

Abstract
The involvement of matrix metalloproteinases (MMPs) has been suggested in cellular mechanisms leading to medulloblastoma (MB), the most common malignant brain tumor in children. A significant association of the expression levels of MMP-9 with survival and M stage suggests that patients with medulloblastoma metastatic disease at diagnosis may benefit from the anti-MMP therapy. Here, we have evaluated the tumorigenicity of medulloblastoma cells after infection with an adenovirus containing a 21 bp siRNA sequence of human MMP-9 gene (Ad-MMP-9). Infection of Daoy medulloblastoma cells with Ad-MMP-9 reduced MMP-9 activity and protein levels compared with parental and Ad-SV controls. Ad-MMP-9 decreased the number of viable Daoy cells in a concentration-dependent manner. FACS analysis indicated Ad-MMP-9 infection caused a dose-dependent cell cycle arrest in the G0/G1 phase. Ad-MMP-9-induced cell cycle arrest appears to be mediated by the ERK MAPK pathway and the cell cycle inhibitors p16INK4 and is phenotypically indistinguishable from senescence. Ad-MMP-9 treatment inhibited medulloblastoma tumor growth in an intracranial model and was mediated by upregulation of p16 expression. These studies validate the usefulness of targeting MMP-9 and provide a novel perspective in the treatment of medulloblastoma.
Keywords: MMP-9, siRNA, medulloblastoma, tumor
INTRODUCTION
Medulloblastoma is an embryonic brain tumor that arises within the external germinal layer (EGL) of the cerebellum. There have been many recent advances in the treatment of medulloblastoma including improved surgical resection techniques, radiation, and chemotherapy (1, 2). The prognosis for patients with these tumors remains variable and is relatively poor in infants and adult patients with metastatic disease. The traditional treatments are also toxic and can lead to long-term disabilities (3). Therefore development of novel therapeutic approaches, such as those aimed at targeting tumor cell invasion and metastasis, is greatly needed.
The underlying molecular mechanisms of brain tumor invasiveness have been found to be closely related to the proteolytic degradation of the extracellular matrix (ECM). The ECM of the brain is mainly comprised of proteins such as fibronectin, laminin, vitronectin, thrombospondin, tenascin, heparin sulfate proteoglycan, and collagen IV (4). Several proteases are thought to be involved in the degradation of ECM components, including matrix metalloproteinases (MMPs). In addition to ECM degradation, MMPs are also capable of releasing growth factors and/or inactive complexes, cleaving growth factor receptors, and activating growth factors excreted as pre-pro-enzymes, such as transforming growth factor (TGF)-α, TGF-β, macrophage-colony stimulating factor (M-CSF), insulin-like growth factor (IGF) and fibroblast growth factor receptor (FGFR)-1 (5–8). Finally, some MMPs have been shown to play a crucial role in tumor invasion. Among the MMPs thought to be involved in cancer, attention has been focused on MMP-2 and MMP-9, which specifically degrade the main structural component of basement membranes, type IV collagen. Studies on regional distribution of MMPs in medulloblastoma tumors indicated that MMP-2 and MMP-9 were strongly expressed (9–11). A number of studies have linked elevated MMP-2 and MMP-9 expression to increased metastasis and advanced tumor stage (12, 13). Notably, MMP-9 is not expressed in normal adult tissues, but is expressed in invasive tumors and represents a key protein in brain tumor progression. MMP-9, MT1-MMP, and MT2-MMP are often and strongly expressed in classical and desmoplastic medulloblastomas and correlates with prognosis in classical medulloblastomas (14).
RNA interference (RNAi) has been shown to be an effective method for inhibiting the expression of a specific gene in human cells via targeting with short duplex RNA (short-interfering RNA or siRNA). Anti-tumor activities have been attained in vivo through siRNA knockdown of several components for tumor cell growth, metastasis, angiogenesis, and chemoresistance (15). Here, we assessed the potential of RNAi-mediated MMP-9 gene knockdown in medulloblastoma cells. In the present study, we constructed a replication-deficient recombinant adenovirus (Ad-MMP-9) to efficiently deliver MMP-9 siRNA targeted to the MMP-9 gene, thereby down regulating MMP-9 expression in a medulloblastoma cell line. Our results show that the downregulation of MMP-9 has a therapeutic effect in inhibiting medulloblastoma cell growth and invasion in vitro and in vivo. We also demonstrate that Ad-MMP-9 promotes p16INK4-mediated senescence and cell cycle arrest in the Daoy medulloblastoma cancer cell line. These results provide insight about the underlying mechanisms of the anti-tumorigenic effects of Ad-MMP-9.
MATERIALS AND METHODS
Cell cultures
We used the Daoy medulloblastoma cell line, which was derived from a tumor in the posterior fossa of a 4 yr-old boy (ATCC #HTB 186). Daoy cells were cultured in Advanced-MEM supplemented with 5% fetal bovine serum, 2 mmol/L L-glutamine, 2 mmol/L sodium pyruvate, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C.
ECM components and antibodies
Laminin, fibronectin and vitronectin were obtained from Sigma (St. Louis, MO). Antibodies for MMP-9, p16, p21, ERK, phospho-ERK and pRb were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), cdk2, cdk4, cyclin D2 and cyclin E from Biomeda (Foster City, CA), αvβ3, α5β1 and β4) were from Chemicon (Temecula, CA)
Adenovirus construction
We constructed two adenoviruses, one carrying siRNA targeting the MMP-9 gene (Ad-MMP-9) and the other carrying a scrambled sequence of the MMP-9 siRNA (Ad-SV). Oligonucleotides were designed using an adenoviral pSuppressor kit (Imgenex, Sorrento Valley, CA) inserted into the suppressor vector under the control of the modified Pol II promoter as per manufacturer’s instructions. This plasmid was cotransfected with the pAd vector backbone in 293 cells. Adenovirus generation, amplification, and titer were performed according to previously described procedures (16). Briefly, viral particles were purified using a cesium chloride density gradient. Viral titers were assessed using the plaque-forming test (PFU) and counting infectious virus particles.
Adenoviral infection
Infection with recombinant viruses was accomplished by exposing cells to adenovirus in serum-free cell culture medium for 30 min followed by addition of serum-containing medium. We used green fluorescent protein–expressing recombinant adenovirus (Ad-SV/GFP) as a control when determining transfection efficiency.
Gelatin zymography
Gelatin zymography was performed as described previously (17).For tumor-conditioned medium, DAOY cells were grown in 6-well tissue culture plates and infected with mock (PBS), 100 MOI of Ad-SV, or 25 to 100 MOI of Ad-MMP-9. After a 36 h incubation period, cells were washed with PBS and cultured overnight in serum-free DMEM/F-12 medium. The total protein concentration of the conditioned media was estimated using BCA reagent (Pierce, Rockford, IL). Equal amounts of protein from various treatments were used to determine gelatinase activity.
Western blotting
Western blot analysis was performed as described previously (17). Briefly, Daoy cells were grown in 100-mm plates and infected with mock, 100 MOI of Ad-SV, or various MOI of Ad-MMP-9 and incubated for 48 h at 37°C. Cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer and protein concentrations were measured using bicinchoninic acid protein assay regents (Pierce, Rockford, IL). For electrophoresis, 30–40 μg of protein in 6X sample buffer were loaded to each well of a SDS-PAGE gel. The blot was blocked and probed overnight with primary antibodies for either MMP-9, ERK, phospho-ERK, p21, p16, pRb, Cdk2, Cdk4, cyclin-D and cyclin-E at 4°C and detected with horseradish peroxidase using ECL.
RT-PCR
Daoy cells were infected as above and after 36 h at 37°C, total RNA was extracted as described by Chomczynski and Sacchi (18). PCR was performed using an RT-PCR kit (Invitrogen, Carlsbad, CA): 35 cycles of denaturation at 94°C for 1 min, annealing at 67°C for 30 s, and extension at 72°C for 90 s. The expected PCR products were visualized using ethidium bromide after resolving on 2% agarose gels. RT-PCR for GAPDH was performed to normalize input RNA We used the following primers: MMP-9, sense: 5’-TGGACGATGCCTGCAACGTG-3’; antisense: 5’-GTCGTGCGTGTCCAAAGGCA-3’; GAPDH, sense: 5’-TGAAGGTCGGAGTCAA CGGATTTGGT-3’, antisense: 5’-CATGTGGGCCATGA GGTCCACCAC-3’.
Cell-proliferation assays
Cell growth rate was determined using a modified MTT assay (R&D Systems, Minneapolis, MN) as a measurement of mitochondrial metabolic activity. Cells were infected with mock, 100 MOI of Ad-SV, or various doses of Ad-MMP-9 and incubated at 37°C. After 0–72 h, MTT reagent was added, cells incubated for 1 h at 37 °C, and the rate of absorbance of formazan (a dye produced by live cells) was measured with a microplate reader at A550.
Flow cytometry
Daoy cells were plated overnight in 100-mm tissue culture plates and infected for 36 h as described above. We used FACS analysis which utilizes propidium iodide staining of nuclear DNA to characterize cell cycle phase (19). Briefly, cells were harvested by trypsinization and stained with propidium iodide (2 mg/mL) in 4 mM sodium citrate containing 3% (w/v) Triton X-100 and RNase A (0.1 mg/mL) (Sigma; St. Louis, MO). Suspensions of 2×106 cells were analyzed by FACS Caliber System (Becton Dickinson Bioscience, San Jose, CA). The percentages of cells in the various phases of the cell cycle were assessed using Cell Quest software (Becton Dickinson Bioscience, San Jose, CA). For integrin analysis cells were incubated with monoclonal anti-integrin antibodies αvβ3 and α5β1, β4 or control mouse IgG in 0.5% BSA for 30 min on ice. After two PBS washes, cells were incubated with FITC-conjugated secondary antibodies in 0.5% BSA for 30 min on ice. Cells were again washed and cell-surface integrins were determined using a flow cytometer. All experiments were performed in triplicate.
Cell adhesion assay
We used the established crystal violet colorimetric method to determine cell adhesion (20). Briefly, 48-well tissue culture plates were coated with purified ECM proteins including laminin (10 μg/mL) fibronectin (10 μg/mL), or vitronectin (5 μg/mL) for 18 h at 4°C. Wells were covered with BSA (10 mg/mL) (fraction V; Sigma, St. Louis MO) in Ca2+ and Mg2+-free PBS for 1 h. BSA was removed and the wells were washed with PBS. Daoy cells were infected with mock, 100 MOI of Ad-SV, or the indicated MOI of Ad-MMP-9 and incubated for 24 h. Cells were harvested by trypsinization, washed and suspended in serum-free medium containing 0.1% BSA at 1×106 cells/mL, then plated onto 96-well cell-culture plates coated with ECM and incubated at 37°C for 2 h and attached cells were stained with 0.1% crystal violet (Sigma) at room temperature for 25 min. Cell adhesion was quantified by counting the average number of cells per five microscopic fields. All experiments were performed in triplicate and the data represent the average of three independent experiments. The student's t test was used to compare treatment groups with control cells with P<0.05 considered significant.
Senescence-associated β-galactosidase activity
SA-β-gal activity was determined using a SA-β-gal staining kit from Cell Signaling (Chemicon International, Temecula, CA). Briefly, Daoy cells (2 × 103 cells/well) were plated in 8-well chamber slides and infected with Ad-MMP-9 as described earlier. After 36 h, we used standard light microscopy to identify senescent cells, which were blue-stained. Five fields were evaluated for each well with three wells per condition (40X magnification).
Cell invasion assay
To gauge the invasive capacity of the tumor cells, we used Transwell cell culture chambers (Corning Costar, Cambridge, MA) as described previously (17). Briefly, Daoy cells were infected with mock, 100 MOI of Ad-SV, or various doses of Ad-MMP-9 for 48 h. 1×106 viable cells from each treatment were allowed ti invade through polycarbonate filters (8-μm pore size) coated with matrigel. The migrating cells on the reverse side of the filter were photographed and counted. Five different fields per filter were analyzed and all experiments were performed in triplicate.
Intracranial tumor model
Daoy cells (1x 105) were stereotactically implanted as described elsewhere with minor modifications (21). Two weeks after tumor cell implantation, the animals were randomized into three groups (6 animals per group). Each mouse received three intratumoral injections on the 14th, 15th and 16th day: group 1, received PBS (6 μL; n = 6), group 2 received 5 × 107 PFU of Ad-SV virus in 6 μl of PBS and group 3, received 5 × 107 PFU of Ad-MMP-9 in 6 μl of PBS. Animals losing ≥ 20% of body weight or having trouble ambulating, feeding, or grooming was sacrificed. Animals were monitored for 180 days when we arbitrarily terminated the experiment. Mouse brains were fixed in 10% formalin and embedded in paraffin. Tissue sections (4 μm-thick) were obtained from the paraffin blocks and stained with H&E using standard histological techniques. Sections were also subjected to immunostaining with antibodies for either MMP-9 or p16. Protein expression was detected using 3,3-diaminobenzidine solution (Sigma, St. Louis, MO). Sections were counterstained with hematoxylin, and negative control slides were obtained by non-specific IgG. Sections were washed and mounted with anti-fade mounting solution (Invitrogen, Carlsbad, CA) and analyzed with an inverted microscope.
Statistical Analyses
All data are expressed as mean ± SD. Statistical analysis was performed using the student’s t test or a one-way analysis of variance (ANOVA). P<0.05 were considered to be significant.
RESULTS
Ad-MMP-9 infection decreased MMP-9 activity and expression
We constructed a recombinant adenovirus encoding siRNA targeted to MMP-9 and then tested its effect on MMP-9 expression and activity. We determined MMP-9 activity, protein and mRNA levels in Daoy medulloblastoma cells infected with various doses of Ad-MMP-9. Gelatin zymography of conditioned media demonstrated that Ad-MMP-9 infection inhibited MMP-9 activity in a dose-dependent manner (Fig. 1A). There was no significant change in MMP-2 expression indicating that this inhibition was specific to MMP-9. Western blot analysis of conditioned media using anti-MMP-9 antibodies demonstrated that Ad-MMP-9 infection decreased MMP-9 protein expression levels in a dose-dependent manner as compared to mock and scrambled vector controls (Fig. 1B). To determine whether decreased production of MMP-9 was caused by gene transcription, we examined MMP-9 transcripts using RT-PCR. As shown in Fig. 1C, the levels of transcripts of MMP-9 in Ad-MMP-9 infected cells were significantly lower compared to cells infected with mock and Ad-SV.
Figure 1. Effect of Ad-MMP-9 on Daoy cells.
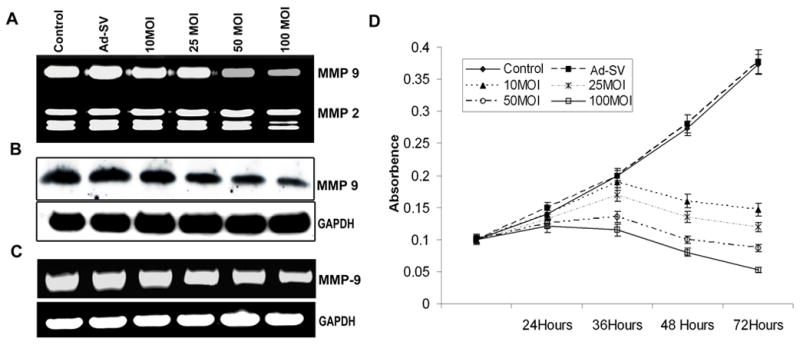
Daoy cells were infected with mock, 100 MOI of Ad-SV or the indicated doses of Ad-MMP-9 for 36 h, and then the conditioned medium was collected. (A) Zymographic analysis for MMP-9 activity in the conditioned medium. (B) Western blot analysis of MMP-9 protein expression in the conditioned medium. (C) Reverse transcription-PCR: Total RNA was extracted as per standard protocols and cDNA was synthesized. The PCR reaction was set up using first stand cDNA as the template for MMP-9. GAPDH served as a loading control. The data shown here are from a typical experiment performed in triplicate. (D) MTT proliferation assay was carried out for Daoy cells, infected with mock, 100 MOI of Ad-SV or the indicated doses of Ad-MMP-9. MTT activities were measured at 550 nm in triplicate at 24h, 36h, 48h and 72 h. and the proliferation curve illustrates average values from a typical experiment. The experiment was repeated three times.
Ad-MMP-9 infection inhibited cell proliferation in Daoy cells
Daoy medulloblastoma cells were infected with various MOI of Ad-MMP-9 for 72 h. Ad-MMP-9 infection led to a dose-dependent decrease in cell proliferation, as shown in Fig. 1D. A 75–80 % inhibition of cell proliferation occurred at an MOI of 50 at 72 hrs time points. This decrease reached almost 90% when Daoy cells were infected with 100 MOI of Ad-MMP-9 compared mock and Ad-SV controls at 72 hr.
Ad-MMP-9 infection blocks medulloblastoma invasion
Matrigel-coated transwell chambers were used in a standard test to study whether infection by Ad-MMP-9 suppressed the invasive capacity of Daoy cells. The results in Fig. 2A demonstrate that Ad-MMP-9 infection for 48 h inhibited the number of Daoy cells invaded through the matrigel in a dose-dependent manner as compared to the mock and Ad-SV controls. Quantification of the invaded cells indicated that 24% and 39% less cells invaded in 10 and 25 MOI Ad-MMP-9-infected Daoy cells than that of the controls. Infection with 50 and 100 MOI of Ad-MMP-9 resulted in a more significant effect on invasion with 59% and 78% inhibition compared to the mock-infected controls (Fig. 2B).
Figure 2. Effect of Ad-MMP-9 on the invasive capacity of Daoy cells through matrigel.
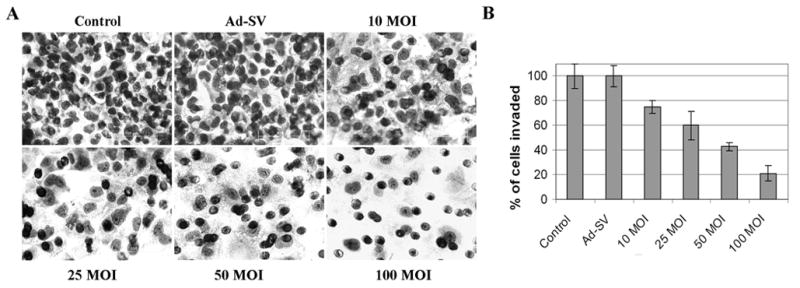
Daoy cells were infected with mock, 100 MOI of Ad-SV or the indicated doses of Ad-MMP-9 and incubated for 48 h. Cells were then allowed to invade transwell inserts containing 12-μm pore polycarbonate membranes pre-coated with matrigel for 24 h at 37°C. Cells were then fixed and stained with Hema-3. (A) Cells that had migrated to the lower side of the membrane were photographed under a light microscope at 20X magnification. (B) Percentages of invading cells were quantified by counting 5 fields in each condition. The data shown here are from a typical experiment performed in triplicate.
Ad-MMP-9 affects medulloblastoma cell adhesion to ECM protein-coated plates
To investigate the impact of MMP-9 inhibition on cell–matrix adhesion, we examined the effect of Ad-MMP-9 infection on Daoy cell attachment onto various extracellular matrix components. MMP-9 inhibition was associated with an overall increase in adhesion to extracellular matrices, an effect that was most evident when cells were allowed to adhere to fibronectin (Fig. 3A). Stimulation of adhesion was dose-dependent in all the matrices and Ad-MMP-9 infection caused a more prominent increase in adhesion on fibronectin compared to the other matrices and controls (Fig. 3B).
Figure 3. Effect of Ad-MMP-9 on Daoy cell adhesion to various matrices.
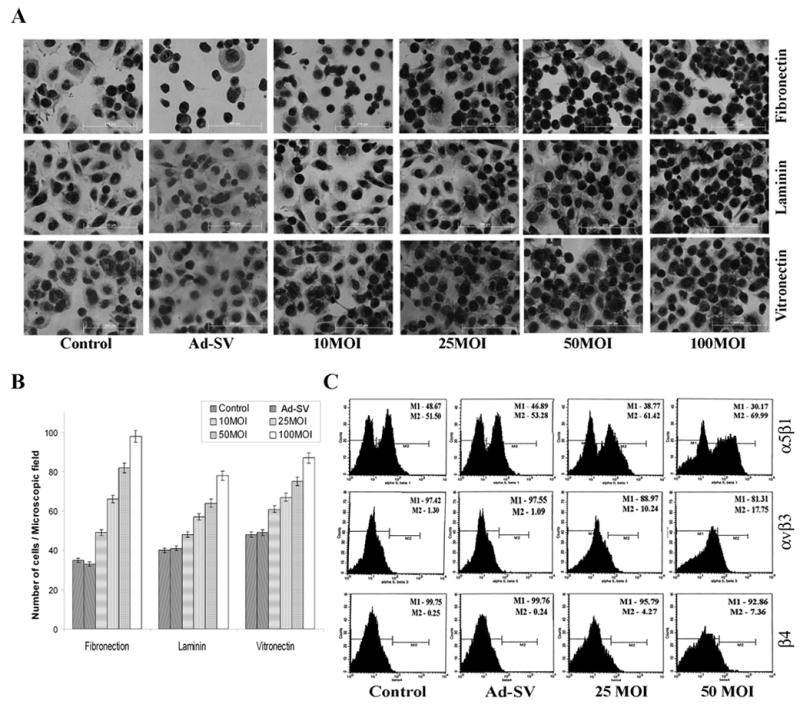
Daoy cells were infected with mock, 100 MOI of Ad-SV or the indicated doses of Ad-MMP-9 for 24 h. (A) Adhesion assays were performed on vitronectin, fibronectin and laminin. (B) Values are reported as percentage of mock control adhesion to vitronectin and represent the mean ± SD of 3 experiments. All experiments were performed in triplicate. P<0.05 is significant. (C) FACS analysis of integrin expression. Daoy cells were washed in blocking solution and incubated with monoclonal anti-integrin antibodies αvβ3 and α5β1, β4 for 30 min. Integrin expression was determined using a FITC-conjugated secondary antibody. The data shown here are from a representative experiment repeated three times with similar results.
Ad-MMP-9 affects expression of αvβ3, α5β1 and β4 integrins
Because cell–matrix interactions are critically determined by integrins, we hypothesized that MMP-9 inhibition could modulate integrin expression. Expression of αvβ3, α5β1 and β4 levels were not significantly different between the mock and Ad-SV control groups. However, the expression of these integrins in Ad-MMP-9 infected Daoy cells was markedly increased in a dose-dependent manner (Fig. 3C).
Ad-MMP-9 infection arrests cells in G0/G1 phase
It is known from previous studies with tumor cells that synthetic MMP inhibitors may induce cell cycle arrest (22, 23). Therefore, we hypothesized that Ad-MMP-9 could cause similar effects. FACS analysis for nuclear DNA content by propidium iodide staining showed that cell growth was arrested in the G0/G1 cell cycle phase. Figure 4A shows that 52–54% of Daoy cells were in G0/G1, 15–14% cells were in S phase, and 18–21% were in G2/M phase in mock and Ad-SV-treated cells. In contrast, Daoy cells infected with 100 MOI of Ad-MMP-9 remained to a high extent, bout 73% in G0/G1 phase. In addition, the number of cells in the G2/M (mitotic/dividing) phase was significantly decreased, thereby resulting in limited cell cycle progression, which in turn translates into a marked decrease in proliferation.
Figure 4. Effect of Ad-MMP-9 on cell cycle and cell cycle regulatory proteins.
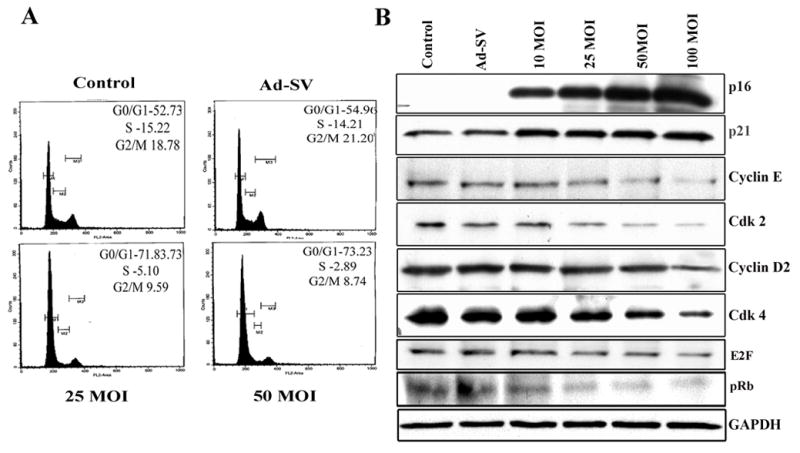
(A) Cell cycle analysis was performed using flow cytometry as detailed in Materials and Methods. The labeled cells were analyzed using a FACS Caliber System and percentage of cells in G0/G1, S and G2/M phases were calculated using Cell Quest software. The data shown here are from a typical experiment repeated three times (B) Immunoblot of cell lysates corresponding to Daoy cells infected with mock, 100 MOI of Ad-SV or the indicated MOI of Ad-MMP-9. Total protein lysates were analyzed for the levels of p21, p16, Cyclin D2, Cyclin E, cdk2, cdk4, pRb and E2F-1. As detailed in Materials and Methods, the cells were infected with mock, 100 MOI of Ad-SV or various doses of Ad-MMP-9 and total cell lysates were prepared for immunoblot analysis. The data shown here are from a representative experiment repeated three times.
Ad-MMP-9 infection induces senescence in Daoy cells
Cellular senescence is characterized by accumulation of lysosomal enzymes. Cells that were treated with Ad-MMP-9 infection and were subsequently growth-arrested, acquired the enlarged and flattened morphology characteristic of cellular senescence. To examine the relative levels of cellular senescence, we assessed the levels of SA-βGal, a well-known marker for cellular senescence (24). As shown in Figure 5A, senescence, as indicated by β-galactosidase staining (blue color), increased in a dose-dependent manner with Ad-MMP-9 infection. We did not observe senescent cells in mock and Ad-SV-treated cells.
Figure 5.
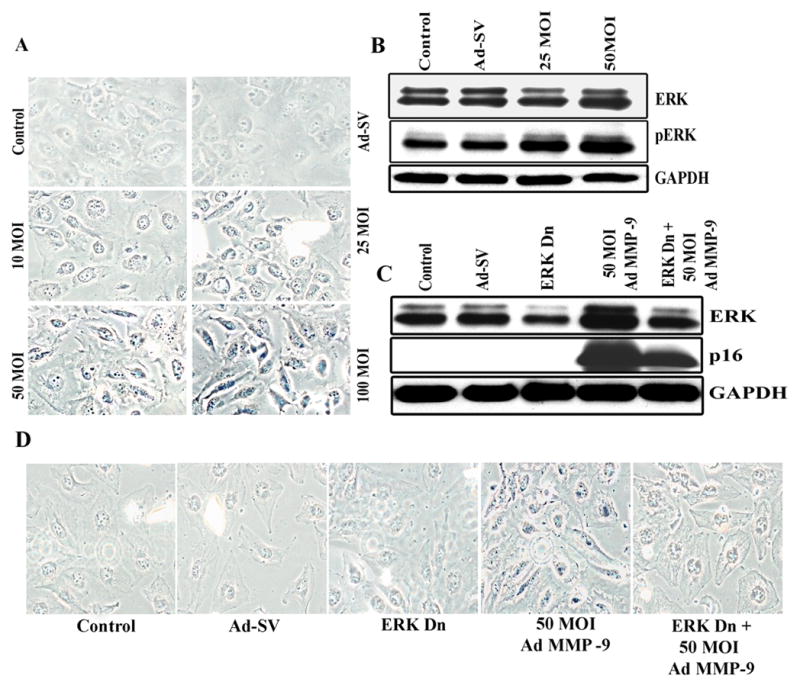
(A). Photographs of Daoy cells infected with Ad-MMP-9 stained for B-gal (pH 6.0) activity. (B). Effect of Ad-MMP-9 treatment on protein expression of ERK1/2 and phospho-ERK levels. Simultaneous treatment of Ad-MMP-9 infection and dominant negative ERK mutant showed inhibition of ERK and p16 expression (C) and senescence marker SA-β-gal (D). The data shown here are from experiments repeated in triplicate
Ad-MMP-9 infection increases p16 and p21 protein expression
To further characterize the nature of the cell cycle arrest caused by Ad-MMP-9 infection, we looked at the expression of several cell cycle regulatory proteins in Ad-MMP-9-infected Daoy cells (Fig. 4B). The changes that distinguish cellular senescence from quiescence are thought to include upregulation of the cell cycle inhibitors p16INK4a and p21CIP (25, 26). We observed a significant induction of p21 with Ad-MMP-9 infection. The expression of p16 protein was at nearly undetectable levels in the mock infected and the Ad-SV-infected cells but increased dramatically with Ad-MMP-9 infection. In contrast, the expression of p16 increased in a dose-dependent manner and as cells reached senescence. These results, along with the accumulation of senescence-associated β-galactosidase, strongly suggest that the cell cycle arrest induced by Ad-MMP-9 is indeed senescence. Using immunoblot analysis, we also assessed the effect of Ad-MMP-9 infection on the protein expression of the cyclins and cdks, which are known to be regulated by p21. Ad-MMP-9 treatment of the cells resulted in a dose-dependent decrease in protein expression of cyclin D2, and cyclin E as well as cdk4 and cdk6.
Ad-MMP-9 inhibits protein expression of pRB and E2F1
Downregulation of cdk4/6 has been shown to be associated with a decrease in the expression of retinoblastoma (pRb) tumor suppressor protein, a key regulator of the G1 → S phase transition in the cell cycle (27). Therefore, we next examined the effect of Ad-MMP-9 on the protein expression of pRb. Immunoblot data revealed that Ad-MMP-9 treatment of cells resulted in a significant decrease in the protein expression of pRb (Fig. 4B). Because pRb controls cell cycle by binding to and inhibiting E2F transcription factors, we assessed the protein expression of E2F transcription factors. As shown in Fig. 4B, Ad-MMP-9 treatment of cells resulted in a dose-dependent decrease in E2F transcription factors.
Ad-MMP-9-induced senescence is mediated by the ERK/mitogen-activated protein (MAP) kinase pathway
The ERK pathway was shown to be responsible for senescence (28). We found that both ERK and phospho-ERK levels were increased in Daoy cells infected with Ad-MMP-9 as compared to the controls (Fig. 5B). To further validate the role of ERK1/2 in Ad-MMP-9-induced cell cycle arrest leading to senescence, we silenced ERK1/2 phosphorylation by transiently transfecting Daoy cells with a dominant negative mutant of ERK (Dn-ERK) prior to Ad-MMP-9 infection. We observed that Ad-MMP-9 did not induce ERK1/2 expression in this condition (Fig. 5C). To determine if p16 upregulation was mediated by ERK activation, we stripped the blot and tested for p16 expression. Figure 5C shows that Dn-ERK transfection reduced p16 expression; thereby suggesting that p16 expression in Ad-MMP-9 treated cells is mediated by ERK. Furthermore, transfection with a dominant negative mutant for ERK (Dn-ERK) led to a decrease of about 50% in the number of SA-β-Gal-positive cells compared to the levels observed without infection with Dn-ERK (Fig. 5D).
Ad-MMP-9 treatment causes loss of tumorigenicity in nude mice
To directly evaluate the role of Ad-MMP-9 on tumor formation in vivo, we injected Daoy cells into nude mice and treated the preformed tumors with intratumoral injections of Ad-MMP-9. Mice treated with mock (PBS) and Ad-SV developed tumors, were symptomatic within four weeks and were sacrificed. In striking contrast, Ad-MMP-9-injected mice survived for 6 months, at which point the animals were sacrificed and their brains examined for tumor growth. Histological examination of the paraffin-embedded tissue sections of the brains from mice that received mock and Ad-SV showed large tumors in the cerebellum. However, H&E staining did not reveal any tumor cells in four out of six mice treated with Ad-MMP-9 (Fig. 6A). Very small tumors were observed in the other two Ad-MMP-9 treated animals.
Figure 6. Tumorigenicity in vivo.
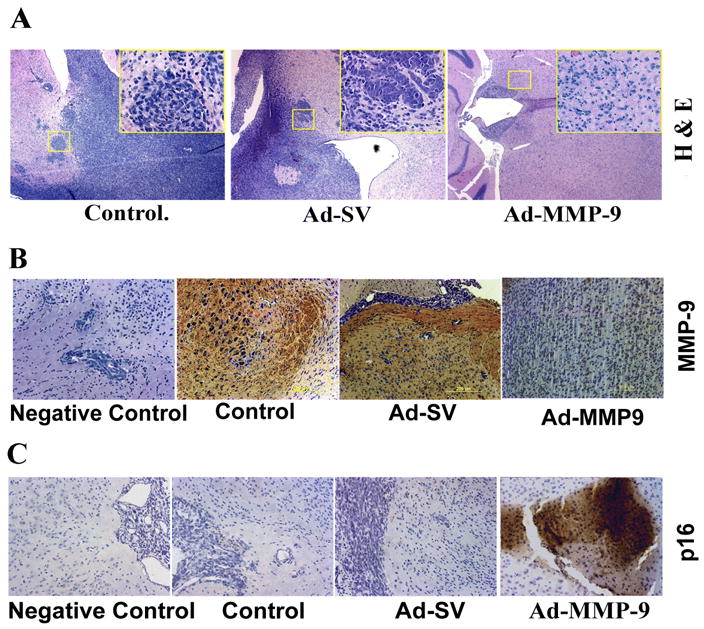
Daoy cells were implanted intracranially in nude mice and treated with intratumoral injections of mock, Ad-SV and Ad-MMP-9 as described in Materials and Methods. (A). H&E staining of brain sections showing neoplastic growth (inset 20X). Immunohistochemical analysis of MMP-9 expression (B), and p16 (C) expression in brain sections as described in methods.
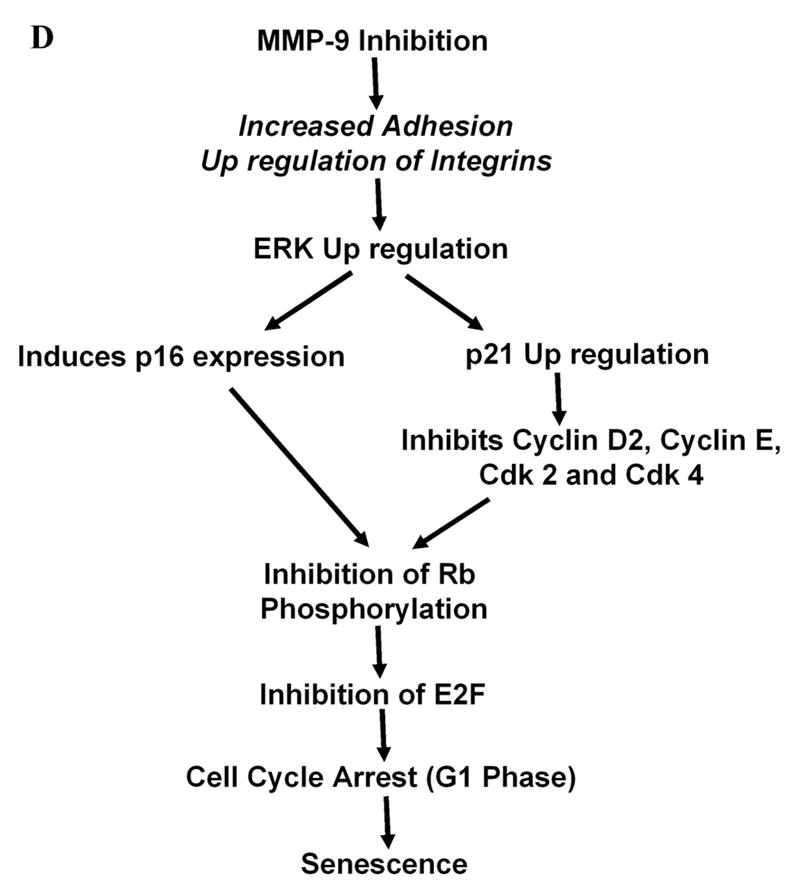
Figure 6D. Proposed schematic model for Ad-MMP-9-mediated cell cycle dysregulation and induction of senescence.
To determine whether MMP-9 was expressed in vivo, brain sections were stained with a monoclonal antibody for human MMP-9. Figure 6B indicates that brain sections from mock and Ad-SV-treated mice showed intense expression of MMP-9. In contrast, MMP-9 expression was not detectable in Ad-MMP-9-treated mice brain sections. Based on the in vitro experiments, we decided to assess p16 expression in brain sections of the mice. We observed a remarkable increase in p16 expression in brain sections from mice that received Ad-MMP-9 treatment. However, we barely found any signal in the control brains sections from the mock and Ad-SV treated mice brain sections (Fig. 6C).
DISCUSSION
A number of studies have demonstrated that the introduction of siRNA into mammalian and human cells causes specific and effective suppression of the corresponding mRNA molecules (29, 30). Therapeutic application of siRNA technology requires an efficient gene delivery system for transduction of siRNA into target cells. Adenoviral vectors have been shown to efficiently transduce genes into many types of cells. The present study describes the profound effects of an adenovirus carrying siRNA against MMP-9 on medulloblastoma tumor growth in vitro and in vivo.
In this study, we have demonstrated that adenovirally transduced siRNA against MMP-9 (Ad-MMP-9) exerted a significant effect on inhibiting MMP-9 mRNA expression using RT-PCR and MMP-9 protein levels by Western blotting. Further, the results demonstrate that Ad-MMP-9 infection causes cell cycle arrest in the G0/G1 phase, senescence in Daoy cells in vivo, and inhibits tumor growth in an intracranial model. Cell cycle arrest occurs as a function of cyclin-dependent kinase inhibitors. In regards to this observation, our results definitively show that MMP-9 inhibition induces the expression of cyclin-dependent kinase (CDK) inhibitors p21 and p16 in a dose-dependent manner. The induction of these CDK inhibitors has been implicated in the initiation and maintenance of cellular senescence (31, 32). p21 inhibits cyclin-dependent protein kinase (33) and the proliferating cell nuclear antigen (PCNA)-dependent DNA replication (34) causing G1 arrest of cell cycle. Expression of the p21 gene suppressed growth of human brain, lung, and colon tumor cells in culture (35).
Cellular senescence is strongly implicated as an important mechanism of tumor suppression and the ability of p53 and p16 to mediate cell cycle arrest is central to their activity in blocking tumor development (36). We show that Ad-MMP-9 infection causes a dramatic increase in p16 expression. Also, p16 is a specific inhibitor of the cyclin D1-dependent kinases cdk4 and cdk6. As expected, the levels of cdk4 and cdk6 were decreased. Normally, in the absence of p16, cyclin D1/cdk4 and cyclin D1/cdk6 complexes phosphorylate and inactivate the Rb protein, permitting E2F-dependent transcription of genes encoding proteins to initiate chromosome replication and ultimately another round of cell division (37). In this study, we demonstrate that Ad-MMP-9 infection decreased phosphorylated Rb levels, thereby activating it, which in turn, resulted in cell cycle arrest. Deregulated activity of the D-type cyclin-dependent kinases cdk4 and cdk6 is widely observed in various tumor cells, illustrating their importance in controlling cell cycle (38). We also show that the brain sections of mice that received Ad-MMP-9 treatment show remarkable levels of p16 expression, which was completely absent in the brain sections of control and Ad-SV-treated mice, suggesting p16-mediated inhibition of tumor growth. Our results confirm earlier studies indicating the involvement of the p16-Rb pathway in medulloblastomas (39).
We next investigated the signaling mechanism mediating senescence upon Ad-MMP-9 infection. Cells acquire increased adhesion to the extracellular matrix while losing cell-cell contacts during the process of senescence. Ad-MMP-9 infection of Daoy cells caused increased adhesion on various ECM proteins and increased expression of αvβ3, α5β1 and β4 integrins. Several studies have suggested a link between integrin-mediated signaling and the Ras-MAP kinase pathway. Specifically, adhesion of cells to ECM components, such as fibronectin, induces activation, tyrosine phosphorylation, and nuclear translocation of MAP kinases through an integrin-dependent mechanism (40), thereby suggesting that integrin-matrix interactions activate a MAP kinase cascade. Interestingly, the ability of oncogenic ras to induce premature senescence depends on the Raf-MEK-ERK pathway that mediates cell proliferation (28). We found that Ad-MMP-9 infection elicited ERK activation. Of the three matrices studied, cell adhesion to fibronectin is greatly increased with Ad-MMP-9 infection with a concomitant increase in its receptor α5β1. p16INK4a was found to sensitize cells to detachment-induced apoptosis by increased transcription of integrin α5β1 (41). The biological and molecular basis of the promotion of adhesion in our study remains to be elucidated. While we found an increase in integrin expression with MMP-9 inhibition, further work is required to determine whether the observed changes in integrin expression are causing sensescence-mediated growth arrest. One possibility is that the cytoskeleton or some other pathway is activated by MMP-9 inhibition. This would explain the increase in adhesion on several matrices. Cytoskeletal reorganization was a key player in induction of anoikis in breast cancer cells (42). Also, cytoskeletal disrupting drugs, such as LatA, induced apoptosis in normal MCF10A cells (43).
Next, our results indicate the ERK pathway plays an important role in the premature senescence that resulted from Ad-MMP-9 infection in Daoy cells. Studies have shown that sustained ERK activation is required to pass the G1-restriction point (44) and that ERK regulates cyclin D1 expression during mid-G1 (45). However, under certain circumstances, the Ras/Raf/MEK/ERK cascade lead to cell cycle arrest instead of proliferation. Ras-induced cell cycle arrest is driven through ERK-mediated upregulation of p53 and p16INK4a activity (28). Ad-MMP-9 infection in Daoy cells caused an increase in ERK and phospho-ERK levels in a dose-dependent manner. Our studies also demonstrate that transfection with a dominant negative ERK construct before Ad-MMP-9 infection inhibited Ad-MMP-9-mediated senescence. We also demonstrate here that induction of p16 expression with Ad-MMP-9 infection is also inhibited by transfection with Dn-ERK. The ERK MAP kinase is known to regulate transcription factors, thereby controlling gene expression (46). In fact, p16INK4a expression can be directly regulated by transcription factors of the ETS family, which in turn, are regulated by ERK (47). Based on the outcome of this study and as shown in the scheme in Figure 6D, we suggest that MMP-9 inhibition induces activation of ERK1/2, which leads to induction of cyclin kinase inhibitor p27/KIP1 and p16, and in turn, inhibits cell cycle regulatory molecules resulting in G1 arrest and senescence. Downregulation of cdk4/6 inhibits pRb, inhibits protein expression of the E2F family, thereby leading to senescence.
Induction of cell cycle arrest and apoptosis represent an established method to treat malignant disorders (48). The influence of synthetic MMP inhibitors on cell cycle and apoptosis, is well documented. The MMP inhibitor batimastat (BB-94) was shown to enhance interferon- γ-induced apoptosis in mice with ovarian cancer (22) and to block ovarian cancer cells in the G0/G1 phase of cell cycle (23). Another MMP inhibitor, AG3340, promoted apoptosis in human prostate and colon carcinoma models (49). GM-6001, a non-specific MMP inhibitor was shown to induce apoptosis in smooth muscle cells (50).
In conclusion, our studies provide the first evidence that MMP-9 inhibition causes ERK-mediated p16 expression resulting in cell cycle arrest in Daoy cells in vitro and in vivo. These findings demonstrate that MMP-9 may play an important role in inhibiting medulloblastoma invasion and tumor growth and identifies MMP-9 as a promising target for adenoviral-mediated, siRNA-based therapy in medulloblastoma.
Acknowledgments
We thank Noorjehan Ali for technical assistance in animal experiments. We also thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for reviewing the manuscript.
Abbreviations
- RNAi
RNA interference
- siRNA
short interfering RNA
- MMP-9
matrix metalloproteinase-9
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
Footnotes
This research was supported by National Cancer Institute Grant CA 75557, CA 92393, CA 95058, CA 116708, N.I.N.D.S. NS47699 and NS57529, and Caterpillar, Inc., OSF Saint Francis, Inc., Peoria, IL (to J.S.R.) and Children’s Miracle Network grant to (S.S.L.).
Reference List
- 1.Taylor RE, Bailey CC, Robinson K, et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581–91. doi: 10.1200/JCO.2003.05.116. [DOI] [PubMed] [Google Scholar]
- 2.Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832–45. doi: 10.1200/JCO.1999.17.3.832. [DOI] [PubMed] [Google Scholar]
- 3.Yang SY, Wang KC, Cho BK, et al. Radiation-induced cerebellar glioblastoma at the site of a treated medulloblastoma: case report. J Neurosurg. 2005;102:417–22. doi: 10.3171/ped.2005.102.4.0417. [DOI] [PubMed] [Google Scholar]
- 4.Wright JW, Kramar EA, Meighan SE, Harding JW. Extracellular matrix molecules, long-term potentiation, memory consolidation and the brain angiotensin system. Peptides. 2002;23:221–46. doi: 10.1016/s0196-9781(01)00599-x. [DOI] [PubMed] [Google Scholar]
- 5.Fowlkes JL, Serra DM, Rosenberg CK, Thrailkill KM. Insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) functions as an IGF-reversible inhibitor of IGFBP-4 proteolysis. J Biol Chem. 1995;270:27481–8. doi: 10.1074/jbc.270.46.27481. [DOI] [PubMed] [Google Scholar]
- 6.Gearing AJ, Beckett P, Christodoulou M, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–7. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 7.Levi E, Fridman R, Miao HQ, Ma YS, Yayon A, Vlodavsky I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc Natl Acad Sci USA. 1996;93:7069–74. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsbrun M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J Biol Chem. 1997;272:31730–7. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- 9.Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Matrix metalloproteinase expression in childhood medulloblastomas/primitive neuroectodermal tumors. In Vivo. 2000;14:667–73. [PubMed] [Google Scholar]
- 10.Rossi M, Rooprai HK, Maidment SL, Rucklidge GJ, Pilkington GJ. The influence of sequential, in vitro passage on secretion of matrix metalloproteinases by human brain tumour cells. Anticancer Res. 1996;16:121–8. [PubMed] [Google Scholar]
- 11.Vince GH, Herbold C, Klein R, et al. Medulloblastoma displays distinct regional matrix metalloprotease expression. J Neurooncol. 2001;53:99–106. doi: 10.1023/a:1012241031138. [DOI] [PubMed] [Google Scholar]
- 12.Hanemaaijer R, Verheijen JH, Maguire TM, et al. Increased gelatinase-A and gelatinase-B activities in malignant vs. benign breast tumors. Int J Cancer. 2000;86:204–7. doi: 10.1002/(sici)1097-0215(20000415)86:2<204::aid-ijc9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Schmalfeldt B, Prechtel D, Harting K, et al. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001;7:2396–404. [PubMed] [Google Scholar]
- 14.Ozen O, Krebs B, Hemmerlein B, Pekrun A, Kretzschmar H, Herms J. Expression of matrix metalloproteinases and their inhibitors in medulloblastomas and their prognostic relevance. Clin Cancer Res. 2004;10:4746–53. doi: 10.1158/1078-0432.CCR-0625-03. [DOI] [PubMed] [Google Scholar]
- 15.Tong AW, Zhang YA, Nemunaitis J. Small interfering RNA for experimental cancer therapy. Curr Opin Mol Ther. 2005;7:114–24. [PubMed] [Google Scholar]
- 16.Mohan PM, Chintala SK, Mohanam S, et al. Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Res. 1999;59:3369–73. [PubMed] [Google Scholar]
- 17.Lakka SS, Gondi CS, Dinh DH, et al. Specific interference of uPAR and MMP-9 gene expression induced by double-stranded RNA results in decreased invasion, tumor growth and angiogenesis in gliomas. J Biol Chem. 2005;280:21882–92. doi: 10.1074/jbc.M408520200. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Healy E, Dempsey M, Lally C, Ryan MP. Apoptosis and necrosis: mechanisms of cell death induced by cyclosporine A in a renal proximal tubular cell line. Kidney Int. 1998;54:1955–66. doi: 10.1046/j.1523-1755.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- 20.Orian-Rousseau V, Aberdam D, Rousselle P, et al. Human colonic cancer cells synthesize and adhere to laminin-5. Their adhesion to laminin-5 involves multiple receptors among which is integrin alpha2beta1. J Cell Sci. 1998;111:1993–2004. doi: 10.1242/jcs.111.14.1993. [DOI] [PubMed] [Google Scholar]
- 21.Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92:326–33. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 22.Burke F, East N, Upton C, Patel K, Balkwill FR. Interferon gamma induces cell cycle arrest and apoptosis in a model of ovarian cancer: enhancement of effect by batimastat. Eur J Cancer. 1997;33:1114–21. doi: 10.1016/s0959-8049(97)88065-3. [DOI] [PubMed] [Google Scholar]
- 23.Erba E, Ronzoni S, Bassano L, Giavazzi R, D'Incalci M. The metalloproteinase inhibitor batimastat (BB-94) causes cell cycle phase perturbations in ovarian cancer cells. Ann Oncol. 1999;10:589–91. doi: 10.1023/a:1026438801080. [DOI] [PubMed] [Google Scholar]
- 24.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–7. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K. Increased activity of p53 in senescing fibroblasts. Proc Natl Acad Sci USA. 1995;92:8348–52. doi: 10.1073/pnas.92.18.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–15. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 28.Lin AW, Barradas M, Stone JC, van AL, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–19. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 30.Lianxu C, Hongti J, Changlong Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarthritis Cartilage. 2006;14:367–76. doi: 10.1016/j.joca.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–7. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- 32.Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 33.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 34.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–7. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 35.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 36.Sharpless NE, DePinho RA. Cancer: crime and punishment. Nature. 2005;436:636–7. doi: 10.1038/436636a. [DOI] [PubMed] [Google Scholar]
- 37.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 38.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 39.Barker FG, Chen P, Furman F, Aldape KD, Edwards MS, Israel MA. P16 deletion and mutation analysis in human brain tumors. J Neurooncol. 1997;31:17–23. doi: 10.1023/a:1005768910871. [DOI] [PubMed] [Google Scholar]
- 40.Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–5. [PubMed] [Google Scholar]
- 41.Plath T, Detjen K, Welzel M, et al. A novel function for the tumor suppressor p16(INK4a): induction of anoikis via upregulation of the alpha(5)beta(1) fibronectin receptor. J Cell Biol. 2000;150:1467–78. doi: 10.1083/jcb.150.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene. 2005;24:8291–303. doi: 10.1038/sj.onc.1208993. [DOI] [PubMed] [Google Scholar]
- 43.Martin SS, Leder P. Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol. 2001;21:6529–36. doi: 10.1128/MCB.21.19.6529-6536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pages G, Lenormand P, L'Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA. 1993;90:8319–23. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–7. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 46.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–15. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 47.Alani RM, Young AZ, Shifflett CB. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc Natl Acad Sci USA. 2001;98:7812–6. doi: 10.1073/pnas.141235398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro GI, Koestner DA, Matranga CB, Rollins BJ. Flavopiridol induces cell cycle arrest and p53-independent apoptosis in non-small cell lung cancer cell lines. Clin Cancer Res. 1999;5:2925–38. [PubMed] [Google Scholar]
- 49.Shalinsky DR, Brekken J, Zou H, et al. Broad antitumor and antiangiogenic activities of AG3340, a potent and selective MMP inhibitor undergoing advanced oncology clinical trials. Ann N Y Acad Sci. 1999;878:236–70. doi: 10.1111/j.1749-6632.1999.tb07689.x. [DOI] [PubMed] [Google Scholar]
- 50.Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139:279–93. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]