

Abstract
The phosphatidylcholine preferring phospholipase C from Bacillus cereus (PC-PLCBc) catalyzes the hydrolysis of phospholipids in the following order of preference: phosphatidylcholine (PC) > phosphatidylethanolamine (PE) > phosphatidylserine (PS). In previous work, mutagenic, kinetic, and crystallographic experiments suggested that varying the amino acids at the 4th, 56th, and 66th positions had a significant influence upon the substrate specificity profile of PC-PLCBc. Here, we report the crystal structures of the native form of several PC-PLCBc variants that exhibited altered substrate specificities for PC, PE, and PS at maximum resolutions of 1.90–2.05 Å. Comparing the structures of these variants to the structure of the wild-type enzyme reveals only minor differences with respect to the number and location of active site water molecules and the side chain conformations of residues at the 4th and 56th positions. These results suggest that subtle changes in steric and electronic properties in the substrate binding site of PC-PLCBc are responsible for the significant changes in substrate selectivity.
Keywords: phosphatidylcholine, phospholipase C, Bacillus cereus, substrate specificity, crystallography
Introduction
Phosphatidylcholine preferring phospholipase C from Bacillus cereus (PC-PLCBc)1 is a monomeric 28.5 kDa phosphodiesterase that contains three zinc ions in its active site. PC-PLCBc hydrolyzes phospholipids to give diacylglycerol and an alkyl phosphate (Figure 1) in the following order of preference: phosphatidylcholine (PC) > phosphatidylethanolamine (PE) > phosphatidylserine (PS) [1]. While there is no significant sequence homology to known eukaryotic gene data bases, antibodies raised towards PC-PLCBc cross-react with proteins in mammalian cells that hydrolyze PC at considerable rates [2]. Investigations focusing on PC-PLCBc are thus of interest as they might lead to information regarding the general structure, function, mechanism, and substrate preference of the phospholipase C class of enzymes. Moreover, inasmuch as the headgroups of PC, PE, and PS each contain an ammonium moiety, structural studies of PC-PLCBc and its variants with these substrates might provide some useful insights regarding the interactions of substituted ammonium ions with proteins [3–6]. Such information would be of general importance because ammonium groups are found in a number of biologically and medicinally active molecules, including histamine, catecholamines, acetylcholine, creatine and various peptides [7, 8].
Fig 1.

Catalytic efficiency (kcat/Km) (mM−1s−1) of wild-type PC-PLCBc and protein variants, E4G, E4Q, and Y56T for substrates C6PC, C6PE, and C6PC. The substrates were at concentrations well below their respective CMC [13, 27].
Studies characterizing the substrate specificity profile of PC-PLCBc have revealed that the spatial orientation of the glycerol side chains on the phosphatidylcholine moiety is an important contributing factor for binding and catalysis; substrates having the S-configuration at the sn-2 center are hydrolyzed 40 times more slowly than the natural R-isomers [9]. The sn-2 ester linkage is also important as replacing it with an ether linkage yields a substrate that is processed 1000 times more slowly [10, 11]. The length of the fatty acid side chains of the glycerol moiety has a modest effect on hydrolysis rates. Namely, PC with six carbon atoms in each of the acyl side chains is processed with slightly greater catalytic efficiency (kcat/Km) than PC with two–four carbon atoms [10, 12]. More relevant to the present work are the different activities of PC-PLCBc toward the three phospholipid substrates PC, PE and PS, which only differ in the structure of the head group. For example, PC-PLCBc hydrolyzes PC, PE, and PS with specificity constants (kcat/Km) in the approximate ratio of 10:7:1, respectively [13].
Crystal structures of native wild-type PC-PLCBc (PDB 1AH7) [14] and its complex with a non-hydrolyzable phosphatidylcholine analogue [15] have been reported. Active site residues Glu4, Tyr56, and Phe66 comprise the substrate head group binding pocket. An oxygen atom of the carboxyl group on the side chain of Glu4 is located 4.8 Å from the nitrogen atom on the choline head group, thus making a polar and/or ionic interaction that stabilizes the net positive charge on the phosphatidylcholine head group (Figure 2). The interaction of Phe66 with the choline head group appears to be mediated by a cation-π interaction as the centroid of the aromatic ring of Phe66 and the ammonium group nitrogen atom are separated by 4.2 Å [8, 16]. The proximity of the centroid of the aromatic ring and the phenolic hydroxyl group of Tyr56 from the nitrogen atom on the choline head group suggests this residue might help stabilize the positive charge on the inhibitor or substrate. However, this simple analysis does not explain the role of residue 56 as a determinant of specificity (vide infra). The phosphodiester group of the inhibitor binds all three zinc ions, whereas the diacylglycerol moiety binds partially within a hydrophobic cleft of the active site. The oxygen atom of the sn-2 carbonyl group forms a hydrogen bond with the backbone nitrogen atom of Asn134, whereas the sn-1 carbonyl oxygen atom is involved in a hydrogen bond network with surrounding water molecules.
Fig. 2.

Residues Glu4, Tyr56, and Phe66 are within close proximity to the choline head group in the structure of wild-type PC-PLCBc complexed to a non-hydrolyzable PC analogue [15]. The distances (in Å) between the binding pocket residues and the PC-derived inhibitor are shown. For Tyr56 and Phe66, the distances displayed are from the centroids of their aromatic rings to the nitrogen atom the PC analogue.
In previous work by our laboratory, a combinatorial library of mutants was recently generated that contained random permutations of Glu4, Tyr56, and Phe66 to identify PC-PLCBc variants with different specificity profiles for PC, PE, and PS [1, 13, 15]. These studies suggested replacing Glu4 with a neutral or positively charged amino acid increased the specificity constant for PS and substantially reduced the specificity constants toward PE and PC. Substitution of Tyr56 by a non-aromatic residue lowered the specificity constant for PC, but increased it for PE and PS. For example, the PC-PLCBc variants E4Q, E4G, and Y56T exhibited a preference for either PS or PE instead of PC, the favored substrate of the wild-type enzyme. Although these variants had significantly different substrate specificity profiles from wild-type, they retained their catalytic activities and were found to have approximately two to six-fold higher specificity constants for their preferred phospholipid substrates compared to that of wild-type (Figure 1). In order to investigate the structural origin of these changes in substrate selectivity on a molecular level, the crystal structures of the E4Q, E4G and Y56T variants of PC-PLCBc were determined and compared with that of wild-type. These results are presented herein.
Materials and methods
Chemicals
Trypsin, trypsin inhibitor, kanamycin, and ampicillin were purchased from Sigma (St. Louis, MO). Amylose resin was purchased from New England Biolabs (Beverly, MA). Q-Sepharose was obtained from GE Healthcare Bio-Sciences (Little Chalfont, U.K.). Unless otherwise noted, all chemicals were purchased from Fisher Scientific (Hampton NH).
Preparation of recombinant PC-PLCBc
Wild-type PC-PLCBc and its variants were expressed and purified as reported [17], and the purified enzyme/variant was dialyzed twice (100-fold) against 0.1 M sodium acetate (EM Science), pH 6.0, at room temperature (rt) and aliquoted into glass test tubes. The tubes were placed on a heating block at 70 °C for 15 min, cooled on ice for 5 min and then centrifuged at 30,000 g for 15 min at 4 °C. The supernatant was then aliquoted into glass test tubes, placed on a heating block at 85 °C for 10 min, and cooled on ice for 5 min prior to centrifuging at 30,000 g for 15 min. After discarding the supernatant, the pellet was suspended in 30 mL water and centrifuged at 30,000 g for 15 min. The pellet was dissolved in 4 mL 6.0 M guanidine HCl (Sigma) containing 0.1 M sodium acetate, pH 6.0, per ~3 mg protein, and the mixture was incubated for 1 h at rt. The sample was dialyzed twice (100-fold) against 1.0 mM dimethyl glutarate (DMG) (Sigma), pH 7.3, 0.1 mM ZnSO4 (Baker) at 4 °C to refold the protein. Following dialysis, the solution was cleared of insoluble protein by centrifugation. Saturated (NH4)2SO4 was then added until the solution became cloudy, and the suspension was centrifuged in a microcentrifuge at 14,000 rpm for 3 min. The protein was resuspended in 30% saturated (NH4)2SO4 to a final concentration of ~4 mg/mL.
X-ray crystallography
Crystals of native E4G, E4Q, and Y56T variants of PC-PLCBc were grown by vapor diffusion over 45% saturated (NH4)2SO4 and all belonged to the P43212 space group. All crystals were cryoprotected in 20% glycerol, 45% (NH4)2SO4 for 2 min and frozen in liquid N2 prior to data collection on an R-Axis 4++ detector. Images were processed and scaled for E4G, E4Q, and Y56T at 20-1.90 Å, 30-2.05 Å, and 20-2.00 Å resolution ranges, respectively, using HKL2000 [18]. CCP4 was employed for identifying a molecular replacement solution [19] from the published native PC-PLCBc structure (PDB 1AH7) [14]. Structures were refined using the CNS suite of programs [20]. Structural manipulation and electron density interpretation were performed using the program, O [21]. Images were created through Pymol [22]. Data collection and refinement statistics are listed in Table 1.
Table 1.
Summary of X-ray diffraction data and refinement statistics. Values in parentheses are for the outer data shell.
| E4G | E4Q | Y56T | |
|---|---|---|---|
| Space Group | P43212 | P43212 | P43212 |
| Unit-cell parameters (Å) | a=89.3, c=72.0 | a=89.4, c=72.0 | a=90.5, c=70.2 |
| Resolution (Å) | 20-1.90 | 30-2.05 | 20-2.00 |
| Unique reflections | 18893 | 18714 | 18461 |
| Completeness (%) | 98.8 | 99.1 | 96.5 |
| Rmerge (%) | 3.6 (16.2) | 3.8 (25.1) | 5.4 (11.0) |
| Average I/σ(I) | 31.1 (6.8) | 24.7 (5.6) | 15.0 (7.5) |
| Rcrys /Rfree (%) | 18.3/20.6 | 18.0/21.2 | 17.9/20.6 |
| Average B factors (Å2) | |||
| Protein Atoms | 20 | 20 | 15 |
| Solvent atoms | 32 | 34 | 27 |
| Zinc ions | 16 | 26 | 14 |
| RMSD bonds (Å) | 0.006 | 0.005 | 0.006 |
| RMSD angles (º) | 1.13 | 1.12 | 1.13 |
| PDB code | 2HUC | 2FFZ | 2FGN |
Results and discussion
Overview of Native Structures of Variants of PC-PLCBc
The backbone atoms of the native structures of the E4G, E4Q, and Y56T PC-PLCBc variants align with the native wild-type structure with root mean square (RMS) deviations of 0.20–0.23 Å. Including all atoms, the E4G, E4Q, and Y56T structures align with the native wild-type structure with RMS deviations of 0.47–0.54 Å, thus indicating that there are no significant differences in the tertiary structures of these proteins. Therefore, changes in substrate specificity that occur upon replacing wild-type residues do not arise from significant changes in the tertiary structures. Moreover, the active site water molecule that is presumed to be the attacking nucleophile in the PC-PLCBc catalyzed reaction was present in all structures [23, 24]. In the wild-type PC-PLCBc structure, this water molecule is located 2.6 Å from the side chain carboxyl group of Asp55, which is believed to be the general base in the hydrolysis reaction. This water molecule is located 2.8, 2.9 and 3.8 Å from the carboxyl group of Asp55 in the corresponding structures of the E4G, Y56T, and E4Q variants. The ubiquitous presence of this water molecule suggests that the mechanism of phosphatidylcholine hydrolysis by the E4G, E4Q, and Y56T variants is similar to wild-type PC-PLCBc. When B-factors (temperature factors) were analyzed and compared, the only significant differences were found for the three active site zinc ions. The zinc ions in the E4G and Y56T structures showed fairly low B-factors (~15.0), whereas those in the E4Q structure had an average value of 26.0. The reason for these differences remains unclear, especially since the E4G, E4Q, and Y56T variants crystallized in the same space group, all data were collected at the same temperature (100 K), the resolution of the data used to solve each structure were nearly identical, and the data suggested the occupancy of the zinc ions in each structure to be at 100% (Table 1). The only significant differences between the structures of the E4G, E4Q, Y56T variants of PC-PLCBc and wild-type are found in the orientations of the side chains of residues 4 and 56, the hydration network in the active site, the net charge in the active site, or the volume of the substrate binding pocket (Figure 4a).
Fig 4.
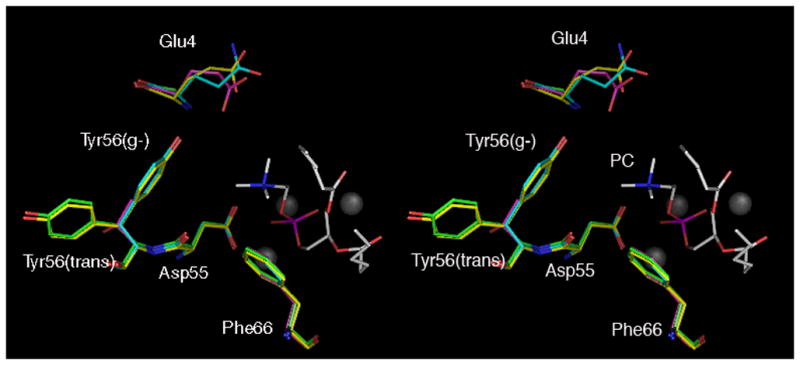

Relaxed stereo views of an alignment of the active sites of selected structures and an example of electron density within the active site. A) Overlay of native E4G (lime), E4Q (cyan), Y56T (magenta), and wild-type (yellow) [14] PLCBc crystal structures. Shown are the three zinc ions (gray spheres), residues identified to be important to substrate specificity (4th, 56th, and 66th positions) and the general base (D55). A PC analgoue (white), taken from the PC/PC-PLCBc complex [15], has been modelled into the active sites. B) Electron density (2Fo-Fc) within the active site of variant E4Q contoured 1.0 σ. Water molecules W4 (attacking nucleophile), W9, and W13 are shown in addition to residues Q4, D55, Y56, F66. The PC analogue, taken from the reported PC/PLCBc complex [15], is also displayed.
Crystal Structure of the Native PC-PLCBc E4G Variant
The backbone atoms in the native structures of wild-type PC-PLCBc and the PS-selective variant E4G align with an RMS deviation of 0.23 Å. With the exception of the Tyr56 side chain, all amino acid side chains in the active site of E4G are oriented in the same way as those in wild-type PC-PLCBc. In the native wild-type structure, electron densities are evident for both the gauche− (g−) and the trans conformations for the χ1 angle of the Tyr56 side chain [25], whereas only the trans conformation for this side chain is observed in the structure of the native E4G variant.
The active site water molecules W1–W9 in the native structure of wild-type PC-PLCBc are shown in Figure 3a. The structure of the complex of wild-type PC-PLCBc with a phosphatidylcholine-derived inhibitor [15] revealed that W1, W2, W3, and W5 are displaced upon phospholipid binding, whereas W4 is the putative nucleophile that attacks phosphorous atom, thereby initiating the chemical hydrolysis of the phosphodiester linkage. The binding site of the E4G variant contains all of the water molecules that are found in the native wild-type structure, although the hydrogen bond contacts to W7 in the two structures are different (Figures 3a,b). Namely, W7 forms hydrogen bonds to Glu4 and Asn147 in the wild-type structure, but it is displaced by about 1.4 Å in the E4G structure and forms a network with W5 and W8. This change in the hydrogen bonding pattern may arise in part because there is no hydrogen bonding partner available at amino acid position 4. Three water molecules W10, W11, and W12 are present in the E4G structure but not in the wild-type structure. The χ1 angle of the Tyr56 side chain in the E4G structure is exclusively trans, so the aromatic ring is oriented away from the active site. W10 then occupies the space that would have been occupied by the Tyr56 side chain in a g(−) conformation, and it forms hydrogen bonds to the backbone carbonyl oxygen atoms of residues Tyr52 and Ala3. Water molecules W11 and W12 occupy the space that was created by replacing Glu4 with a glycine residue. W11 forms hydrogen bonds to W4, W12, and the backbone nitrogen atoms of Ala3 and Gly4, and W12 forms additional hydrogen bonds to W6 and the backbone carbonyl oxygen atom of Val11.
Fig. 3.

Cartoon representation of the polar and hydrogen bonding interactions with distances in Å of water molecules, W, with residues in the substrate binding pockets in the native structures of wild-type PLCBc and the E4G, E4Q and Y56T variants. A) Interactions in the crystal structure of native wild-type PC-PLCBc; water molecules, W1–W3 and W5 are displaced upon binding of a PC analogue. B) Interactions in the crystal structure of the native E4G variant of PC-PLCBc showing the three additional water molecules (W10, W11, and W12) relative to wild-type. C) Interactions in the crystal structure of the native E4Q variant of PC-PLCBc showing three water molecules (W13–W15) that are not present in the wild-type structure; three water molecules (W5, W7, and W8) found in the wild-type structure are not present. and D) Interactions in the crystal structure of the native Y56T variant of PC-PLCBc showing three water molecules (W10, W15, and W16) that are not found in the wild-type structure; two water molecules (W5 and W7) found in the wild-type structure are absent.
Crystal Structure of the Native PC-PLCBc E4Q Variant
The backbone atoms in the native structures of wild-type PC-PLCBc and the PS selective protein variant E4Q align with an RMS deviation of 0.20 Å, so these two structures are also very similar. There are, however, significant differences in the conformations of the side chains of residues at the 4th and 56th positions (Figure 4a,b). Namely, the side chain of Tyr56 in the E4Q structure resides exclusively in the g(−) conformation oriented toward the substrate binding pocket, while in the wild-type enzyme both the g(−) and the trans conformations are populated. The polar interactions and orientations of the side chains of the Gln4 residue in the E4Q structure and the Glu4 residue in the native structure of wild-type PC-PLCBc are somewhat different (Figures 3a,c). In both structures, the Oε1atom of the Glu4 or Gln4 side chain forms a hydrogen bond to the Nδ2 atom of Asn147. In the wild-type structure, the Glu4 Oε2 atom forms a hydrogen bond to water molecule W7, but there is no corresponding hydrogen bond in the E4Q structure because W7 is absent. Relative to the wild-type structure, the E4Q variant lacks two other active site water molecules, W5 and W8. On the other hand, three water molecules W13, W14, and W15 are found in the E4Q structure but not the native structure of wild-type PC-PLCBc. W13 forms a hydrogen bonding network with water molecules W9 and W4, whereas W14 is involved in a network with water molecules W1–W3, each of which is coordinated with one or more zinc ions; W15 forms a hydrogen bond with the side chain carboxyl group of Glu146.
Crystal Structure of the Native PC-PLCBc Y56T Variant
The backbone atoms in the native structures of the PE selective variant Y56T and wild-type PC-PLCBc align with an RMS deviation of 0.22 Å. The replacement of Tyr56 by threonine results in a significant decrease in the specificity constant toward PC, while the specificity constants toward PE and PS are roughly two-fold higher (Figure 1). Comparison of the wild-type and Y56T structures shows that there are no significant differences in the conformations of any of the active site residues except for the side chain of Glu4, the carboxyl group of which lies approximately 1 Å closer to the substrate binding pocket in Y56T compared to wild-type (Figure 3d). This change brings the Glu4 side chain within hydrogen bonding distance of W4, the putative attacking nucleophile. Temperature factors measured for Glu4 in the wild-type and Y56T variant structures are similar, and although they are relatively high (29 Å2 and 35 Å2, respectively) compared to the average B-factors for all atoms (22 Å2 and 25 Å2 respectively), the electron density surrounding Glu4 is strong. The hydroxyl group of Thr56 is directed away from the active site, so unless the orientation of the Thr56 side chain changes upon binding, there is no evidence to suggest it interacts with substrate. The Y56T structure contains three active site water molecules, W10, W15 and W16 that are not found in the wild-type structure, although W10 and W15 are present in the E4G and E4Q structures. W10 in the Y56T variant forms hydrogen bonds with W16 and the backbone carbonyl oxygen atoms of Tyr52 and Ala3. W16 is unique to the Y56T variant and makes a hydrogen bond contact with the backbone carbonyl oxygen atom of Ala3.
Conclusion
The E4G, E4Q, and Y56T variants of PC-PLCBc exhibit specificity constants for PC that are 2 to 10-fold lower than wild-type, whereas the specificity constants of these variants for either PE or PS are 2 to 6-fold higher than wild-type (Figure 1). These findings suggest that the binding pocket of wild-type PC-PLCBc has evolved to preferentially bind and hydrolyze PC. In order to probe how single mutations in the active site binding pocket of PC-PLCBc can lead to significant changes in the substrate specificity profiles, the native structures of the E4G, E4Q, and Y56T variants were solved. Superimposing these structures with that of the native wild-type PC-PLCBc shows that the four structures are virtually identical with respect to alignment of the backbone atoms. Temperature factors are nearly identical for backbone and solvent atoms in all structures, although the B-factors for the zinc ions in the E4Q structure are approximately two-fold higher than in the structures of the E4G and Y56T variants. Perhaps the presence of W14, which is exclusively found in the E4Q structure, and its interaction with water molecules W1-W3 has some affect on the B-factors of these three zinc ions. The three active site zinc ions and the water molecule W4, which is thought to be the attacking nucleophile in the enzymatic reaction, are all conserved in the native structures of E4G, E4Q, and Y56T, suggesting that the mechanisms of PE and PS hydrolysis are similar to that proposed for PC [12, 23, 24, 26, 27]. Because replacing the wild-type residues at the positions 4 or 56 does not appear to alter the overall tertiary structure of the enzymes, changes in substrate specificity must be due to more subtle steric and/or electronic perturbations in the substrate binding pocket.
Both E4G and E4Q show a marked preference for PS, but their specificity profiles are different as E4Q has an approximately 16-fold higher specificity constant toward PE than E4G. There is one significant difference in the substrate binding pockets of E4G and E4Q that may be relevant in this context. In particular, the χ1 angle for the side chain of Tyr56 in the native structure of E4Q is g(−) and directed toward the substrate binding pocket, whereas in the E4G structure the conformation of this side chain is trans and positioned away from the active site. In the native wild-type structure, Tyr56 adopts both the g(−) and the trans conformations, whereas in the complex of wild-type PC-PLCBc with a PC-derived inhibitor (Figure 2) [15], Tyr56 lies exclusively in the g(−) conformation. The conformational preference of the Tyr56 side chain may thus be catalytically relevant, so the fact that Tyr56 appears to adopt different preferred conformations in the E4G and E4Q structures may be reflected in their specificity profiles.
The native structures of wild-type PC-PLCBc and its Y56T variant, which exhibits increased catalytic efficiencies for PE and PS (Figure 1) are similar, yet there are some notable differences in the substrate binding pockets of these two enzymes. Namely, replacing Tyr56 with the smaller Thr residue results in extra space in the binding site, which is occupied by W16. The side chain of Glu4 is then positioned approximately 1 Å closer to the substrate binding pocket in Y56T than it is in the native structure of wild-type PC-PLCBc. In previous work, we showed that the Y56S, Y56V, and Y56A variants exhibit specificity profiles similar to that of Y56T [13], suggesting that the threonine hydroxyl group itself plays little role in the altered selectivity of Y56T relative to wild-type.
The differences in hydration network in the active sites of E4G, E4Q, and Y56T may also be partially responsible for the observed substrate specificity profiles. Although there is presently no clear basis for speculating how changes in the hydrogen bonding networks in these native structures might affect substrate specificity, it is perhaps noteworthy that the headgroups of PE and PS are capable of forming hydrogen bonds whereas the headgroup of PC is not. Each of the three PC-PLCBc variants has one water molecule that is unique to their active sites: These are W11 in E4G, W13 in E4Q, and W16 in Y56T. These water molecules reside within 4 Å of the space that is occupied by the choline head group in the crystal structure of the complex between wild-type PC-PLCBc and a PC-derived inhibitor (Figure 2) [15]. If PE and PS are recognized by the PLCBc variants Y56T, E4G, and E4Q in a manner similar to how wild-type recognizes PC, it is likely these active site water molecules are not displaced upon binding of substrate and, therefore, may play a role in binding and hence may influence the specificity profiles.
Inasmuch as the headgroups of PC, PE and PS differ in size, charge, and hydrogen bonding capability, it was not surprising that replacing the binding site residues at the 4 and 56 positions of wild-type PC-PLCBc to give the E4G, E4Q, and Y56T variants led to dramatically altered substrate specificities. We have now verified that these changes in specificity constants did not arise from significant dissimilarities in tertiary structure as the three dimensional structures of wild-type PC-PLCBc and its E4G, E4Q, and Y56T variants are very similar. It thus appears that replacing the wild-type amino acid residues at positions 4 and 56 of PC-PLCBc, which have ionizable functional groups, with amino acids lacking such groups affects primarily the hydration networks and the electronic and steric properties within the substrate binding pockets. Such changes would certainly be expected to play a role in at least the binding of the structurally different substrates PC, PE and PS. Further insights regarding the structural origin of the specificity profiles of wild-type PC-PLCBc and its E4G, E4Q, and Y56T variants might be obtained from studies of complexes of E4G, E4Q, and Y56T with substrate analogues, and these efforts are ongoing.
Acknowledgments
We wish to thank the Robert A. Welch Foundation, the National Institutes of Health, and the Texas Advanced Research Program for financial support of this research. We are also grateful to Professors John Tesmer (Professor of Life Sciences Institute and Pharmacology, Medical School, University of Michigan) and Jon Robertus (The University of Texas, Austin) for their guidance and assistance with the crystallographic data.
The abbreviations used are
- PC-PLCBc
phosphatidylcholine preferring phospholipase C of Bacillus cereus
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PS
phosphatidylserine
- CMC
critical micelle concentration
- C6PC
1,2-dihexanoyl-sn-glycero-3-phosphatidylcholine
- C6PE
1,2-dihexanoyl-sn-glycero-3-phosphatidylethanolamine
- C6PS
1,2-dihexanoyl-sn-glycero-3-phospho-L-serine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martin SF, Follows B, Hergenrother P, Trotter B. The choline binding site of phospholipase C (Bacillus cereus): insights into substrate specificity. Biochemistry. 2000;39:3410–3415. doi: 10.1021/bi9919798. [DOI] [PubMed] [Google Scholar]
- 2.Clark M, Shorr R, Bomalaski J. Antibodies prepared to Bacillus cereus phospholipase C crossreact with a phosphatidylcholine preferring phospholipase C in mammalian cells. Biochem Biophys Res Commun. 1986;140:114–119. doi: 10.1016/0006-291x(86)91065-x. [DOI] [PubMed] [Google Scholar]
- 3.Raine AR, Yang CC, Packman LC, White SA, Mathews FS, Scrutton NS. Protein recognition of ammonium cations using side-chain aromatics: a structural variation for secondary ammonium ligands. Protein Sci. 1995;4:2625–2628. doi: 10.1002/pro.5560041222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 5.Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, Axelsen PH, Silman I, Sussman JL. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci U S A. 1993;90:9031–9035. doi: 10.1073/pnas.90.19.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dennis M, Giraudat J, Kotzyba-Hibert F, Goeldner M, Hirth C, Chang JY, Lazure C, Chretien M, Changeux JP. Amino acids of the Torpedo marmorata acetylcholine receptor alpha subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry. 1988;27:2346–2357. doi: 10.1021/bi00407a016. [DOI] [PubMed] [Google Scholar]
- 7.Scrutton NS, Raine AR. Cation-pi bonding and amino-aromatic interactions in the biomolecular recognition of substituted ammonium ligands. Biochem J. 1996;319:1–8. doi: 10.1042/bj3190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 9.Snyder WR. Bacillus cereus phospholipase C: carboxylic acid ester specificity and stereoselectivity. Biochim Biophys Acta. 1987;920:155–160. doi: 10.1016/0005-2760(87)90255-4. [DOI] [PubMed] [Google Scholar]
- 10.El-Sayed MY, DeBose CD, Coury LA, Roberts MF. Sensitivity of phospholipase C (Bacillus cereus) activity to phosphatidylcholine structural modifications. Biochim Biophys Acta. 1985;837:325–335. doi: 10.1016/0005-2760(85)90056-6. [DOI] [PubMed] [Google Scholar]
- 11.Massing U, Eibl H. Substrates for phospholipase C and sphingomyelinase from Bacillus cereus. Cambridge Univeristy Press; Cambridge: 1994. [Google Scholar]
- 12.Martin SF, Pitzer G. Solution conformations of short-chain phosphatidylcholine. Substrates of the phosphatidylcholine-preferring PLC of Bacillus cereus. Biochim Biophys Acta. 2000;1464:104–112. doi: 10.1016/s0005-2736(99)00252-7. [DOI] [PubMed] [Google Scholar]
- 13.Antikainen NM, Hergenrother PJ, Harris MM, Corbett W, Martin SF. Altering substrate specificity of phosphatidylcholine-preferring phospholipase C of Bacillus cereus by random mutagenesis of the headgroup binding site. Biochemistry. 2003;42:1603–1610. doi: 10.1021/bi0267285. [DOI] [PubMed] [Google Scholar]
- 14.Hough E, Hansen LK, Birknes B, Jynge K, Hansen S, Hordvik A, Little C, Dodson E, Derewenda Z. High-resolution (1.5 A) crystal structure of phospholipase C from Bacillus cereus. Nature. 1989;338:357–360. doi: 10.1038/338357a0. [DOI] [PubMed] [Google Scholar]
- 15.Hansen S, Hough E, Svensson LA, Wong YL, Martin SF. Crystal structure of phospholipase C from Bacillus cereus complexed with a substrate analog. J Mol Biol. 1993;234:179–187. doi: 10.1006/jmbi.1993.1572. [DOI] [PubMed] [Google Scholar]
- 16.Burley SK, Petsko GA. Weakly polar interactions in proteins. Adv Protein Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 17.Martin SF, Spaller MR, Hergenrother PJ. Expression and site-directed mutagenesis of the phosphatidylcholine-preferring phospholipase C of Bacillus cereus: probing the role of the active site Glu146. Biochemistry. 1996;35:12970–12977. doi: 10.1021/bi961316f. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.N. Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Cryst D Biol Cryst. 1994;1:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst D Biol Cryst. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 22.Delano WL. Delano Scientific LLC; San Carlos: 2004. [Google Scholar]
- 23.Hergenrother PJ, Martin SF. Phosphatidylcholine-Preferring Phospholipase C from B. Cereus. Function, Structure, and Mechanism. Topics in Current Chemistry. 2000;211:132–165. [Google Scholar]
- 24.Martin SF, Hergenrother P. General base catalysis by the phosphatidylcholine-preferring phospholipase C from Bacillus cereus: the role of Glu4 and Asp55. Biochemistry. 1998;37:5755–5760. doi: 10.1021/bi972948k. [DOI] [PubMed] [Google Scholar]
- 25.Gibson SE, Guillo N, Tozer MJ. Towards Control of chi-Space: Conformationally constrained analogues of Phe, Tyr, Trp and His. Tetrahedron. 1999;55:585–615. [Google Scholar]
- 26.Martin SF, Hergenrother PJ. Catalytic cycle of the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. Solvent viscosity, deuterium isotope effects, and proton inventory studies. Biochemistry. 1999;38:4403–4408. doi: 10.1021/bi9821216. [DOI] [PubMed] [Google Scholar]
- 27.Hergenrother P, Martin SF. Determination of the kinetic parameters for phospholipase C (Bacillus cereus) on different phospholipid substrates using a chromogenic assay based on the quantitation of inorganic phosphate. Anal Biochem. 1997;251:45–49. doi: 10.1006/abio.1997.2251. [DOI] [PubMed] [Google Scholar]