

Abstract
Tiron and N-acetyl-L-cysteine (NAC) have been recognized as potential antioxidants capable of inhibiting apoptosis induced by reactive oxygen species (ROS). Although the ROS-scavenging function of tiron and NAC is clear, the mechanism for their regulation of apoptosis is still elusive. Here we demonstrate that tiron increases nuclear factor-kappaB (NF-κB)/DNA binding and as a result enhances NF-κB transcriptional activity. In contrast, NAC inhibits NF-κB activation by reducing Inhibitor of kappaB Kinase (IKK) activity. Moreover, the expression of an NF-κB target gene, the chemokine CXCL1, is promoted by tiron and suppressed by NAC. Finally, tiron confers an anti-apoptotic function, while NAC imparts a pro-apoptotic function in melanoma cells. These functions correlate with the alteration of mitochondrial membrane potential but not ROS production or induction of activating protein-1 (AP-1). This study underscores the potential benefits of regulating NF-κB activity in melanoma cells as a therapeutic approach.
Keywords: Tiron, N-acetyl-L-cysteine, Reactive oxygen species, NF-κB, Melanoma, Apoptosis
Introduction
Recent studies have suggested that the resistance of human melanoma to apoptosis is an important mechanism underlying this cancer’s aggressiveness and its poor response to chemotherapeutic agents [1]. A class of potent agents identified for regulating the apoptotic processes in many tumor cells include reactive oxygen species (ROS). Some reports support the notion that ROS, acting as pro-survival, anti-apoptotic factors, contribute to cell transformation through protection of tumor cells from apoptosis [2, 3]. However, there has been controversy regarding the role of ROS in cancer cell survival since some in vivo studies revealed that chemotherapeutic agents or radiation induced ROS leads to an induction of apoptosis and a consequent suppression of tumorigenesis [4, 5]. Moreover, there are other related studies suggesting that ROS do not contribute to the regulation of apoptosis [6, 7]. The inconsistency among these studies reflects the fact that the knowledge about ROS function in tumor survival or death is still incomplete.
Mitochondria play a central role in the regulation of cellular apoptosis via Bcl-2 family members, including Bcl-2, Bad, or Bax. Bcl-2 is located in the outer mitochondrial membrane and acts to promote cell survival, whereas Bad or Bax show proapoptotic effects, either through interaction with Bcl-2 or through direct interaction with the mitochondria. The expression of Bcl-2 family members is regulated transcriptionally by activated NF-κB family members, which in turn control mitochondria-mediated apoptosis and, as a result, have profound effects on cell survival [8–11]. The NF-κB family is comprised of five subunits, Rel (c-Rel), p65 (RelA), RelB, p50, and p52. The NF-κB protein is comprised of two subunits and the predominantly active form in many cell types is the p65/p50 heterodimer complex, while the p50/p50 homodimer is mainly expressed in resting cells and its function is not fully understood [12]. NF-κB complexes are sequestered in the cytoplasm through their association with an inhibitor of κB protein (IκB). When the cell is exposed to activating signals, such as tumor necrosis factor-α, the IκB protein is phosphorylated by IκB kinase (IKK), ubiquitinated, and then broken down in the 26 S proteasome. This frees the NF-κB to translocate into the nucleus, where it binds to the NF-κB element in the promoter/enhancer regions of specific genes to activate transcription. Proteasome inhibitors, such as PS-341 (Bortezomib), abolish IκB degradation and maintain stable NF-κB/IκB complexes in the cytoplasm, thus inhibiting the transcriptional activity of NF-κB [13]. This results in a substantial anti-tumor activity in a variety of tumor cell lines.
Whether ROS have the capacity to induce NF-κB transcriptional activity has been controversially reported. The observation that inducers of NF-κB such as tumor necrosis factor-α (TNFα), interleukin-1 (IL-1), lipopolysaccharide (LPS), UV and ionizing radiation initiate an increase in ROS, prompted speculation that ROS may function as common mediators of NF-κB activation [2, 14–18]. However, inhibition of NF-κB activity by the ROS scavenger N-Acetyl-L-cysteine (NAC) has been shown to occur through an ROS-independent mechanism [19, 20]. In contrast, another ROS inhibitor, tiron, attenuates bortezomib-induced cell death through an ROS-dependent mechanism [4, 21], although it is unclear whether NF-κB activity has also been modified by this reagent.
Previously, our studies have shown that constitutive up regulation of NF-κB activity is associated with anti-apoptotic characteristics of several melanoma cell lines and melanoma tissues. Whether ROS play a role in the constitutive activation of NF-κB in melanoma and whether ROS scavengers directly or indirectly modify NF-κB activity of melanoma cells have not been investigated and are, therefore, the aims of this study. We show here that both tiron and NAC regulate the apoptosis network in melanoma cell lines via NF-κB, mitochondrial dependent, but AP-1 independent pathways, hence by-passing the need for ROS involvement.
Materials and Methods
Chemicals and reagents
Tiron and N-acetyl-L-cysteine were purchased from Sigma-Aldrich (St. Louis, MO). BMS-345541 (4(2′-aminoethyl) amino-1,8-dimethylimidazo (1,2-a)-quinoxaline)-4,5-Dihydro-1,8-dimethylimidazo (1,2-a) quinoxalin-4-one-2-carboxylic acid was provided by the Bristol-Myers Squibb Pharmaceutical Research Institute. BMS-345541 was dissolved in dimethyl sulfoxide (DMSO) to make up a 50 mM stock solution for in vitro experiments. Antibodies to IKKα, IKKβ, Bcl-2 and Bax were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Dihydroethidine, 3,3′dihexyloxacarbocyanine iodide, and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) were purchased from Molecular Probes (Eugene, OR). The melanoma cell lines, SK-MEL-5, SK-MEL-28, A375 and Hs294T, originally established from human metastatic melanoma, were purchased from American Type Culture Collection and cultured in DMEM:Ham’s F-12 medium containing 10% fetal bovine serum (FBS), 2 mM of L-glutamine, 100 μM of MEM non-essential amino acids (Invitrogen Corporation, Carlsbad, CA), and 1 mM of sodium pyruvate (Sigma-Aldrich, St. Louis, MO). NF-κB and AP-1 luciferase reporter gene vectors were purchased from BD Biosciences Clontech (Palo Alto, CA). The pRSV β-galactosidase reporter vector was purchased from Promega Corporation (Madison, WI).
Assessment of Cell Death
Apoptotic and necrotic cell death were assessed independently. Cytological characterization of apoptotic cells was performed by staining cells with propidium iodide (PI). Cells were permeabilized with digitonin to allow PI to enter cells and stain all nuclei. Briefly, floating cells and trypsinized adherent cells were collected, centrifuged, and resuspended in staining buffer (1×PBS, 200 μM digitonin and 50 μM PI). Fragmented and condensed nuclei, characteristic features of apoptosis, were counted using a fluorescent microscope. To assess necrosis, trypan blue staining of melanoma cells was performed and the round, trypan blue-stained nuclei were scored as necrotic. Trypan blue enters cells and stains nuclei when the plasma membrane permeability barrier fails, a hallmark characteristic of necrosis [22]. The percentage of apoptotic cells or necrotic cells within the cell population was determined by microscopy. At least 100 cells were counted from multiple fields of view for each sample. Additionally, cell surface phosphatidylserine as an apoptosis marker was stained using a FACS Annexin V-fluorescein isothiocyanate (FITC) apoptosis Detection kit (R&D System, Minneapolis, MN) and analyzed by flow cytometry as per the manufacturer’s instructions.
Preparation of Subcellular Fractions
Mitochondria were isolated from cultured melanoma cells using Mitochondria Isolation Kit (Pierce, Rockford, IL) according to the manufacturer’s instruction for the Reagent-based method. Briefly, 2×107 cells were collected, washed with 1×PBS buffer and resuspended in 0.8 ml of Reagent A on ice for 2 min. 10 μl of Reagent B was added and samples were kept on ice, with vortexing 5 sec every minute for 5min. After addition of 0.8 ml of Reagent C, the mixture was centrifuged at 700 × g for 10 min to remove the cell debris and nuclei. The supernatant was then centrifuged at 3,000 × g for 15 min. The mitochondria pellet was washed with Reagent C. The purity of mitochondria was determined by Western blotting with Hsp70 and β-actin. Hsp70 positive and β-actin negative mitochondria preparations were determined to be pure. The supernatant (cytosolic fraction) was further cleared by centrifugation at 12,000 × g for 15 min.
Mitochondria Membrane Potential (Δψm) Assay and ROS Analysis
To determine the change in mitochondria membrane potential (Δψm), the purified mitochondria were resuspended in buffer (225 mM mannitol, 75 mM sucrose, 10 mM KCl, 10 mM Tris-HCl, 5 mM KH2PO4, pH 7.2) and incubated with 80 nM DiOC6 for 15 min at 37°C, followed by FACS analysis with 488 nm excitation and 530 nm emission filters. To evaluate the generation of ROS, cells were washed and incubated in PBS containing either 5 μM dihydroethidine or 5 μM CM-H2DCFDA at 37 °C for 15 min. In the presence of ROS, dihydroethidine is oxidized to red fluorescent ethidium and CM-H2DCFDA is oxidized to become fluorescent. Cells were harvested by trypsinization, and washed with cold PBS solution three times. ROS was quantitated by FACS analysis with 488 nm excitation/585 nm emission filters for dihydroethidine and with 492 nm excitation/520 nm emission filters for fluorescent CM-H2DCFDA.
Immunoprecipitation and Kinase Assay in vitro
To analyze the IKK activity, cytoplasmic extracts were prepared from cells cultured for 24 h in serum-free medium. Cells were mechanically released from tissue culture plates by scraping in cold PBS according to standard protocols. Cells were collected by centrifugation (800 × g), and then resuspended in lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NP40, 1 mM DTT, and 0.5 mM phenylmethylsulfonyl fluoride) with Complete Protease Inhibitors (Sigma-Aldrich, St. Louis, MO). 400 μg aliquots of cytoplasmic extracts were incubated with 1 μg each of IKKα and IKKβ polyclonal antibodies and 60 μg aliquots of Protein A/G agarose-conjugated beads for 4 h at 4°C. After washing twice with wash buffer (50 mM HEPES, pH 7.0, 250 mM NaCl, 5 mM EDTA, and 0.1% NP40) and once with kinase buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 2 mM MnCl2, 25 mM β-glycerophosphate, 4 mM NaF, 0.1 mM sodium orthovanadate, and 1 mM DTT), the beads were mixed with 20 μl of kinase buffer containing 100 μM ATP, 5 μCi of γ-32P labeled ATP, and 1 μg of glutathione S-transferase (GST) fused protein of IκBα (amino acids 1–54) as a substrate of the IκB kinase and incubated at 30°C for 30 min. The reaction mixtures were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and phosphorylated IκB was detected by autoradiography. Blots of the same membrane were probed with the anti-IKKα/β specific polyclonal antibodies to quantitate and normalize the IKK activities to the total IKK protein immunoprecipitated.
Western Blot Analysis
Proteins from cell extracts were separated by 12% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. Nonspecific binding was blocked with 5% milk in TBST buffer (4 mM Tris base, 100 mM NaCl, pH 7.5, 0.05% Tween-20), incubated with the indicated primary antibodies with a dilution of 1:1000 at room temperature for 2h or 4°C overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibody for 1h. Blots were visualized by enhanced chemiluminescence assay.
Transfection and Luciferase Reporter Activity Assay
In 6-well tissue culture plates, 2×105 melanoma cells per well were co-transfected with 1 μg vector DNA of NF-κB-luciferase or AP-1-luciferase reporter gene and 0.2 μg vector DNA of pRSV β-galactosidase reporter using FuGENE 6 Transfection Reagent (Roche Diagnostics Corporation, Indianapolis, IN) following the manufacturer’s protocol. Extracts from cultured cells were prepared using a Dual-Light kit (Tropix Inc., Bedford, MA) according to the manufacturer’s instruction. The activities of luciferase and β-galactosidase were detected from a 10-sec constant light emission by the Monolight TM 3010 luminometer (BD Biosciences Pharmingen, San Diego, CA). Luciferase activity was normalized to β-galactosidase activity.
Enzyme-linked immunosorbent assay (ELISA)
To quantify the CXCL expression level in melanoma cell culture medium, 1.5×105 SK-MEL-5 cells/well were seeded in 6-well plates in duplicate in DMEM:Ham’s F-12 medium containing 10% FBS. After overnight incubation, the cell monolayers were washed with serum-free culture medium and then incubated with the serum-free media containing different concentrations of tiron or NAC for an additional 24 h at 37°C, followed by collecting the supernatants. After being cleared by centrifugation, supernatant aliquots were subjected to CXCL1 ELISA assay (R&D Systems Inc., Minneapolis, MN) according to the manufacturer’s protocol.
Establishment of Stable Bcl-2 Expressing Melanoma Cell Line
Bcl-2 expression vector pcDNA3-Bcl-2 was linearized and transfected into SK-MEL-5 cells. Empty pcDNA3 vector was also used for transfection as a vector control. Stable cell lines were selected with 1.2 mg/ml G418 in culture media containing 10% FBS for three weeks. The expression of human Bcl-2 protein was examined by Western Blot with specific monoclonal antibody to Bcl-2 (sc-7382, Santa Cruz Biotechnologies).
Electrophoretic Mobility Shift Assay (EMSA)
Cultured cell monolayers were scraped from culture dishes and the pellets were incubated in lysis buffer (10 mM HEPES, pH 7.9, 10 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 5 mM β-mercaptoethanol, 1 μM PMSF, and protease inhibitor cocktail purchased from Roche Applied Science, Indianapolis, IN) for 30 min at 4°C followed by centrifugation at 600 ×g for 5 min. After removing the supernatant, the remaining nuclear pellets were resuspended in hypertonic buffer (20 mM HEPES, pH 7.9, 450 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.5 mM DTT, 5 mM β-mercaptoethanol, 1% NP-40, 1 μM PMSF and protease inhibitor cocktail), rotated for 30 min at 4°C, followed by centrifugation at 16,000 ×g for 10 min. The supernatant was collected and the protein concentration was determined with Bio-Rad Protein Assay Reagent. Double-stranded NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′, Promega, Madison, WI) was labeled with γ-32P (Amersham Pharmacia Biotech), and 105 cpm of labeled NF-κB oligonucleotides were used in binding reactions with 5 μg nuclear extract at room temperature for 30 min as per the manufacturer’s instructions (Promega). The protein/DNA complexes were resolved in a 3.7% non-reduced polyacrylamide gel and visualized by autoradiography. Specificity of the DNA/protein complex was examined by adding 100-fold excess unlabeled oligonucleotide or a non-competitive oligonucleotide OCT-1 (Promega, Madison, WI). Supershift analysis was performed with addition of 1μg anti-NF-κB antibody (p65 or p50, Santa Cruz, CA) to the binding reaction for 30 min before addition of radio-labeled probe to identify the components of the protein/DNA complex.
Statistical Analysis
The results are expressed as mean ± S.D. from at least three independent experiments. Statistical analysis was performed using the unpaired Student’s t-test. A probability of less than 0.05 was considered significant.
Results
Tiron displays an anti-apoptotic effect while NAC has a pro-apoptotic effect on melanoma cells
Tiron is a vitamin E analog and NAC is a non-toxic dietary glutathione precursor. Both are antioxidant compounds that have been reported to protect cancer cells from ROS-induced apoptosis [4, 23]. To examine whether either tiron or NAC directly modulates apoptosis in melanoma cells, we first exposed SK-MEL-5 melanoma cells to increasing concentrations of tiron (0, 0.1, 1.0 and 10 mM) or NAC (0, 0.1, 1.0 and 10 mM) in serum-free medium for 24 h. After harvest, SK-MEL-5 cells were stained with either Propidium Iodide (PI) or trypan blue and morphological evaluation of apoptotic or necrotic cells was performed by microscopy (Fig. 1A). The percentage of apoptotic or necrotic cells was scored as described in “Materials and Methods”. The results showed a protective role for tiron in the cultured melanoma cells. In contrast, 1 mM NAC initiated 22 ± 8.7% apoptosis and 10 mM NAC treatment resulted in 78 ± 14% apoptosis in SK-MEL-5 cells. Nevertheless, no significant difference in the percentage of necrotic cells was observed between NAC and tiron treated samples (Fig. 1B, 1C). To further confirm that the apoptotic process was involved in the above cell death, SK-MEL-5 melanoma cells pre-treated with tiron (0, 1.25, 2.5, 5 and 10 mM), NAC (0, 1.25, 2.5, 5 and 10 mM) or PBS buffer control were subjected to Annexin V-FITC staining, followed by flow cytometry analysis for quantitating the apoptotic and necrotic cell populations in these cell samples. The results showed that the treatment with tiron had no effect on SK-MEL-5 cell apoptosis, whereas NAC treatment resulted in an increase in the apoptotic cell population (Fig. 1C). These data are consistent with the previous morphological observations and demonstrate that NAC, but not tiron, induces apoptosis in melanoma cells. To clarify whether this apoptotic process is cell line-specific or drug-specific, similar cytometry analysis were performed on three additional melanoma cell lines (Hs 294T, A375 and SK-MEL-28) after treatment with either 2 mM NAC or 10 mM tiron for 24 h. The results showed again that NAC, but not tiron, was a specific inducer of apoptosis in all the melanoma cell lines investigated (Fig. 1D). To extend this finding, SK-MEL-5 cells were co-exposed to BMS-345541[24], an IKKβ inhibitor that induces melanoma cell apoptosis at the concentration of 10 μM, and either tiron (10 mM) or NAC (1 mM) for 24h followed by the quantification of percentage of apoptotic cells. The results showed that tiron protected melanoma cells from BMS-345541-induced apoptosis by reducing 26% of apoptotic cell population (reduced from 52 ± 1.7% in control to 38 ± 4.2% in tiron treated cultures), while NAC increased the apoptotic cell population by 54% (from 52 ± 1.7% in control to 80 ± 6.2% in NAC treated cultures) (Fig. 1E). Together with other previous findings [19, 21], our results suggest that tiron exhibits anti-apoptotic effects, while NAC acts as a pro-apoptotic factor for melanoma cells.
Fig. 1.
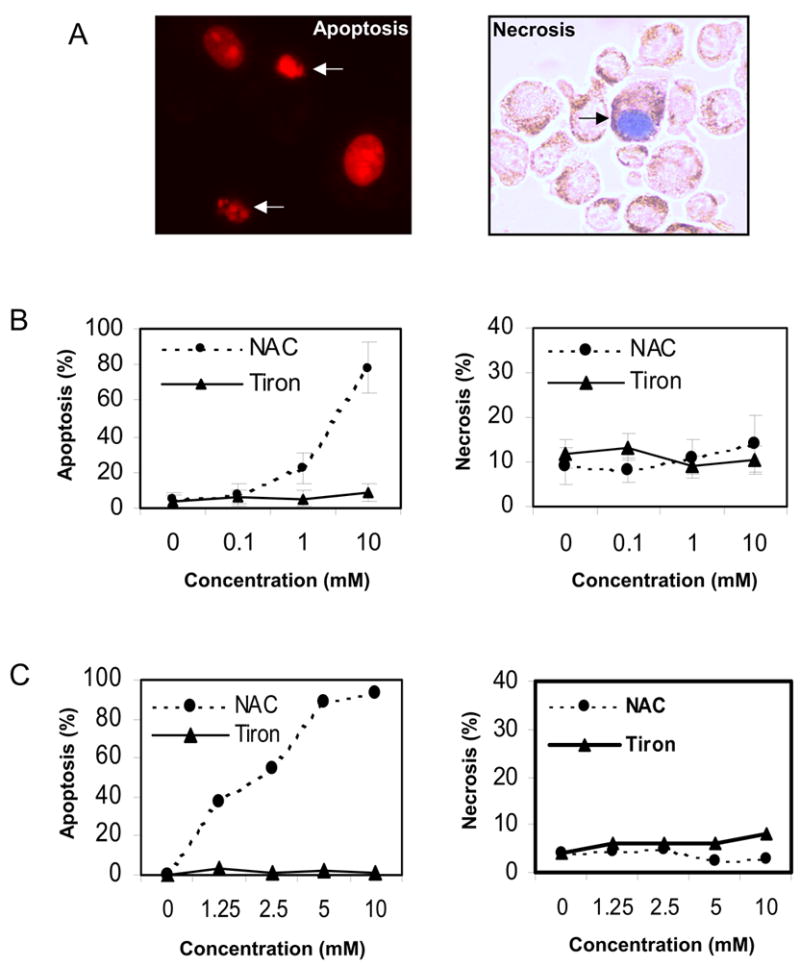
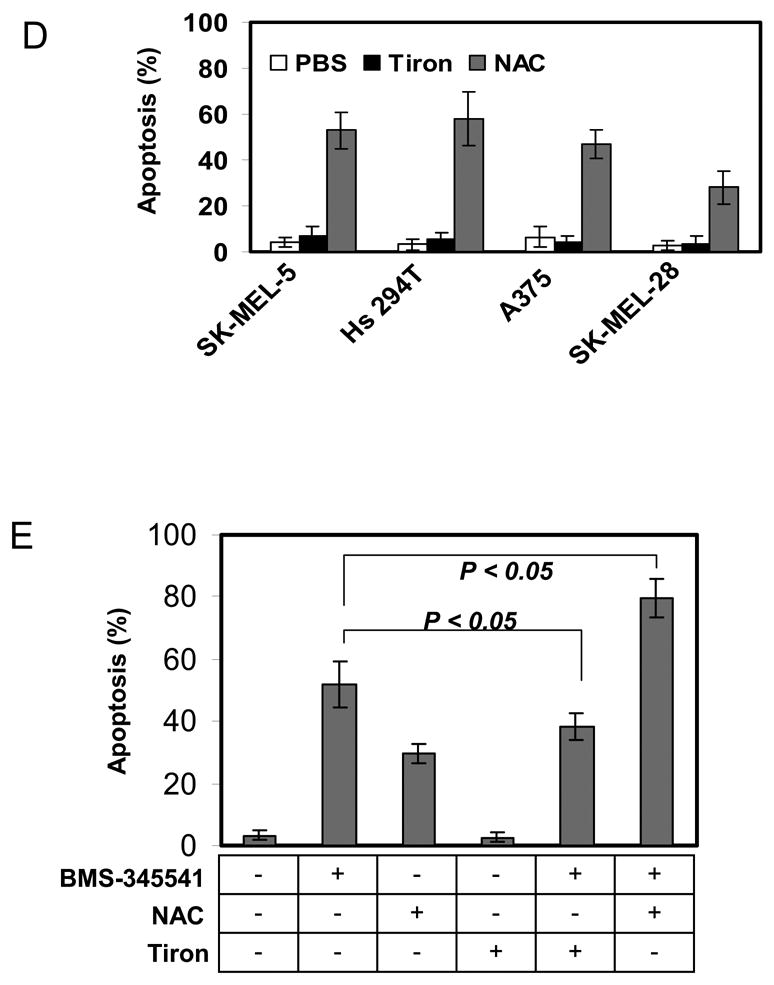
NAC, but not tiron, induces apoptosis in cultured melanoma cells. A, Propidium Iodide (PI) staining for typical apoptotic (arrow) and non-apoptotic nuclei (left panel). Trypan blue staining for necrotic (arrow) and non-necrotic cells (right panel). B, 2×105 SK-Mel-5 cells were cultured in serum-free medium in 6-well plates and treated with the indicated concentrations of NAC and tiron for 24h. Subsequently, cells were harvested and scored for apoptosis (left panel) or necrosis (right panel) as described in “Materials and Methods.” C, 1×106 SK-Mel-5 cells were cultured in serum-free medium in 10-cm culture dishes and treated with the indicated concentrations of NAC and tiron. After 24h treatment, cells were harvested and subjected to Annexin V-FITC and trypan blue exclusion staining. Apoptosis and necrosis were analyzed by flow cytometry and normalized to untreated controls. D, melanoma cell lines (SK-MEL-5, Hs294T, A375 and SK-MEL-28) were exposed to 10 mM tiron or 2 mM NAC for 24h. Apoptotic cells were determined using PI staining. E, tiron protected, while NAC enhanced, the BMS-345541-induced cell death after SK-MEL-5 cells were exposed to tiron (10 mM) or NAC (2 mM) and/or BMS-345541 (10 μM) for 24h. The induction of apoptotic cells between different treatments was analyzed using the unpaired Student’s t test. A probability (p) of less than 0.05 was taken as statistically different.
Tiron and NAC regulate melanoma cell survival or death through mitochondria dependent and ROS independent mechanisms
To clarify whether ROS contribute to apoptosis in melanoma cells, SK-MEL-5 cells were first exposed to 10 mM tiron or 2 mM NAC for 24 h, then subjected to 10 μM BMS-345541 for 24 h. These SK-MEL-5 cells were harvested for the determination of BMS-345541 induced ROS production (Fig. 2A, B). To evaluate the general profiles of ROS production in these cells, two indicative reagents, Dihydroethidine and CM-H2DCFDA, were utilized in the assays for the detection of intracellular superoxide, hydrogen peroxide and other ROS, respectively (Fig. 2A, B). The results showed that BMS-345541 increased ROS production by 1.3 and 3.3 fold with Dihydroethidine and CM-H2DCFDA assays, respectively. However, the pre-treatments of SK-MEL-5 cells with tiron or NAC alleviated BMS-345541 induced ROS production. Obviously, these data suggest that the differential effect of tiron and NAC on SK-MEL-5 cell apoptosis, as shown previously in figure 1E, can not be attributed to the abilities of these two antioxidants to reduce intracellular ROS. Based on these findings, we speculated that in addition to being antioxidants, tiron and NAC possess other features, which may regulate melanoma cell apoptosis in a ROS-independent manner. Since mitochondrial functional integrity is critical for cell apoptosis and survival, and Bcl-2 protein is important to maintain mitochondrial normal function [24–26], we next assayed mitochondrial membrane potential and examined mitochondrial Bcl-2 expression in SK-MEL-5 cells after tiron or NAC treatments. The data show that NAC greatly reduced mitochondrial membrane potential in a concentration dependent fashion, while tiron had no effect on the membrane potential (Fig. 2C). In addition, tiron did not interfere with the expression of Bcl-2 in cultured SK-MEL-5 cell mitochondria (compare lanes 1 and 2 in Fig. 2D), whereas the treatment of NAC alone markedly down-regulated mitochondrial Bcl-2 expression (compare lanes 1 and 3, Fig. 2D). When BMS-345541 was introduced into the culture media, the expression of Bcl-2 was suppressed and was no longer detectable by Western blot (Fig. 2D, lane 4). Surprisingly, such an inhibition of Bcl-2 expression by BMS-345541 was countered moderately by the pre-treatment with 10 μM tiron (Fig. 2D, lane 5), but not with NAC pre-treatment (Fig. 2D, lane 6). As Bcl-2 is an important anti-apoptotic factor, we speculated that the differential effects of tiron and NAC on mitochondrial Bcl-2 expression may account for the lower or higher apoptosis rates induced by these antioxidants, respectively (Fig. 1B to 1E). To test this notion, we established a stable SK-MEL-5 cell line, which expressed a high level of recombinant human Bcl-2 protein (Fig. 2E, left panel), and added 0 to 10 mM NAC to induce apoptosis. The increased expression of Bcl-2 reduced the apoptotic response to 2.5 and 5 mM NAC treatment by 61% (from 41±6.7% to 16±5.0% apoptotic cells, p=0.007) and 35% (from 71±9.5% to 46±9.6% apoptotic cell percentage, p=0.03), respectively (Fig. 2E, right panel) as compared to that of pcDNA3 empty vector transfected control SK-MEL-5 cells. These data clearly reveal that the loss of Bcl-2 is an important cause of SK-MEL-5 apoptosis during NAC treatment. In contrast, tiron does not interfere with Bcl-2 expression and therefore shows no pro-apoptotic effect. Furthermore, to rule out a direct action of these two antioxidants on mitochondrial integrity, mitochondria fractions isolated from SK-MEL-5 were incubated with 1 or 10 μM NAC or tiron for 30 min, and the release of cytochrome-c into the supernatant was analyzed by Western blot as previously described [27]. As shown in Figure 2F, in comparison with 200 μM Ca2+ positive control, no release of mitochondrial cytochome-c was detected in PBS, NAC (1 and 10 mM) or tiron (1 and 10 mM) treated mitochondria samples, indicating that neither NAC nor tiron is capable of directly affecting the mitochondrial integrity. These results demonstrate that, in spite of antioxidant properties, the differential outcome of tiron and NAC on the apoptotic pathway in melanoma cells is predominantly dependent on their individual effect on Bcl-2 expression, which consequently affects the mitochondrial integrity. The data also suggest that the intimate association of Bcl-2 protein with mitochondrial membrane is important for melanoma cell mitochondrial integrity, cellular apoptosis, as well as cell survival.
Fig. 2.
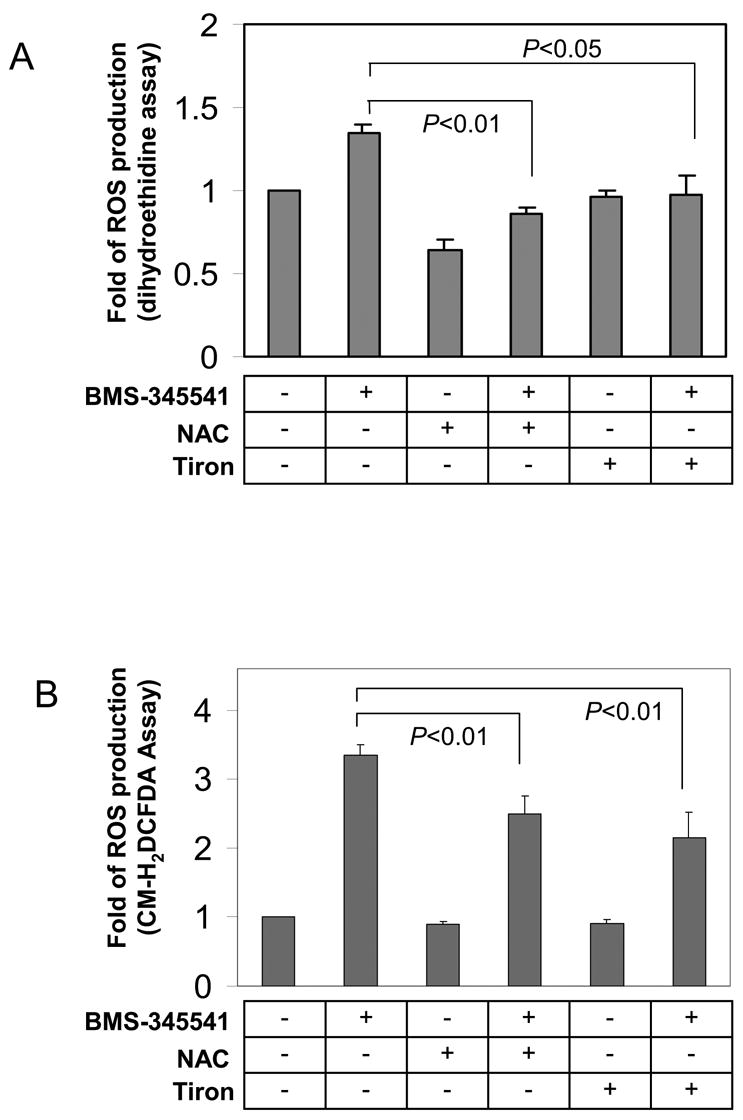
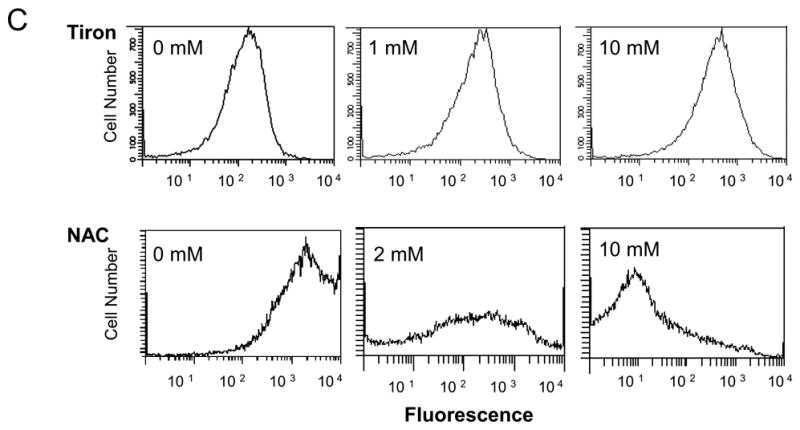
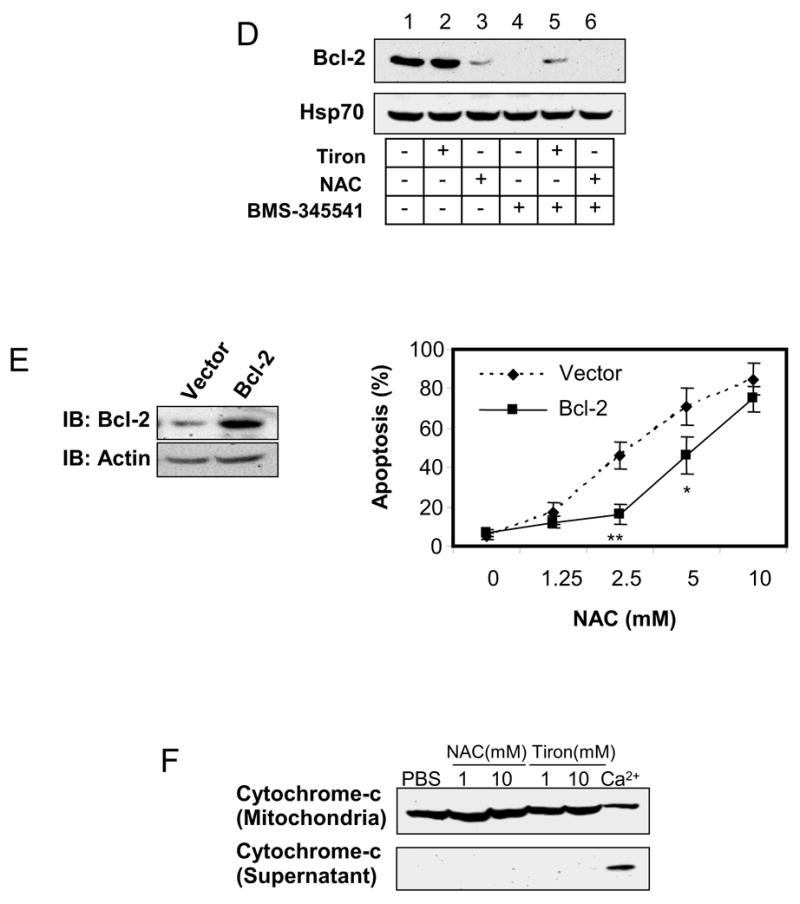
Different effects of tiron and NAC on mitochondria-dependent melanoma cell apoptosis. A, ROS generation is abrogated in melanoma cells by antioxidants in dihydroethidine assay. SK-Mel-5 cells were exposed to 10 mM tiron or 2 mM NAC for 24 h followed by additional treatment with or without 10μM BMS-345541 for 24h. Cellular ROS were determined as described in “Materials and Methods”. The changes of ROS production under each treatment were expressed as fold of non-treatment group. The mean ± SD of three independent experiments are indicated and statistical analyses were performed by Student’s t test. B, ROS generation is abrogated in melanoma cells by antioxidants in CM-H2DCFDA assay. SK-Mel-5 cells were treated similarly as described in Figure 2A and underwent CM-H2DCFDA assay for quantification of intracellular ROS. The fold differences of ROS in the cells with these treatments were calculated and the mean ± SD of three independent experiments are shown. The statistical analyses were performed by Student’s t test. C, NAC, but not tiron, dissipated mitochondrial membrane potential. 2×106 SK-Mel-5 cells were cultured in a 10-cm culture dish in serum-free medium containing the indicated concentrations of tiron (upper panels) and NAC (lower panels). After 24 h treatment, cells were harvested and subjected to determination of mitochondrial membrane potential by flow cytometry. D, SK-Mel-5 cells (2×106) were exposed to 10 mM tiron or 2 mM NAC and/or 10 μM BMS-345541 as indicated for 24h. Mitochondria were isolated and subjected to Western blotting for Bcl-2 and Hsp70 proteins. Hsp70 was used as mitochondrial protein loading control. E, stable SK-MEL-5 cell lines transfected with pcDNA3-Bcl-2 or pcDNA3 vector were established and recombinant Bcl-2 expression was highly increased in the formal vector transfected cells (left panel). The two cell lines were exposed to 0 to 10 mM NAC for 24 h and subjected to PI staining. The apoptotic populations of the two cell lines were evaluated from three independent experiments and the mean ± SEM of percentage of apoptotic cells is shown. *: p<0.05, **: p<0.01. F, the purified mitochondria were incubated with PBS, tiron (1 and 10 mM) or NAC (1 and 10 mM) at room temperature for 30 min. The supernatant was assessed for cytochrome-c release. Treatment of mitochondrial preparations with 200 μM Ca2+ was used as a positive control. The experiments were repeated two times in duplicates. The results shown are from a representative experiment (Fig D, E and F).
The regulation of NF-κB, but not AP-1 is involved in ROS scavenger induced melanoma cell apoptosis and survival
The NF-κB mediated signal transduction pathway has been demonstrated as a critical regulatory mechanism for pro- or anti-apoptotic events in many cell lines. To illustrate whether NF-κB also regulates mitochondria-dependent apoptosis in SK-MEL-5 cells, we transiently transfected SK-MEL-5 cells with an NF-κB-responsive luciferase reporter gene and examined the effect of tiron or NAC on NF-κB transcriptional activity. Interestingly, tiron markedly elevated NF-κB activity in a concentration-dependent manner, whereas NAC reduced NF-κB activity (Fig. 3A, left panel). Furthermore, such differential changes in NF-κB activity were concomitant with different CXCL1 production from SK-MEL-5 cells treated with tiron or NAC. As shown in Figure 3A, right panel, CXCL1 secretion into the culture medium was significantly increased in response to tiron-induced NF-κB activity and was linearly decreased in response to NAC inhibition of NF-κB activity. Since IKK is a key upstream mediator of the NF-κB signal transduction pathway, we exposed SK-MEL-5 cells to 1, 5, and 10 mM of either tiron or NAC for 24 h and determined the IKK activity of the immunoprecipitated IKK complexes from each cell lysate. The results clearly indicate a robust inhibition of IKK activity in SK-MEL-5 cells treated with 5 and 10 mM NAC (Fig. 3B, right panel). Surprisingly, treatment with tiron did not significantly alter the IKK activity in SK-MEL-5 cells (Fig. 3B, left panel). These results suggest that NAC acts as an inhibitor for IKK activity, while tiron has no effect on IKK activity. Since the strong induction of NF-κB luciferase activity by tiron was not accompanied by an equally strong induction of IKK activity, further studies using a DNA-protein binding assay were performed to explore the mechanism of tiron induced NF-κB activation. Nuclear proteins were extracted from cultured SK-MEL-5 cells that were exposed to tiron for 24 h and the NF-κB/DNA binding activity was analyzed by EMSA. As shown in Figure 3C, the untreated sample displays a faint nuclear NF-κB band, which represents a basal level of NF-κB/DNA binding in SK-MEL-5 cells and is expressed constitutively. When these cells were treated with tiron, NF-κB/DNA binding was increased in a concentration-dependent manner (Figure 3C, lanes 1–4). Further EMSA experiments using SK-MEL-5 nuclear fractions derived from cells treated with 10 μM tiron confirmed that the binding complex was of NF-κB origin and comprised mainly of NF-κB p50 and p65 elements, since it was depleted by the competitive double-stranded NF-κB oligo (Figure 3C, compare lanes 5 and 6), unaffected by the non-competitive OCT-1 double-stranded oligo (Figure 3C, lane 7), and shifted by anti-human NF-κB p65 and p50 antibody or the combination of these two antibodies (Figure 3C, lanes 8, 9, 10).
Fig. 3.
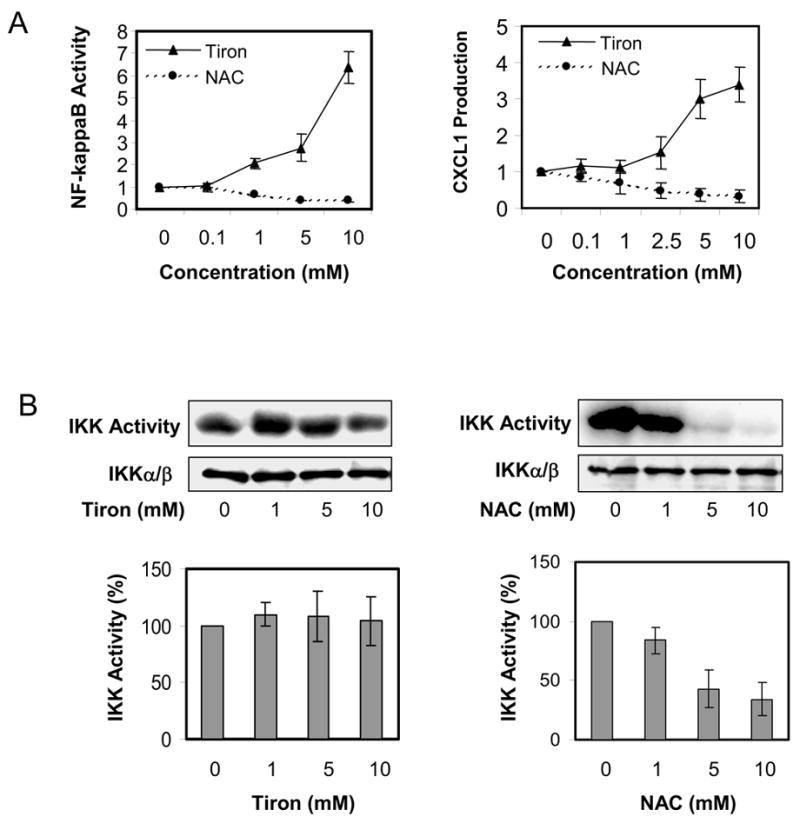
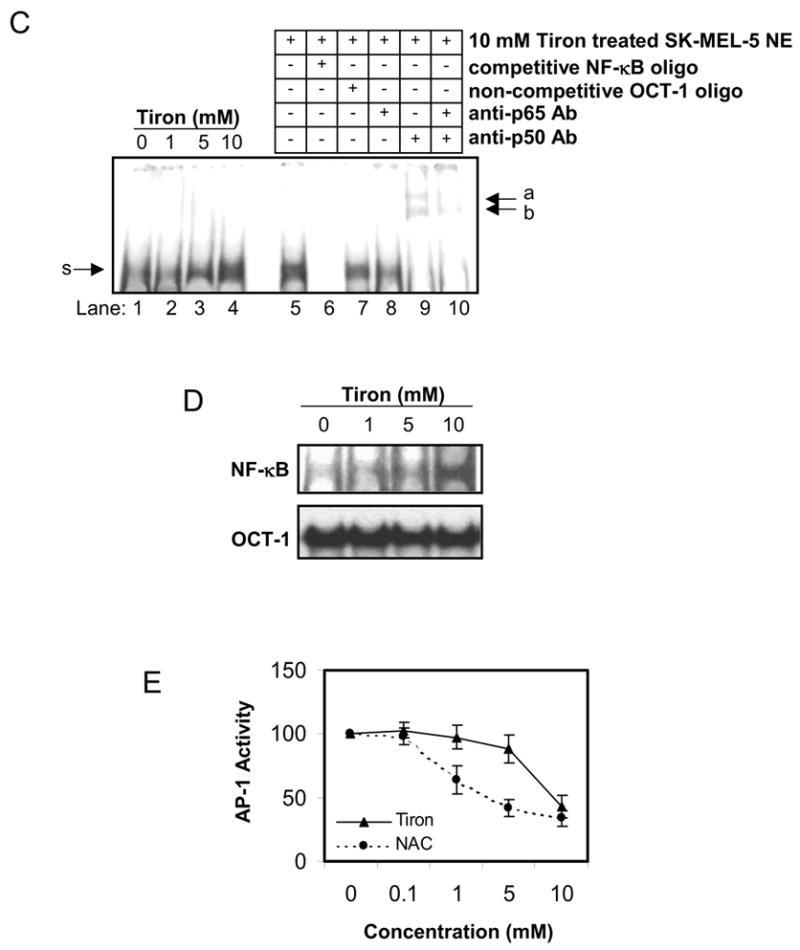
Regulation of tiron or NAC on NF-κB pathway. A, NF-κB reporter luciferase activity and CXCL1 secretion are induced by tiron and suppressed by NAC. SK-Mel-5 cells were co-transfected with an NF-κB transcriptional luciferase reporter vector and a pRSV β-galactosidase reporter vector for 24 h followed by treatment with increasing concentrations of tiron or NAC (left panel) for an additional 24 h. Subsequently, luciferase activity was determined as described in “Materials and Methods.” For assessment of NF-κB target gene expression, release of the CXCL1 chemokine into the medium of cultured SK-MEL-5 cells exposed to tiron or NAC was measured using ELISA assay (right panel). Both luciferase activity and CXCL1 production are expressed as mean ± S.D. and the experiments were repeated three times with consistent results. B, IKK activity is down-regulated by NAC, but not tiron. SK-MEL-5 cells were treated with the indicated concentrations of either tiron or NAC for 24 h. The cellular IKKα/β proteins were immunoprecipitated and subjected to IKK activity assay and immunoblotting for IKKα/β proteins (upper panel) as described in “Materials and Methods.” The mean ± S.D. of IKK activity from three independent experiments is shown in the lower panel. C, tiron increased cellular NF-κB DNA binding in vivo. Nuclear extracts (NE) were prepared from the SK-MEL-5 cells treated 24 h with the indicated concentrations of tiron (lanes 1–4). Nuclear NF-κB proteins binding to labeled NF-κB oligonucleotide probe was assessed by EMSA as described in “Materials and Methods.” The specificity of the protein/DNA complex was identified by competition with NF-κB oligonucleotides and by super-shift analysis using specific NF-κB antibodies (p50 or/and p65) (lanes 5–10). (s), specific NF-κB band. (a, b), shifted bands. D, tiron directly increased the binding affinity between NF-κB complex and DNA. Five microgram of SK-MEL-5 nuclear extract were incubated with γ-32P labeled NF-κB oligonucleotides. The indicated concentrations of tiron were added to the “cell-free” mixture and NF-κB/DNA binding activity was assessed by EMSA. The binding of nuclear protein and OCT-1 oligo with the addition of different concentrations of tiron serves as loading control. Data shown here are representative of three independent experiments. E, the AP-1 activity profile in melanoma cells treated with either tiron or NAC. SK-MEL-5 cells were co-transfected with AP-1 luciferase reporter and β-galactosidase reporter. After 24 h transfection, cells were subsequently treated with different concentrations of either tiron or NAC for an additional 24 h. Cellular luciferase activity was determined as described in “Materials and Methods.” Luciferase activity in the treated cells was normalized to that of control cells. Mean ± S.D. of luciferase activity from three experiments is shown.
It is interesting to note that tiron increases NF-κB binding to its target DNA. We speculate two possible explanations for this result. First, tiron may facilitate NF-κB translocation into the nucleus, and secondly, tiron may enhance NF-κB binding to its target DNA. Both possibilities could result in activation of NF-κB associated gene transcription. To clarify this issue, tiron was added directly into a cell-free NF-κB/DNA binding reaction mixture containing equal amounts of SK-MEL-5 cell nuclear extract and labeled oligonucleotide. The result demonstrated that 5–10 mM tiron potentiated NF-κB/DNA binding activity (Fig. 3D). Furthermore, the direct binding enhancement by tiron at the studied concentrations is specific for NF-κB complex because no such affect was observed on the binding of nuclear protein to OCT-1 oligo (Fig. 3D).
Lastly, although the NF-κB signal has been recognized as a key regulator in the network of apoptosis, there is an extensive debate over the rival roles between NF-κB and AP-1 on apoptosis [28]. To elucidate whether the change of AP-1 profile is linked to apoptosis in melanoma cells, SK-MEL-5 cells transfected with an AP-1-responsive luciferase reporter gene were exposed to tiron or NAC treatment for 24 h. The results showed that AP-1 promoter activity was suppressed by both tiron and NAC treatment in a concentration dependent manner (Fig. 3E). Therefore, the profile of AP-1 transcriptional activity does not appear to be directly correlated to the differential effects of tiron or NAC on the apoptosis of SK-MEL-5 cells observed in Figure 1. Taken together, the above data suggest that in addition to being a ROS-scavenger, both tiron and NAC act on a second target—the NF-κB signaling pathway regulating melanoma cell death.
Discussion
In this study we have shown that the antioxidant tiron plays a novel role of enhancing NF-κB/DNA binding in melanoma cells. This effect makes tiron a unique ROS scavenger that can function as a “chemical enhancer” for NF-κB dependent gene transcription with anti-apoptotic effect, even when the upstream components of this pathway are interrupted by the IKK inhibitor BMS-345541. In contrast, another antioxidant, NAC, acts negatively to regulate the NF-κB pathway by inhibiting IKK activity, leading to decreased expression of NF-κB regulated anti-apoptotic genes, such as Bcl-2, and disruption of mitochondrial function as well as induction of apoptosis in melanoma cells. As both tiron and NAC inhibited ROS formation, but differentially affected the fate of cultured melanoma cells, this study underscores the importance of the anti-apoptotic function of NF-κB activation in melanoma cells and reveals that modification at different levels of the NF-κB pathway by chemical reagents may have a potent effect on the expression of NF-κB regulatory genes, and this effect may impose significant impact on tumor cell apoptosis.
Previously, we have shown that constitutive activation of NF-κB leads to melanoma tumor progression in both in vivo and ex vivo models [10, 29, 30]. We have also shown that blocking the NF-κB pathway by treating melanoma cells either with the IKKβ inhibitor BMS-345541 or with the proteasome inhibitor PS-341 is an effective strategy to inhibit melanoma growth [21, 24, 31]. Although the relationship between NF-κB activation and escape from apoptosis is established in melanoma models, a recent study by Fribley et al. in head and neck squamous cell carcinoma (HNSCC) cells revealed that inhibition of NF-κB activation by PS-341 alone could not sufficiently result in apoptosis. The study suggested another mechanism, specifically the induction of ROS, played a more important role in PS-341 induction of squamous cell carcinoma apoptosis [21]. Further comparisons with studies on vascular endothelial and fibroblast cell lines indicated that stress-induced ROS either activate NF-κB through the activation of PKC pathway or directly modulate the expression of genes such as IL-6, which is a NF-κB regulated gene, or through the NF-κB independent ERK and p38 MAPK pathway [32, 33]. Based on these studies, the regulation of NF-κB activation by ROS is rather cell line dependent. Consequently, it is not surprising that inconsistency exists among several studies regarding the effects of ROS scavengers on NF-κB activation [15, 32, 34–36]. In analysis of the experimental results, it is interesting to note that BMS-345541 is capable of inducing an increase of ROS in SK-MEL-5 melanoma cells, especially with the detection by CM-H2DCFDA assay, in addition to selectively inhibiting IKK activity [37]. In agreement with our previous report [24], BMS-345541 induced apoptosis in SK-MEL-5 cells, as shown in Figure 1E. By performing studies with two different ROS scavengers, we show that both NAC and tiron inhibit BMS-345541 induced ROS formation, but these reagents have differential effects on NF-κB activation and this in turn leads to the difference in cell fate, where tiron is anti-apoptotic and NAC is pro-apoptotic. Thus, melanoma represents a tumor type in which NF-κB largely regulates the apoptotic fate, whereas ROS show little, if any, regulatory effect on apoptosis.
The bulk of information about ROS regulation of NF-κB activation was based on studies in which ROS scavengers were utilized, and these anti-ROS reagents were considered to act exclusively on the inhibition of intracellular ROS. Recent studies revealed that these scavengers also regulate signal transduction pathways and these regulatory mechanisms are not completely compatible with their antioxide functions. For example, NAC has long been recognized as a ROS inhibitor and its effect on negative regulation of NF-κB was considered a secondary consequence of the compromised ROS formation. This notion has been challenged by Hayakawa et al. in a study on Jurkat T cells showing that the NAC inhibition of NF-κB activation is not mediated by ROS, but results from a reduction in the affinity of TNF to its receptor. As a consequence, TNF induced NF-κB activation was significantly decreased [15]. Similarly, the pro-survival effect of tiron may also be mediated by ROS-independent mechanisms. Fernandez et al. recently showed that in a broad spectrum of cancer cell lines including melanoma, tiron selectively blocks the effect of the proteasome inhibitor PS-341 and restores the function of the proteasome by preventing the accumulation of proteasome targets and processing of apoptotic effectors [38]. This study suggests a close relationship between the affects of tiron and NF-κB activation in melanoma cells, because proteasome processing is a critical regulator of NF-κB activation, which is related to multidrug resistance and melanoma growth [10, 39, 40]. Indeed, our EMSA assay showed that the addition of tiron in cell culture enhances NF-κB/DNA binding in a concentration dependent manner. Furthermore, this enhanced binding is paralleled with increased CXCL-1 production. This effect of tiron on NF-κB/DNA binding was confirmed particularly in a cell-free NF-κB/DNA binding EMSA, where the involvement of ROS is unlikely. In addition, our results reveal that the effect of tiron is relatively specific for NF-κB since the binding of nuclear extracts to an OCT-1 DNA probe was not affected. These data suggest a unique biochemical feature for tiron acting directly at the transcription level to augment NF-κB activity.
Further analysis of our results also revealed the following possible mechanisms for tiron or NAC on the regulation of SK-MEL-5 cell survival or death. First, the survival or apoptotic death of this melanoma cell line is attributable in large part to the status of mitochondrial function that are maintained exquisitely by the Bcl-2 family of proteins [9, 11, 25]. In our study, tiron did not interfere directly with mitochondrial integrity, membrane potential or mitochondria-associated Bcl-2 expression. However, it partially countered BMS-345541 induced suppression of mitochondrial Bcl-2 expression. This effect on Bcl-2 and perhaps other NF-κB regulated inhibitors of apoptosis [41] apparently rescued a fraction of SK-MEL-5 cells from BMS-345541 induced apoptosis. In contrast, as shown by this study, NAC suppressed NF-κB activation through inhibition of IKK kinase activity, thus compromising Bcl-2 expression and enhancing apoptosis in melanoma cells.
Although there is a debate for the rival role between NF-κB and AP-1 in the regulation of apoptosis [28], our signaling profiles of melanoma cells in response to treatment with tiron or NAC suggest that NF-κB, not AP-1, is most likely responsible for the BMS-345541 induction of melanoma cell apoptosis. According to the above analysis, we have summarized with a schematic illustration the proposed mechanisms associated with the functions of tiron and NAC in Figure 4. SK-MEL-5 cell responses to the treatment with these three reagents are summarized in Table 1.
Fig. 4.
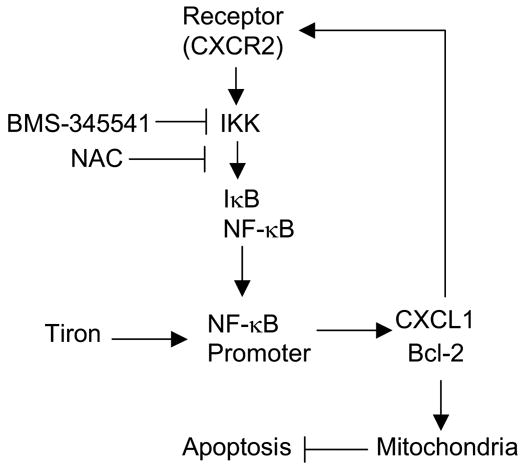
Schematic of targeting the NF-κB pathway in melanoma cells. Constitutive activation of IKK/NF-κB via chemokine CXCL1 forms an “autocrine-loop” that confers autonomous growth capacity to melanoma cells via induction of Bcl-2 to stabilize mitochondria membrane, allowing cells to escape from apoptosis. The inhibition of IKK activity by either NAC or BMS-345541 results in mitochondria-mediated apoptosis. In contrast, tiron increases NF-κB/DNA binding activity, which promotes NF-κB transcriptional activity, and up-regulates down-stream anti-apoptotic gene expression, consequently, maintains mitochondrial membrane stability and protects cells from apoptosis.
Table 1.
Summary of the molecular alteration in SK-MEL-5 melanoma cells with treatments of tiron, NAC and BMS-345541.
| Responder | Tiron | NAC | BMS-345541 |
|---|---|---|---|
| NF-κB | ↑ | ↓ | ↓ |
| CXCL1 | ↑ | ↓ | ↓ |
| IKK | — | ↓ | ↓ |
| Bcl-2 | — | ↓ | ↓ |
| Δψm | — | ↓ | ↓ |
| ROS | ↓ | ↓ | ↑ |
| AP-1 | ↓ | ↓ | ↑ |
|
| |||
| Apoptosis | ↓ | ↑ | ↑ |
Note: ↑ increase; ↓ decrease; — no significant change.
Given the role of constitutive NF-κB signaling in the development of cancer and in the tumor resistance against chemotherapy, much effort has been directed at the identification and characterization of drugs that can modulate this pathway [9, 10, 31, 42]. Elucidating the mechanism by which antioxidants directly modulate NF-κB activation will extend our understanding of apoptosis-resistance in melanoma cells, as well as the molecular mechanisms of antioxidants on NF-κB signaling in mammalian cells.
Conclusions
The main finding of this study is that the antioxidants tiron and NAC regulate apoptosis in melanoma cells through an NF-κB mediated event that also affects mitochondrial function. In contrast, the actions of AP-1 or ROS are less likely associated with the regulation of apoptosis by these antioxidants in melanoma.
Acknowledgments
These studies were supported by grants from Department of Veterans Affairs Career Scientist Award and Merit Award (to A. R.), CA116021 (to A.R.), the NIH sponsored VICC Cancer Center Grant CA68485 and 5P30 AR41943 to the VUMC Skin Disease Research Center. We thank Dr. James R. Burke, Bristol-Myers Squibb Pharmaceutical Research Institute, Princeton, New Jersey, for providing BMS-345541 and Dr. Lee Yong, Department of Surgery and Pharmacology, University of Pittsburgh School of Medicine, for pcDNA3-Bcl-2 vector. We thank Catherine E. Alford, VA Flow Cytometry, Special Resource Center, Nashville, Tennessee for assistance on Flow Cytometry analysis. We also thank Nicole F. Neel, Katayoun I. Amiri and Ross M. Potter of Editor’s Club of Vanderbilt University School of Medicine for critical reading of the manuscript.
Abbreviations
- NAC
N-acetyl-L-cysteine
- IKK
IκB kinase
- NF-κB
nuclear factor-κB
- IκB
inhibitor of κB
- BMS
Bristol-Myers Squibb
- CXCL
CXC chemokine ligand
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 2.Dhar A, Young MR, Colburn NH. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol Cell Biochem. 2002;234–235:185–193. [PubMed] [Google Scholar]
- 3.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 4.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 5.Lebedeva IV, Su ZZ, Sarkar D, Gopalkrishnan RV, Waxman S, Yacoub A, Dent P, Fisher PB. Induction of reactive oxygen species renders mutant and wild-type K-ras pancreatic carcinoma cells susceptible to Ad.mda-7-induced apoptosis. Oncogene. 2005;24:585–596. doi: 10.1038/sj.onc.1208183. [DOI] [PubMed] [Google Scholar]
- 6.Callapina M, Zhou J, Schmid T, Kohl R, Brune B. NO restores HIF-1alpha hydroxylation during hypoxia: role of reactive oxygen species. Free Radic Biol Med. 2005;39:925–936. doi: 10.1016/j.freeradbiomed.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Lin S, Fujii M, Hou DX. Rhein induces apoptosis in HL-60 cells via reactive oxygen species-independent mitochondrial death pathway. Arch Biochem Biophys. 2003;418:99–107. doi: 10.1016/j.abb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP, Bours V. NF- kappa B2/p100 induces Bcl-2 expression. Leukemia. 2003;17:1349–1356. doi: 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- 9.Fahy BN, Schlieman MG, Mortenson MM, Virudachalam S, Bold RJ. Targeting BCL-2 overexpression in various human malignancies through NF-kappaB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother Pharmacol. 2005;56:46–54. doi: 10.1007/s00280-004-0944-5. [DOI] [PubMed] [Google Scholar]
- 10.Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2:664–674. doi: 10.1038/nri887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlowski RZ, Baldwin AS., Jr NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 12.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 13.Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419–1428. [PubMed] [Google Scholar]
- 14.Briganti S, Picardo M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J Eur Acad Dermatol Venereol. 2003;17:663–669. doi: 10.1046/j.1468-3083.2003.00751.x. [DOI] [PubMed] [Google Scholar]
- 15.Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K. Evidence that reactive oxygen species do not mediate NF-kappaB activation. Embo J. 2003;22:3356–3366. doi: 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reelfs O, Tyrrell RM, Pourzand C. Ultraviolet a radiation-induced immediate iron release is a key modulator of the activation of NF-kappaB in human skin fibroblasts. J Invest Dermatol. 2004;122:1440–1447. doi: 10.1111/j.0022-202X.2004.22620.x. [DOI] [PubMed] [Google Scholar]
- 17.Vile GF, Tanew-Ilitschew A, Tyrrell RM. Activation of NF-kappa B in human skin fibroblasts by the oxidative stress generated by UVA radiation. Photochem Photobiol. 1995;62:463–468. doi: 10.1111/j.1751-1097.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]
- 18.Murley JS, Kataoka Y, Cao D, Li JJ, Oberley LW, Grdina DJ. Delayed radioprotection by NFkappaB-mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162:536–546. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 19.Qanungo S, Wang M, Nieminen AL. N-Acetyl-L-cysteine enhances apoptosis through inhibition of nuclear factor-kappaB in hypoxic murine embryonic fibroblasts. J Biol Chem. 2004;279:50455–50464. doi: 10.1074/jbc.M406749200. [DOI] [PubMed] [Google Scholar]
- 20.Morini M, Cai T, Aluigi MG, Noonan DM, Masiello L, De Flora S, D’Agostini F, Albini A, Fassina G. The role of the thiol N-acetylcysteine in the prevention of tumor invasion and angiogenesis. Int J Biol Markers. 1999;14:268–271. doi: 10.1177/172460089901400413. [DOI] [PubMed] [Google Scholar]
- 21.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieminen AL, Gores GJ, Wray BE, Tanaka Y, Herman B, Lemasters JJ. Calcium dependence of bleb formation and cell death in hepatocytes. Cell Calcium. 1988;9:237–246. doi: 10.1016/0143-4160(88)90004-8. [DOI] [PubMed] [Google Scholar]
- 23.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 Targets Inhibitor of {kappa}B Kinase and Induces Apoptosis in Melanoma: Involvement of Nuclear Factor {kappa}B and Mitochondria Pathways. Clin Cancer Res. 2006;12:950–960. doi: 10.1158/1078-0432.CCR-05-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raisova M, Hossini AM, Eberle J, Riebeling C, Wieder T, Sturm I, Daniel PT, Orfanos CE, Geilen CC. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol. 2001;117:333–340. doi: 10.1046/j.0022-202x.2001.01409.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res. 2001;61:7339–7348. [PubMed] [Google Scholar]
- 27.Boya P, Morales MC, Gonzalez-Polo RA, Andreau K, Gourdier I, Perfettini JL, Larochette N, Deniaud A, Baran-Marszak F, Fagard R, Feuillard J, Asumendi A, Raphael M, Pau B, Brenner C, Kroemer G. The chemopreventive agent N-(4-hydroxyphenyl)retinamide induces apoptosis through a mitochondrial pathway regulated by proteins from the Bcl-2 family. Oncogene. 2003;22:6220–6230. doi: 10.1038/sj.onc.1206827. [DOI] [PubMed] [Google Scholar]
- 28.Shin MH, Jang JH, Surh YJ. Potential roles of NF-kappaB and ERK1/2 in cytoprotection against oxidative cell death induced by tetrahydropapaveroline. Free Radic Biol Med. 2004;36:1185–1194. doi: 10.1016/j.freeradbiomed.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Richmond A. Constitutive IkappaB kinase activity correlates with nuclear factor-kappaB activation in human melanoma cells. Cancer Res. 2001;61:4901–4909. [PubMed] [Google Scholar]
- 30.Haghnegahdar H, Du J, Wang D, Strieter RM, Burdick MD, Nanney LB, Cardwell N, Luan J, Shattuck-Brandt R, Richmond A. The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukoc Biol. 2000;67:53–62. doi: 10.1002/jlb.67.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004;64:4912–4918. doi: 10.1158/0008-5472.CAN-04-0673. [DOI] [PubMed] [Google Scholar]
- 32.Ogata N, Yamamoto H, Kugiyama K, Yasue H, Miyamoto E. Involvement of protein kinase C in superoxide anion-induced activation of nuclear factor-kappa B in human endothelial cells. Cardiovasc Res. 2000;45:513–521. doi: 10.1016/s0008-6363(99)00364-8. [DOI] [PubMed] [Google Scholar]
- 33.Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res. 2001;89:661–669. doi: 10.1161/hh2001.098873. [DOI] [PubMed] [Google Scholar]
- 34.Chang JW, Kim CS, Kim SB, Park SK, Park JS, Lee SK. C-reactive protein induces NF-kappaB activation through intracellular calcium and ROS in human mesangial cells. Nephron Exp Nephrol. 2005;101:e165–172. doi: 10.1159/000087940. [DOI] [PubMed] [Google Scholar]
- 35.Postea O, Krotz F, Henger A, Keller C, Weiss N. Stereospecific and redox-sensitive increase in monocyte adhesion to endothelial cells by homocysteine. Arterioscler Thromb Vasc Biol. 2006;26:508–513. doi: 10.1161/01.ATV.0000201039.21705.dc. [DOI] [PubMed] [Google Scholar]
- 36.Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem J. 2002;367:729–740. doi: 10.1042/BJ20020752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez Y, Miller TP, Denoyelle C, Esteban JA, Tang WH, Bengston AL, Soengas MS. Chemical blockage of the proteasome inhibitory function of bortezomib: impact on tumor cell death. J Biol Chem. 2006;281:1107–1118. doi: 10.1074/jbc.M511607200. [DOI] [PubMed] [Google Scholar]
- 39.Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med. 2003;81:235–245. doi: 10.1007/s00109-003-0422-2. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz R, Davidson T. Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology (Williston Park) 2004;18:14–21. [PubMed] [Google Scholar]
- 41.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 42.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]