

Abstract
Background:
Immunoglobulin E (IgE)-mediated responses contribute to allergy and asthma. Little is understood regarding the relationship of tissue IgE to systemic IgE, inflammation or clinical outcomes.
Objectives:
To evaluate local IgE expression and cellular inflammation in proximal and distal lung of normal subjects and subjects with asthma of varying severity and relate those tissue parameters to systemic IgE levels, atopy, lung function and history of severe exacerbations of asthma.
Methods:
Tissue from over 90 subjects with eosinophilic (SAeo+) and non-eosinophilic (SAeo−) severe asthma, mild asthma and normal subjects were immunostained for IgE, signal-amplifying isoform of IgE receptor (FcεRIβ) and markers of mast cells, eosinophils and lymphocytes. Tissue expression of IgE, FcεRIβ, cellular inflammation, serum IgE and atopy were compared. Regression models were used to determine the relationship of local and systemic IgE to lung function and severe exacerbations of asthma.
Results:
Mast cell-bound IgE was present along airways, but absent in lung parenchyma. While the groups were similar in systemic/serum IgE and atopy, local/tissue IgE was highest in SAeo+ and correlated with eosinophils and lymphocytes (rs=0.52; p<0.0001 and rs=0.23; p=0.03, respectively). Higher local IgE was associated with better lung function, but also with more severe exacerbations of asthma.
Conclusion:
Local IgE appears to be primarily a component of responses within the mucosal immune compartment and is related to cellular inflammation, lung function and clinical outcomes in asthma.
Clinical Implications:
Local/airway IgE-related processes rather than systemic markers of atopy may be relevant in determining clinical outcomes in asthma.
Capsule Summary:
The study reports mucosal distribution of mast cell-bound IgE in human lung and suggests that local IgE and related responses rather than systemic/serum IgE and atopy are more relevant in determining clinical outcomes in asthma.
Keywords: Immunoglobulin E, mucosal immune response, asthma exacerbation, mast cells, eosinophils, lymphocytes
INTRODUCTION
Immunoglobulin E (IgE) has been associated with allergic diseases and asthma. Recent studies of the effects of anti-IgE therapy have confirmed that it plays a role in these diseases1-5. Despite this, questions regarding the role of IgE in immune processes related to asthma and other diseases remain. For instance, the majority of investigations related to allergy in humans have focused on systemic markers, i.e. serum IgE and hypersensitivity reactions in the skin. Yet, how these systemic markers relate to local/mucosal IgE in asthma or in other diseases is unclear. While it is appreciated that mucosal IgE can be induced and produced locally as a component of specific mucosal responses in both subjects with and without systemic markers of atopy, it is unknown whether systemic IgE could modify or even amplify tissue IgE expression and IgE binding to mast cells6-10.
The studies addressing local IgE in nasal mucosa from patients with allergic and idiopathic rhinitis reported that IgE+ cells were increased in subjects with either allergic or idiopathic rhinitis as compared to normal non-atopic subjects6-10. Similar studies of proximal airways in asthma reported that there were more B cells with IgE-related mRNA transcripts and greater numbers of high-affinity IgE receptor (FcεRI)-bearing cells in proximal airways of atopic and non-atopic asthma subjects and in atopic controls than in nonatopic controls11, 12.
While the expression of IgE (protein) in nasal mucosa was reported to associate with increased eosinophils, expression of IgE in lung or its relationship to asthma severity or cellular inflammation has not been studied10. Eosinophilia in asthma has long been associated with symptoms, asthma exacerbations and, less consistently, with lung function13-15. Anti-IgE therapy reportedly reduces tissue eosinophilia and frequency of asthma exacerbations, suggesting that there may be a relationship between IgE, tissue eosinophils and asthma exacerbations1-4. However, lung function in asthma was marginally affected by anti-IgE therapy, suggesting that further studies to understand the relationships of local and systemic IgE to airway inflammation and lung function in asthma are needed. Therefore, the local expression of IgE and the signal-amplifying tetrameric isoform of FcεRI in various lung regions (both not hitherto reported) were evaluated and compared to systemic/serum IgE and skin reactivity to aeroallergens. Both local and systemic measures of IgE were then compared to the distribution of airway inflammatory cells (lymphocytes, eosinophils and mast cells), the history of severe exacerbations of asthma and to lung function. It was hypothesized that the local IgE expression in the lung will vary by asthma severity and be associated with an “asthma-like” inflammatory cell environment and that the association of local IgE expression with lung function and severe exacerbations of asthma will be stronger than that seen with systemic/serum IgE.
Some of the results of these studies have been previously reported in abstract form16.
METHODS
Subjects
Subjects' asthma severity was determined using previously published criteria14. Severe asthma subjects were further sub-grouped into those with tissue eosinophilia (SAeo+; >22 eosinophils/mm2 of tissue, equivalent to ≥ 2 × standard deviation from the mean value measured in normal controls) or without (SAeo−), as previously reported14. The details are in the online repository (OLR).
This study also included 5 healthy organ donors' lung samples. The lungs, which were procured but not used as transplants, were obtained through NDRI (National Disease Research Interchange, Philadelphia, PA), processed and analyzed similar to other tissue samples. The study was approved by the National Jewish Institutional Review Board and all subjects gave informed consent.
Atopic status, lung function, rhinitis symptoms and asthma exacerbation data
Subjects underwent allergen skin testing and lung function measurements according to standard protocols (details in OLR). The history of seasonal or allergic rhinitis symptoms (AR; yes/no) and the history of severe exacerbations of asthma were collected by questionnaire. The history of severe exacerbation of asthma was defined as any previous admittance to an intensive care unit (ICU) due to an asthma attack or the requirement for assisted ventilation due to an asthma attack.
Sample collection, immunostaining and tissue analysis
Bronchoscopy and tissue processing were performed as previously described14. Serum samples for IgE measurements were collected and stored at −80°C.
Tissue sections were immunostained for IgE, β-chain of FcεRI, mast cell tryptase, CD3 (lymphocytes) and eosinophil major basic protein using peroxydase-based detection system (details in OLR).
Inflammatory cell counts were determined per area of tissue in proximal and distal airway regions and alveolar tissue as previously described (details in OLR)17.
Statistical Analysis
The nonparametric Kruskal-Wallis test was used to compare severity groups. When an overall significant difference was detected for a particular variable (p<0.05), pairwise group tests were performed using Wilcoxon Rank-Sum tests. Serum IgE, normalized by log transformation, was analyzed by ANOVA. Categorical variables (sex, atopy) were compared by Pearson's chi-squared test. Spearman's correlations (rs) were used to measure the strength of association between 88 pairs of variables; the false discovery rate (FDR) procedure was employed to maintain the false discovery rate at 5%18. Only the correlations that remained significant after the procedure were reported.
Standard multiple regression was used to model pulmonary function parameters as outcomes and IgE level (measured as 3 different variables and considered as separate models: IgE+ cell number, percentage and IgE serum concentration), mast cells, lymphocytes, eosinophils and asthma severity as predictors19. The slope of the IgE variable, indicating the average change in pulmonary function per unit increase in IgE, was of primary interest.
In addition to pulmonary function, two exacerbation outcomes (ever required assisted ventilation and ever stayed in ICU) were evaluated using logistic regression. Predictors used in these models were IgE level (the same 3 variables considered as for pulmonary function, in separate models), mast cells, lymphocytes, eosinophils and FEV1. Odds ratios (OR) for exacerbations were determined and reported in relationship to increases in IgE. In all models, insignificant variables (p>0.2) other than IgE were removed using backward selection.
RESULTS
Subjects
Forty-seven subjects with severe asthma [21 SAeo+ (81% atopic) and 26 SAeo− (75% atopic)], 20 subjects with mild asthma (MA, 85% atopic) and 23 normal subjects (NC, 77% atopic) were evaluated (Table E1 in OLR, including data on asthma exacerbations). The atopic status among the four groups was similar (p=0.87). Fourteen SAeo+ and 19 SAeo− were on oral corticosteroids alone or in addition to high dose inhaled corticosteroids, while 7 subjects in each group were on high dose inhaled corticosteroids only.
The lung transplant tissue specimens were from healthy organ donors, without a history of smoking, who died of causes unrelated to lung and whose organs were procured within 24h of mechanical ventilation. The subjects in this group were significantly older than the subjects in other groups (age range 50–78 years; overall p<0.0001).
Serum IgE, atopy, rhinitis symptoms and asthma
When all four subject groups were analyzed, similar to atopic status, there was no difference in mean serum IgE levels (p=0.61; Figure E1 in OLR) or in percent of subjects with elevated (>100 IU/ml) serum IgE levels (p=0.68). This lack of difference was not unexpected as normal subjects were included in this study regardless of their atopic status and/or AR. Although the association between AR and atopy or elevated serum IgE was marginal (p=0.04 and p=0.06, respectively), it was still substantially stronger than between asthma and those markers (p=0.79 and p=0.27, respectively).
Expression of IgE and FcεRIβ in proximal airways and relation to inflammatory cells
The cell-associated expression of IgE and FcεRIβ was evaluated in proximal airways of subjects from all groups. A similar analysis was performed in transbronchial biopsy tissue samples from the subset of 16 severe asthmatics and 4 mild asthmatic subjects who, along with endobronchial biopsy, underwent concomitant transbronchial biopsy. This subset of subjects was not significantly different in age, gender or therapy from others in the respective groups (data not shown). Small airway tissue was identified in 11 SA (5 SAeo+ and 6 SAeo−), while peripheral alveolar tissue was present in samples from 6 severe (1 of whom also had small airway tissue) and 4 mild asthmatic subjects. Proximal and distal airway regions and peripheral alveolar tissue were also analyzed in lung tissue samples from the 5 NC organ donors.
Positive immunostaining for IgE and FcεRIβ was observed as membrane staining on cells identified as mast cells (co-localized with tryptase staining; Figure 1). Not all mast cells stained positive for IgE and FcεRIβ, nor were both markers always expressed on the same mast cell. Thus, the results for tissue IgE and FcεRIβ expression were also analyzed as the percentage of the mast cell population with positive staining for IgE or FcεRIβ (%MCIgE+ and %MCFcεRIβ+). The distribution of IgE+ mast cells (range 0–71/mm2) differed among the 4 groups (overall p=0.01; Figure 3). Despite the general decrease in total mast cell counts in severe asthma (see Figure 2), SAeo+ had the highest absolute number of IgE+ mast cells among the groups, which was significantly higher than in SAeo− (p=0.009), but similar to MA and NC (p=0.11 and p=0.13, respectively). Additionally, the %MCIgE+ was highest in SAeo+ (overall p=0.008; compared to SAeo− p=0.002; MA p=0.03; NC p=0.03).
Figure 1.
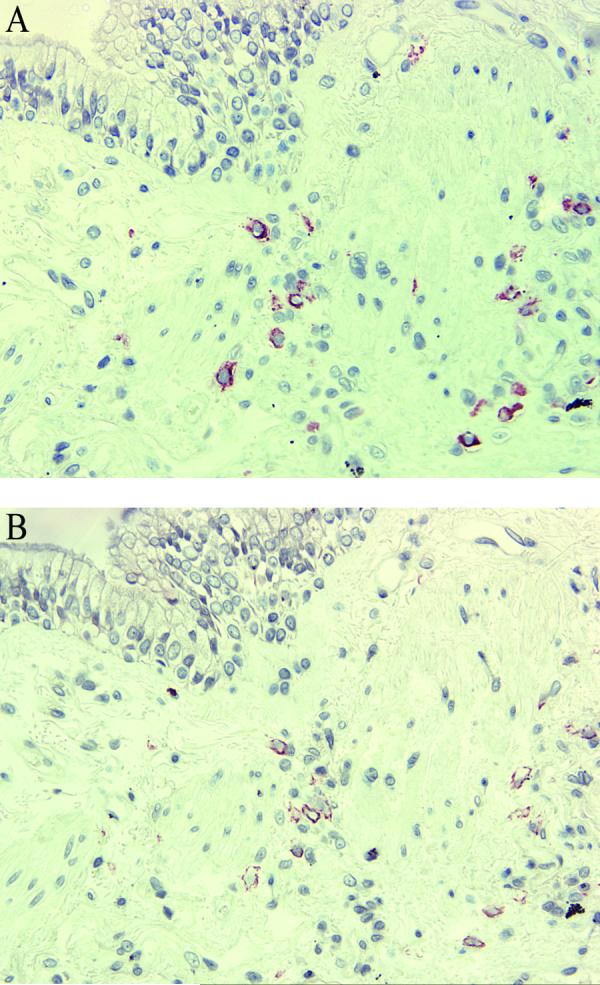
Immunostaining for mast cell tryptase and IgE in consecutive tissue sections. Tryptase-positive mast cells in small airway inner and outer wall (A) co-localize with IgE+ cells (B). Original magnification: X200.
Figure 3.

Percentages of mast cells expressing IgE in proximal airway (inner wall) and distal airway regions (inner and outer wall and alveolar attachments) of 11 subjects with severe asthma. Lines connect data within subjects. Capital letters identify subjects as atopic (A) or not (NA), while numbers indicate the serum IgE (kU/l).
Figure 2.

IgE expression and inflammatory cell counts (eosinophils, mast cells and lymphocytes) in proximal airways of subjects with severe asthma with or without persistent eosinophilia (SAeo+ and SAeo−, respectively), mild asthma and in normal subjects. Each column presents median (rectangular bar) and interquartile range (span of “error” bars). Horizontal lines connect groups that are significantly different (p≤0.05).
In contrast, the groups were similar in both numbers of FcεRIβ+ cells [overall p=0.12; SAeo+ median (IQR) = 0/mm2(0–15); SAeo− 0/mm2(0–0); MA 2/mm2(0–10); NC 0//mm2(0–3)] and in the %MCFcεRIβ+ (overall p=0.2; data not shown). Although staining for FcεRIβ could not always be co-localized with IgE+ cells, IgE+ and FcεRIβ+ cells, as well as the %MCIgE+ and %MCFcεRIβ+ were correlated (rs=0.56; p<0.0001 and rs=0.54; p<0.0001, respectively).
The number of IgE+ cells and %MCIgE+ correlated with eosinophils (rs=0.50; p<0.0001 and rs=0.52; p<0.0001, respectively). That relationship was not secondary to a relationship between mast cells and eosinophils (rs=0.11; p=0.31). FcεRIβ+ cells and %MCFcεRIβ+ also correlated with eosinophils (rs=0.34; p=0.007 and rs=0.34; p=0.008, respectively).
Although lymphocyte numbers were also higher in SAeo+ than in all other groups (overall p=0.003, p<0.05 for each intergroup comparison), they correlated to a lesser degree with IgE+ cells and %MCIgE+ (rs=0.37; p=0.0004 and rs=0.23; p=0.03, respectively) than did eosinophils. The dose of oral steroids did not significantly correlate with the number of IgE+ cells, %MCIgE+ or serum IgE (rs=−0.07; p=0.52, rs=−0.008; p=0.94 and rs=0.12; p=0.32, respectively)). Similarly, there was no significant correlation with the number of lymphocytes or eosinophils rs=0.15; p=0.15 and rs=0.14; p=0.20, respectively). In contrast, oral steroid dose negatively correlated with the number of mast cells (rs=−0.48; p<0.0001).
Expression of IgE in distal lung
IgE+ cells were detected in small airways of 5 out of 7 severe asthmatics with IgE+ cells in proximal airways (Figure 3). Four severe asthmatics with no proximal IgE+ cells had none in distal airway regions. Similarly, among the 5 NC organ donors (4 of 5 whom had no IgE in proximal airway), none was detected in distal airways (data not shown). IgE+ cells in peripheral alveolar tissue were not found in any subject, regardless of whether IgE+ cells were present in proximal airways (data not shown).
There were no significant relationships between IgE expression and eosinophils or lymphocytes in distal airways, but the small numbers limit meaningful comparisons (data not shown).
Relationship between local IgE and serum IgE
Statistical analysis indicated a positive relationship between local/airway IgE expression and serum IgE levels. Both the number of IgE+ cells and %MCIgE+ in proximal airways correlated with serum IgE (rs=0.39; p=0.0005 and rs=0.41; p=0.0002, respectively; Figure 4). The %MCIgE+ and serum IgE were also correlated in distal airways (rs=0.72; p=0.01), while there were no significant correlations in peripheral alveolar tissue where there were no IgE+ cells. Serum IgE did not correlate with tissue lymphocytes, eosinophils or mast cells (rs≤0.13; p≥0.25).
Figure 4.

Correlation between serum IgE and percentages of mast cells expressing IgE in proximal airways (all subjects included). Vertical dotted line marks normal serum IgE. Atopic subjects (positive skin reaction to aeroallergens) are marked with hollow circles. Nonatopic subjects (filled circles) that had mast cell-bound IgE in proximal airways are identified as severe asthmatics (SA) or normal controls (NC).
Relationship of local IgE and serum IgE with lung function and severe exacerbations of asthma
Regression models, adjusted for asthma severity and evaluating lung function measures as outcomes and proximal airway IgE and serum IgE as predictors, demonstrated that FEV1 was positively predicted by %MCIgE+. A 10% increase in %MCIgE+ was associated with an average increase in predicted FEV1 of 2% (p=0.008). Residual volume (RV % predicted) was negatively predicted by %MCIgE+ and IgE+ cell counts. A 10% increase in %MCIgE+ was associated with an 8% decrease in residual volume, while a 10-cell increase in IgE+ cells was associated with a 15% decrease (both p=0.03). Serum IgE was marginally associated with FEV1 (p=0.05) and insignificantly with RV (p=0.76). Figure 5 demonstrates the relationships between FEV1 and %MCIgE+ and between RV and %MCIgE+. Separate estimates were derived for mild and severe asthma subjects by including interaction terms in the models. Although the interactions were not significant (p=0.12 for FEV1 and p=0.70 for RV), the graphs indicate that the observed significant relationships between lung function and %MCIgE+ were driven more by relationships in severe asthma than in mild asthma.
Figure 5.

Regression models evaluating lung function parameters FEV1 and RV as outcomes, and the percentage of mast cells with membrane-bound IgE (%MCIgE+) in proximal airway submucosa as a predictor. Separate estimates were derived for subjects with severe asthma (filled circles and full line) and mild asthma (hollow circles and dotted line). The interactions in either model were not significant (p=0.12 and p=0.70, respectively).
In contrast to lung function, there was a positive relationship between a history of asthma-related assisted ventilation and both the number of IgE+ cells and %MCIgE+ (OR=2 for a 10-cell increase in IgE+ cells, p=0.07; OR=1.2 for 5% increase in %MCIgE+, p=0.08), controlling for mast cell and lymphocyte counts. However, there was no relationship with serum IgE (OR=1.00, p=0.83).
DISCUSSION
This study is the first to present a comprehensive evaluation of local IgE expression throughout the human lung in tissue samples from over 90 subjects with and without asthma, as well as the first evaluation of tissue expression of the tetrameric/signal-amplifying isoform of the high affinity IgE receptor. The results demonstrate preferential expression of IgE in the airway submucosa as compared to the lung parenchyma and only a limited relationship of local/mucosal IgE expression to systemic IgE levels. Further, a stronger relationship of local/mucosal IgE than of serum/systemic IgE existed with eosinonophilic and lymphocytic tissue inflammation, lung function and severe exacerbations of asthma.
Although the four subject groups were similar regarding systemic IgE and atopy (i.e. total serum IgE and skin reactivity to aeroallergens), they differed in local/airway mucosa IgE expression. The local IgE-related markers (mast cell-bound IgE, FcεRIβ) were present in proximal airways of both normal subjects and subjects with asthma. However, local IgE expression was highest in the airways of severe asthma subjects with persistent eosinophilia and was correlated with tissue eosinophils.
While the degree of tissue IgE expression differed among the groups and was associated with the inflammatory process, the general pattern of IgE expression was similar in lungs of normal and asthmatic subjects. Mast cell-bound IgE was primarily present in proximal airways. A subset of subjects with proximal airway IgE also had IgE detectable in small airways, but no subjects had IgE expression limited to the small airways. In addition, no subjects, regardless of proximal airway or serum IgE or atopic status, had IgE detectable in the lung parenchyma, despite the presence of mast cells. This pattern suggests that IgE is distributed along mucosal surfaces, extending from proximal toward distal airways, but not into the lung parenchyma (away from mucosal surfaces). As no mast cells in the parenchyma (even in atopic subjects with increased serum IgE) demonstrated IgE binding, the presence of elevated serum IgE may be insufficient to induce high IgE binding to these mast cells. Thus, the IgE process along mucosal surfaces appears not to be influenced by systemic IgE but rather induced and regulated by local mucosal responses to environmental antigens. Indeed, the larger size (similar to IgM, i.e. larger than IgG or IgA) and heavy glycosylation of the molecule may limit IgE diffusion in tissue. That may serve to keep the IgE in sites where it is produced - in and along mucosal surfaces. The very low concentrations of IgE in serum (∼1/104 of IgG; 0.004% of serum immunoglobulins) also suggest that immune surveillance by IgE occurs primarily in tissue20. Restricted diffusion may lead to increased concentrations of locally produced IgE, which along with the high affinity of the FcεRI for IgE, may be necessary to ensure high binding of IgE to mast cells. This may also prolong the IgE half-life in tissue as compared to serum (∼14 days vs. 3 days, respectively) and thus provide long-lasting IgE-mediated responses within mucosa. There is evidence that receptors for IgG and IgE compete for the γ chain dimer, which is a common component of both immunoglobulins' receptors and whose availability in mast cells is limited21. As IgG easily diffuses in tissue, tissue IgG concentrations may be comparable to serum concentrations. Thus, unless local/tissue IgE concentrations are several orders higher than normally seen in serum, the disparity between IgG and IgE concentrations in tissue may result in relatively low IgE binding to mast cells even when accounting for the difference in affinity of respective receptors, perhaps explaining the lack of positive staining for IgE on parenchymal mast cells20, 22. Whether this leads to differences in regulation of local versus systemic reactions remains to be evaluated, but will be difficult to study in humans23.
The proposal that these mucosal processes are separate from systemic IgE is consistent with the concept of separate mucosal and systemic immune compartments, investigated in animal models, but still difficult to address in humans24-27. The mucosal IgE-mediated process, as this and other studies reported, can exist without systemic manifestations of atopy and increased serum IgE. Powe and colleagues reported specific binding of grass pollen allergen to IgE-expressing cells in sections of nasal mucosa from nonatopic subjects with idiopathic rhinitis. They went on to suggest “entopy” as the term describing an allergic disease limited to the mucosa and without systemic manifestations of atopy9, 10. Similarly, Ying et al. suggested that neither atopy nor increased serum IgE are necessary for local IgE-related mRNA expression in asthma12. Taken together, all these data support a role for IgE primarily in the mucosal compartment.
Despite these conclusions, serum IgE did correlate with the degree of local IgE expression. Although one cannot exclude that some of the locally produced IgE reaches the circulation, in keeping with the previous discussion about separate mucosal and systemic immune compartments, it is likely that the increase in serum IgE reflects induction of IgE at a systemic level, as a consequence of mucosal processes. Specifically, induction of local IgE synthesis and binding to local mast cells, seen in airways of both normal and asthmatic subjects, may indicate an effort by the mucosal immune compartment to activate alternative responses, including induction of local IgE, to enhance the local mucosal defense. In this regard, the percentage of IgE-positive mast cells in apparently healthy normal subjects in this study ranged from 0–96%, with IgE present also in proximal airways of 2 nonatopic normal subjects. Frequent breaches of the mucosal defense and perpetual systemic exposures to environmental antigens may elicit systemic immune responses, including increased serum IgE. Increased serum IgE might then reflect a deficient local mucosal responses26. In this study, perhaps due to inclusion of normal subjects regardless of their allergic characteristics (normal non-atopic subjects without any allergic rhinitis symptoms may be different), the responses at the systemic level (atopy and serum IgE) did not differentiate normal from asthmatic subjects. Interestingly, common allergic rhinitis symptoms (with or without asthma) rather than the more complex syndrome of asthma had stronger association with systemic markers of allergy. This suggests that the induction of systemic responses to environmental antigens alone (regardless whether due to a breach of the mucosal barrier in the nose/sinuses or in the lower airways) is not sufficient to lead to the development of asthma. It is possible that in normal subjects, unlike in subjects with asthma, the redundancy of the repertoire of alternative mechanisms along mucosal surfaces is still sufficient to appropriately compensate for deficient mucosal protection and thus to adequately maintain homeostasis along mucosal surfaces. That may prevent triggering of presently unknown immunological processes at local and/or systemic level that could result in development of asthma, as discussed in our recent study28. Accordingly, local IgE may indeed be only an element and marker of particular alternative pathways, which may be induced to enhance local mucosal protection.
The expression of tetrameric signal-amplifying isoform of FcεRI (FcεRIβ) consistently correlated with tissue IgE, but was present at a lower level that did not differentiate the groups. Previous studies suggested an increase in overall FcεRI expression in atopic and nonatopic asthma and in atopic controls as compared to nonatopic controls11. As in humans both trimeric and tetrameric isoforms of FcεRI are functional, it is possible that high IgE binding to mast cells induces the FcεRIβ isoform to a limited degree. Additional studies comparing the trimeric FcεRI and FcεRIβ isoforms' expression in human mast cells as related to IgE (and perhaps concomitant IgG) binding are necessary to resolve this issue29, 30.
Local IgE expression strongly correlated with eosinophils and, to a lesser degree, with lymphocytes. Both lymphocytes and IgE were significantly increased in severe asthmatics with persistent eosinophilia as compared to other groups, including eosinophil-negative severe asthmatics. Also, total mast cells in severe asthmatics with persistent eosinophilia were higher as compared to eosinophil-negative severe asthmatics. As a high proportion of these mast cells were IgE-positive, and previous reports suggest that IgE binding prolongs mast cell survival, it is possible that the relative resilience of mast cells in severe asthmatics with persistent eosinophilia is related to an IgE-inducing mucosal response, high IgE binding to mast cells and higher mast cell activity14, 31, 32.
Regression models examining the relationships of tissue IgE with lung function suggest that mucosal IgE-related immune processes in asthma are less detrimental to lung function than other defense mechanisms. Thus, the ability to elicit local immune responses that include IgE, particularly in severe asthma, may have better outcomes than alternatives. Indeed, IgE-mediated reactions and eosinophilia are otherwise appropriate responses against parasites.
In contrast, the positive association between local IgE expression and a history of asthma-related assisted ventilation suggests that higher expression of IgE in the airways may predispose to severe exacerbations of asthma. Although in these models eosinophils were not a significant contributor, eosinophils have been associated with severe exacerbations of asthma13-15. A recent study suggested that anti-IgE therapy in mild asthma reduced both IgE-positive cells and eosinophils in the tissue3. This may be part of the mechanism by which anti-IgE therapy reduces asthma exacerbations in moderate and severe asthma1, 2, 4. Interestingly, although anti-IgE therapy significantly decreased eosinophilic inflammation and tissue and serum IgE levels, these changes did not improve lung function3. While these complex but consistent relationships suggest that increased IgE binding to mast cells and concomitant acute reactions are marginally related to very severe exacerbations, the relation of mast cell IgE to lung function also suggests that persistent engagement of Th2 pathways in asthma may preserve airflow through reparative/pro-fibrotic effects15, 33-35
Although analyzing human tissue samples offers indispensable information about the in vivo tissue environment, conclusions about the causality of the processes cannot be made. Also, this study did not evaluate the presence of IgE-producing plasma cells in the submucosa. Thus, the connection with local production of IgE cannot be made6, 7. The limited sensitivity of IgE detection in GMA-embedded tissue resulted in identification of mast cells with high IgE expression only and prevented detection of other cells that bind/express IgE at lower levels, such as other mast cells, eosinophils, dendritic cells, B cells etc.
In conclusion, this study reports that IgE expression in human lung follows a distribution pattern typical for a mucosal immune response. The IgE process may be primarily locally induced and regulated, can exist with or without systemic IgE or atopy, but is unlikely to be locally induced or amplified by systemic/serum IgE. The local IgE process may represent an alternative homeostatic mechanism to maintain mucosal defense in both normal subjects and subjects with asthma. It is prominently active in severe asthma with eosinophilia, but not in severe asthma without eosinophilia, and appears to be associated with better lung function, but more severe exacerbations of the disease. A better understanding of airway mucosal immunity, as it relates to IgE and asthma is needed.
Supplementary Material
Acknowledgment
The authors thank Ashley Busacker and Jill Ketzer for their valuable technical support.
Abbreviations
- FcεRIβ
Tetrameric, signal-amplifying isoform of the high affinity IgE receptor
- FEV1%
Forced expiratory volume in one second, percent of predicted
- FVC%
Forced vital capacity, percent of predicted
- ICU
Intensive care unit
- IgE
Immunoglobulin E
- IQR
Interquartile range
- MA
Subjects with mild asthma
- %MCIgE+
Percentage of mast cells staining positive for IgE
- %MCFcεRIβ+
Percentage of mast cells staining positive for FcεRIβ
- NC
Normal control subjects
- NDRI
National Disease Research Interchange, Philadelphia, PA
- OLR
Online repository
- OR
Odds ratio
- AR
Seasonal/allergic rhinitis symptoms
- RV%
Residual volume, percent of predicted
- SAeo+
Subjects with severe asthma with eosinophilia
- SAeo−
Subjects with severe asthma without eosinophilia
Footnotes
Supported by: NIH grants HL-64087, AI-40600, RR-00051 and ALA of Colorado, Oklahoma and Alaska and Genentech Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Soler M, Matz J, Townley R, Buhl R, O'Brien J, Fox H, et al. The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur Respir J. 2001;8:254–61. doi: 10.1183/09031936.01.00092101. [DOI] [PubMed] [Google Scholar]
- 2.Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108:184–90. doi: 10.1067/mai.2001.117880. [DOI] [PubMed] [Google Scholar]
- 3.Djukanovic R, Wilson SJ, Kraft M, Jarjour NN, Steel M, Chung KF, et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–93. doi: 10.1164/rccm.200312-1651OC. [DOI] [PubMed] [Google Scholar]
- 4.Bousquet J, Cabrera P, Berkman N, Buhl R, Holgate S, Wenzel S, et al. The effect of treatment with omalizumab, an anti-IgE antibody, on asthma exacerbations and emergency medical visits in patients with severe persistent asthma. Allergy. 2005;60:302–8. doi: 10.1111/j.1398-9995.2004.00770.x. [DOI] [PubMed] [Google Scholar]
- 5.Ong YE, Menzies-Gow A, Barkans J, Benyahia F, Ou TT, Ying S, et al. Anti-IgE (omalizumab) inhibits late-phase reactions and inflammatory cells after repeat skin allergen challenge. J Allergy Clin Immunol. 2005;116:558–64. doi: 10.1016/j.jaci.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 6.KleinJan A, Vinke JG, Severijnen LW, Fokkens WJ. Local production and detection of (specific) IgE in nasal B-cells and plasma cells of allergic rhinitis patients. Eur Respir J. 2000;15:491–7. doi: 10.1034/j.1399-3003.2000.15.11.x. [DOI] [PubMed] [Google Scholar]
- 7.Smurthwaite L, Walker SN, Wilson DR, Birch DS, Merrett TG, Durham SR, et al. Persistent IgE synthesis in the nasal mucosa of hay fever patients. Eur J Immunol. 2001;31:3422–31. doi: 10.1002/1521-4141(200112)31:12<3422::aid-immu3422>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 8.Takhar P, Smurthwaite L, Coker HA, Fear DJ, Banfield GK, Carr VA, et al. Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J Immunol. 2005;174:5024–32. doi: 10.4049/jimmunol.174.8.5024. [DOI] [PubMed] [Google Scholar]
- 9.Powe DG, Huskisson RS, Carney AS, Jenkins D, Jones NS. Evidence for an inflammatory pathophysiology in idiopathic rhinitis. Clin Exp Allergy. 2001;31:864–72. doi: 10.1046/j.1365-2222.2001.01106.x. [DOI] [PubMed] [Google Scholar]
- 10.Powe DG, Jagger C, Kleinjan A, Carney AS, Jenkins D, Jones NS. ‘Entopy’: localized mucosal allergic disease in the absence of systemic responses for atopy. Clinical & Experimental Allergy. 2003;33:1374–9. doi: 10.1046/j.1365-2222.2003.01737.x. [DOI] [PubMed] [Google Scholar]
- 11.Humbert M, Grant JA, Taborda-Barata L, Durham SR, Pfister R, Menz G, et al. High-affinity IgE receptor (FcepsilonRI)-bearing cells in bronchial biopsies from atopic and nonatopic asthma. Am J Respir Crit Care Med. 1996;153:1931–7. doi: 10.1164/ajrccm.153.6.8665058. [DOI] [PubMed] [Google Scholar]
- 12.Ying S, Humbert M, Meng Q, Pfister R, Menz G, Gould HJ, et al. Local expression of epsilon germline gene transcripts and RNA for the epsilon heavy chain of IgE in the bronchial mucosa in atopic and nonatopic asthma. J Allergy Clin Immunol. 2001;107:686–92. doi: 10.1067/mai.2001.114339. [DOI] [PubMed] [Google Scholar]
- 13.Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–21. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 14.Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–8. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 15.Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol. 2004;113:101–8. doi: 10.1016/j.jaci.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 16.Balzar S, Chu HW, Wenzel S. Local airway tissue IgE and Fc epsilon RI beta expression in severe asthmatics and normal subjects in relation to inflammation and serum IgE [abstract] Am J Respir Crit Care Med. 2004;169:A816. [Google Scholar]
- 17.Balzar S, Wenzel SE, Chu HW. Transbronchial biopsy as a tool to evaluate small airways in asthma. Eur Respir J. 2002;20(2):254–9. doi: 10.1183/09031936.02.00261102. [DOI] [PubMed] [Google Scholar]
- 18.Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1–8. doi: 10.1152/ajpregu.2000.279.1.R1. [DOI] [PubMed] [Google Scholar]
- 19.Kleinbaum DG, Kupper LL, Muller KE, Nizam A. Applied Regression Analysis and Multivariable Models. 3rd edition Duxbury Press; CA: 1998. [Google Scholar]
- 20.Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, et al. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 21.Dombrowicz D, Flamand V, Miyajima I, Ravetch JV, Galli SJ, Kinet JP. Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. J Clin Invest. 1997;99:915–25. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–34. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 23.Daeron M, Malbec O, Latour S, Arock M, Fridman WH. Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest. 1995;95:577–85. doi: 10.1172/JCI117701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–6. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 25.Macpherson AJ, Uhr T. Compartmentalization of the mucosal immune responses to commensal intestinal bacteria. Ann N Y Acad Sci. 2004;1029:36–43. doi: 10.1196/annals.1309.005. [DOI] [PubMed] [Google Scholar]
- 26.Akadegawa K, Ishikawa S, Sato T, Suzuki J, Yurino H, Kitabatake M, et al. Breakdown of mucosal immunity in the gut and resultant systemic sensitization by oral antigens in a murine model for systemic lupus erythematosus. J Immunol. 2005;174:5499–506. doi: 10.4049/jimmunol.174.9.5499. [DOI] [PubMed] [Google Scholar]
- 27.Zambrano JC, Carper HT, Rakes GP, Patrie J, Murphy DD, Platts-Mills TA, et al. Experimental rhinovirus challenges in adults with mild asthma: response to infection in relation to IgE. J Allergy Clin Immunol. 2003;111:1008–16. doi: 10.1067/mai.2003.1396. [DOI] [PubMed] [Google Scholar]
- 28.Balzar S, Strand M, Nakano T, Wenzel SE. Subtle immunodeficiency in severe asthma: IgA and IgG2 correlate with lung function and symptoms. Int Arch Allergy Immunol. 2006;140:96–102. doi: 10.1159/000092252. [DOI] [PubMed] [Google Scholar]
- 29.Lin S, Cicala C, Scharenberg AM, Kinet JP. The Fc(epsilon)RIbeta subunit functions as an amplifier of Fc(epsilon)RIgamma-mediated cell activation signals. Cell. 1996;85:985–95. doi: 10.1016/s0092-8674(00)81300-8. [DOI] [PubMed] [Google Scholar]
- 30.Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–72. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 31.Asai K, Kitaura J, Kawakami Y, Yamagata N, Tsai M, Carbone DP, et al. Regulation of mast cell survival by IgE. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 32.Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, et al. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–11. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 33.Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, et al. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32:1558–65. doi: 10.1046/j.1365-2222.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhou X, Trudeau JB, Schoonover KJ, Lundin JI, Barnes SM, Cundall MJ, et al. Interleukin-13 augments transforming growth factor-beta1-induced tissue inhibitor of metalloproteinase-1 expression in primary human airway fibroblasts. Am J Physiol Cell Physiol. 2005;288:C435–42. doi: 10.1152/ajpcell.00035.2004. [DOI] [PubMed] [Google Scholar]
- 35.Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med. 2005;171:431–9. doi: 10.1164/rccm.200407-949OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.