ. Inhibition of Pulmonary and Skeletal Metastasis by a Transforming Growth Factor-{beta} Type I Receptor Kinase Inhibitor. Cancer Res. 2006;66:6714–6721. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 33.Yue J, Mulder KM. Activation of the mitogen-activated protein kinase pathway by transforming growth factor-beta. Methods Mol.Biol. 2000;142:125–131. doi: 10.1385/1-59259-053-5:125. [DOI] [PubMed] [Google Scholar]
- 34.Cross TG, Scheel-Toellner D, Henriquez NV, Deacon E, Salmon M, Lord JM. Serine/threonine protein kinases and apoptosis. Exp.Cell Res. 2000;256:34–41. doi: 10.1006/excr.2000.4836. [DOI] [PubMed] [Google Scholar]
- 35.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 36.Sun L. Tumor-suppressive and promoting function of transforming growth factor beta. Front Biosci. 2004;9:1925–1935. doi: 10.2741/1382. [DOI] [PubMed] [Google Scholar]
- 37.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2005;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Hsu S, Huang F, Hafez M, Winawer S, Friedman E. Colon carcinoma cells switch their response to transforming growth factor beta 1 with tumor progression. Cell Growth Differ. 1994;5:267–275. [PubMed] [Google Scholar]
- 39.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nature Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 40.Dumont N, Arteaga CL. Targeting the TGF beta signaling network in human neoplasia. Cancer Cell. 2003;3:531–536. doi: 10.1016/s1535-6108(03)00135-1. [DOI] [PubMed] [Google Scholar]
- 41.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massague J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J.Clin.Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nam JS, Suchar AM, Kang MJ, Stuelten CH, Tang B, Michalowska AM, Fisher LW, Fedarko NS, Jain A, Pinkas J, Lonning S, Wakefield LM. Bone sialoprotein mediates the tumor cell-targeted prometastatic activity of transforming growth factor beta in a mouse model of breast cancer. Cancer Res. 2006;66:6327–6335. doi: 10.1158/0008-5472.CAN-06-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 44.Moustakas A, Heldin CH. Non-Smad TGF-{beta} signals. J.Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 45.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J.Biol.Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 46.Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol.Biol.Cell. 2001;12:3328–3339. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.