

Abstract
To study drug-receptor interactions, new thio-derivatives of salvinorin A, an extremely potent natural κ-opioid receptor (KOR) agonist, were synthesized. Obtained compounds were examined for receptor binding affinity. Analogs with the same configuration at carbon atom C-2 as in natural salvinorin A showed higher affinity to KOR than their corresponding epimers.
Salvinorin A (1) is a neoclerodane diterpenoid with a strong hallucinogenic activity. It has been shown to have a high affinity and selectivity to kappa-opioid receptors (KOR)1. Salvinorin A is isolated from the psychoactive plant Salvia divinorum as a major secondary metabolite, and makes an attractive lead compound for drug development due to its strong effects on human mood and low toxicity.
In the last three years numerous derivatives and analogs of salvinorin A were synthesized showing a broad range of KOR affinities2. Synthesis of new analogs of salvinorin A is important for generating new data on the structure of the receptor binding site and for possible changing of a pharmacological profile of action from agonist to antagonist. The wealth of the experimental data collected so far allows for structure-activity relationship conclusions pointing out the crucial importance of the C-2 configuration. Structural modifications of salvinorin A skeleton and functional groups in the other than C-2 positions did not provide compounds with higher affinity. In the course of our work on the molecular mechanism of interaction of salvinorin A to KOR we examined the effects of cysteine-substitution mutagenesis on the binding of salvinorin A along with analogs containing free sulfhydryl group at carbon C-23. These studies prompted us to find a convenient way to synthesize of 2-thioanalogs of salvinorin A and B.
Recently we reported the synthesis of sulfur analog of salvinorin A, in which the oxygen atom at the side chain at carbon atom C-2 was substituted by a sulfur atom2c. We now report the new method of the synthesis of sulfur analogs of salvinorin A and B epimeric at the C-2 stereogenic center. In our method salvinorin B (2) is transformed to 2β-chloroderivative (3) with 65% yield using mild condition of chlorination of alcohols with CCl4-Ph3P (Appel reaction). It is an improvement over previously used conditions (SOCl2/Py) which gives 3 with only 29% yield2c. After 2 hours of refluxing of 2 with CCl4-Ph3P the only product formed was 3 with 80% conversion of starting material. The prolonged heating (6 h) moved the reaction to completion but a small amount (14%) of the product of epimerization at carbon C-8 (4) appeared (Scheme 1).
Scheme 1.
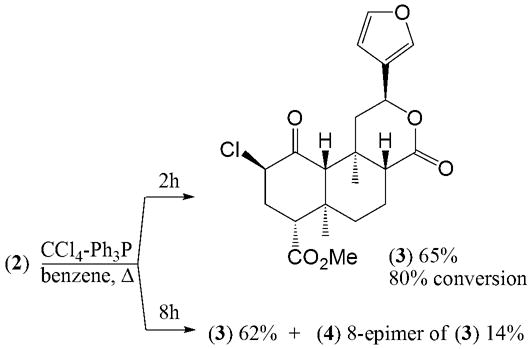
Synthesis of 2β-chloroderivatives
Epimerization at C-8 is well known in salvinorin chemistry, although it was previously observed repeatedly in basic and once in acidic conditions2e, 2j-n ,4. The nucleophilic substitution of the chlorine atom in 3 by sulfur-containing reagents may be achieved with inversion or retention of configuration at C-2. Reaction of 3 with sodium hydrogen sulfide (NaSH) in methanol and dimethylformamide produces a thiol 5, while the reaction with potassium thioacetate in acetone2c provides sulfur analog of salvinorin A (6) (Scheme 2).
Scheme 2.
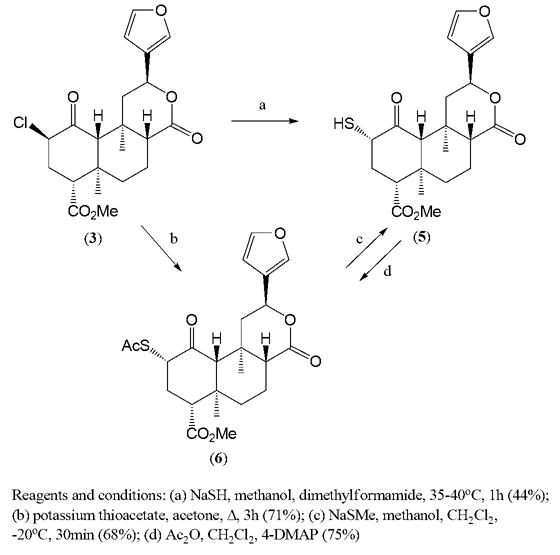
2α-Sulfur analogs of salvinorin A
The formation of thiol 5 from 3 is accompanied with several side products. To avoid this, the deacetylation of thioacetate 6 with NaSMe was chosen as cleaner and more effective method of the synthesis of 5. Deacetylation with sodium thiomethoxide5a proved to be advantageous in our case over the other known methods of hydrolysis of thioacetate group5b-f. The structure of compound 5 was confirmed by spectroscopic analysis (NMR) and high resolution mass spectrometry (HRMS)6 as well as by chemical re-acetylation to thioacetate 6. 1HNMR spectrum of 5 showed the characteristic doublet of doublets of H-2 due to the coupling with two protons at C-3. The large coupling constants for H-2 and H-3 protons of 5 (J=6 Hz and 12Hz) implied the β-orientation of H-2 and hence the desired α-configuration at C-2. The presence of thiol group was also confirmed by characteristic color reaction with Ellman’s reagent (DTNB).
The thioacetate 7 was obtained from triflated salvinorin B by reaction with potassium thioacetate in acetone at −20°C7. In these conditions the nucleophilic substitution occurred with inversion of configuration producing 2β-epimer (7) of compound 6. Deacetylation of 7 in analogous conditions8 described above for 6 yielded 2β-thiosalvinorin B (8) (Scheme 3).
Scheme 3.
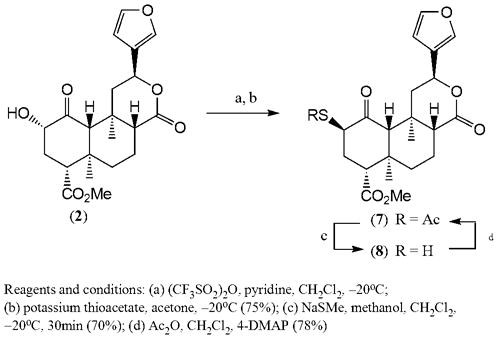
2β-Sulfur analogs of salvinorin A
Conducted NMR and HRMS analyses confirmed the structure of compounds 7 and 89. In this case the coupling constants between H-2 and H-3 were smaller (J=2Hz and 5Hz for 7) and corresponded well with calculated values for the dihedral angles H2α-C2-C3-H3α and H2α-C2-C3-H3β indicating the β-configuration at C-2.
Compounds 5–8 were evaluated for affinity to κ-opioid receptor (KOR) at the NIMH-sponsored Psychoactive Drug Screening Program at University of North Carolina at Chapel Hill using radioligand binding assays. The assays were conducted according to the procedure described earlier1,2o. The results are presented in Table 1.
Table 1.
Binding affinity of compounds 1 and 5–8 to cloned rKOR (competitive binding in the presence of [3H]U69,593) and hKOR mediated activation of intracellular calcium mobilization in HEK-293 cells.
| Compounds | Kia (nM) | EC50b (nM) | Emax (%) |
|---|---|---|---|
| 1 | 0.91 | 2.82±1.70 | 100 |
| 5 | 62 | 287±85 | 89±14 |
| 6 | 18.4±7.9 | 4.77±2.72 | 107±4 |
| 7 | 151±53 | 123±30 | 106±1 |
| 8 | 546±140 | >2000 | 71±12 |
Values are means of three experiments, standard deviation is given in parentheses
In vitro effective concentration
In conclusion, we were able to obtain new sulfur analogs of salvinorin A and B with “natural” (α) and inverted (β) configurations at carbon atom C-2. Thioanalogs with the same configuration at C-2 as in natural salvinorin A showed higher affinity to KOR than their corresponding epimers. Readily available 2β-chlorosalvinorin B (3) may serve as a convenient intermediate for the synthesis of various new analogs modified at C-2 position, retaining α-configuration of natural salvinorin A.
Supplementary Material
Figure 1.
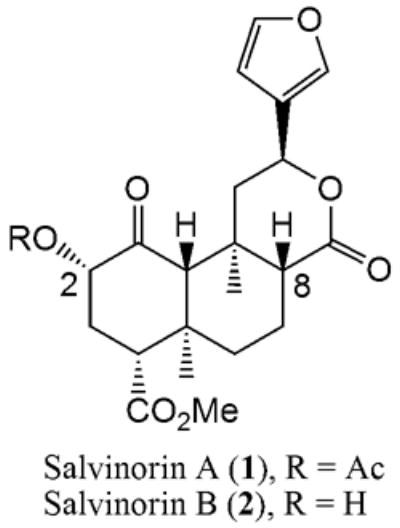
Salvinorins A and B
Acknowledgments
Authors would like to acknowledge Dr. Jeremy Stewart for his valuable suggestions and Kelly Thomas and Lukasz Kutrzeba for help in extraction of salvinorin A from plant material.
This work was supported by NIH grants P20 RR 021929-01 (Center of Research Excellence in Natural Products Neuroscience), and R01 DA017204, and conducted in a facility constructed with support from research facilities improvement program grant C06 RR-14503-01 from National Center for Research Resources, National Institutes of Health, Bethesda, MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Lee DYW, He M, Liu-Chen L-Y, Wang Y, Li J-G, Xu W, Ma Z, Carlezon WA, Jr, Cohen B. Biorg Med Chem Lett. 2006;16:5498. doi: 10.1016/j.bmcl.2006.08.051. [DOI] [PubMed] [Google Scholar]; (b) Beguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, Carlezon WA, Cohen BM. Biorg Med Chem Lett. 2006;16:4679. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]; (c) Stewart DJ, Fahmy H, Roth BL, Yan F, Zjawiony JK. Arzneimittel Forschung Drug Research. 2006;56:269. doi: 10.1055/s-0031-1296720. [DOI] [PubMed] [Google Scholar]; (d) Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]; (e) Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Biorg Med Chem Lett. 2006;16:3170. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]; (f) Prisinzano T. 2006058264. US Patent Appl. 2006; Chem Abstr. 2006;144:292904. [Google Scholar]; (g) Zjawiony J, Fahmy H, Stewart DJ, Roth B. PCT Int. Appl. WO 2006012643, 2006. Chem Abstr. 2006;144:164284. [Google Scholar]; (h) Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:107. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Beguin C, Carlezon W, Cohen BM, He M, Lee DY-W, Richards MR. PCT Int. Appl. WO 2005089745, 2005. Chem Abstr. 2005;143:326476. [Google Scholar]; (j) Lee DYW, He M, Kondaveti L, Liu-Chen LY, Ma Z, Wang Y, Chen Y, Li JG, Beguin C, Carlezon WA, Cohen B. Biorg Med Chem Lett. 2005;15:4169. doi: 10.1016/j.bmcl.2005.06.092. [DOI] [PubMed] [Google Scholar]; (k) Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA, Cohen B. Biorg Med Chem Lett. 2005;15:3744. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]; (l) Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. J Med Chem. 2005;48:4765. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]; (m) Beguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DYW, Carlezon WA, Cohen BM. Biorg Med Chem Lett. 2005;15:2761. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]; (n) Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J Pharmacol Exp Ther. 2004;308:1197. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]; (p) Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Biorg Med Chem Lett. 2004;14:5099. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 3.Yan F, Mosier PD, Westkaemper RB, Stewart J, Zjawiony JK, Vortherms TA, Sheffler DJ, Roth BL. Biochemistry. 2005;44:8643. doi: 10.1021/bi050490d. [DOI] [PubMed] [Google Scholar]
- 4.(a) Valdez LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda MJ. J Org Chem. 1984;49:4716. [Google Scholar]; (b) Koreeda M, Brown L, Valdes LJ., III Chem Lett. 1990:2015. [Google Scholar]; (c) Valdez LJ, III, Chang H-M, Visger DC, Koreeda M. Org Lett. 2001;3:3935. doi: 10.1021/ol016820d. [DOI] [PubMed] [Google Scholar]
- 5.(a) Wallace OB, Springer DM. Tetrahedron Lett. 1998;39:2693. [Google Scholar]; (b) Yelm KE. Tetrahedron Lett. 1999;40:1101. [Google Scholar]; (c) Mastalerz H, Zhang G, Kadow J, Fairchild C, Long B, Vyas DM. Org Lett. 2001;3:1613. doi: 10.1021/ol015727m. [DOI] [PubMed] [Google Scholar]; (d) Iimura S, Manabe K, Kobayashi S. Org Lett. 2003;5:101. doi: 10.1021/ol026906m. [DOI] [PubMed] [Google Scholar]; (e) MacCoss RN, Henry DJ, Brain CT, Ley SV. Synlett. 2004;4:0675. [Google Scholar]; (f) Holmes BT, Snow AW. Tetrahedron. 2005;61:12339. [Google Scholar]
- 6.NMR and mass spectral data were obtained for all final products purified by preparative HPLC (C18 column, MeCN-water). 2-thiosalvinorin B (5): white solid, m.p. 201–203°C, [α]25D -54 (c 0.05, CHCl3), 1H NMR (400 MHz, CD2Cl2): δ 1.07 (3H, s, H-19), 1.44 (3H, s, H-20), 1.56 (4H, m, H-6b, H-7b, H-11b, SH), 1.75 (1H, m, H-6a), 2.08 (2H, m, H-7a, H-8), 2.20 (1H, m, H-3b), 2.24 (1H, s, H-10), 2.47 (1H, ddd, J 3, 7, 14 Hz, H-3a), 2.52 (1H, dd, J 5, 13 Hz, H-11a), 2.79 (1H, dd, J 3, 13 Hz, H-4), 3.68 (3H, s, OCH3), 3.75 (1H, dd, J 6, 12 Hz, H-2), 5.53 (1H, dd, J 5, 12 Hz, H-12), 6.42 (1H, s, H-14), 7.44 (2H, m, H-15, H-16); 13C NMR (125 MHz, CDCl3): δ 15.6 (C-20), 16.9 (C-19), 18.4 (C-7), 36.0 (C-9), 37.3 (C-3), 38.4 (C-6), 42.8 (C-5), 43.9 (C-11), 49.3 (C-2), 51.6 (C-21), 52.1 (C-8), 56.0 (C-4), 66.1 (C-10), 72.1 (C-12), 108.3 (C-14), 125.3 (C-13), 139.1 (C-16), 143.5 (C-15), 170.8 (C-17), 172.4 (C-18), 202.8 (C-1); HRESIMS m/z [M+H]+ 407.1590 (calcd for C21H26O6S 406.1450).
- 7.Procedure for synthesis of 2-epi-2-thiosalvinorin A (7): To suspension of (2) (1 mmol) in CH2Cl2 (5 mL) at 0°C were added excess of pyridine (1 mL) and trifluoromethanesulfonic anhydride (1.2 mmol), the reaction mixture was stirred for 20 min. The reaction solution was washed with 1N HCl and brine, dried (Na2SO4) and solvents were evaporated in vacuum. A mixture of obtained extract and potassium thioacetate (5 mmol) were placed to dry acetone (10 mL) and stirred at −20°C under argon for 1 h. The reaction mixture was allowed to warm up to room temperature, concentrated in vacuum, diluted with water and extracted with chloroform. The combined organic extracts were dried (Na2SO4), concentrated in vacuum to give semisolid product which was purified by column chromatography (hexane/EtOAc, 2:1). Yield 75% starting from 2, white solid, m.p. 192–194°C, [α]25D -64 (c 0.05, CHCl3), 1H NMR (400 MHz, CDCl3); δ 1.15 (3H, s, H-19), 1.44 (3H, s, H-20), 1.62 (4H, m, 2H-6, H-7b, H-11b), 2.09 (3H, m, H-3b, H-7a, H-8), 2.33 (1H, s, H-10), 2.38 (3H, s, COCH3), 2.55 (1H, dd, J 5, 13 Hz, H-11a), 2.70 (2H, m, H-3a, H-4), 3.72 (3H, s, OCH3), 4.27 (1H, dd, J 2, 5 Hz, H-2), 5.54 (1H, dd, J 5, 11, H-12), 6.40 (1H, s, H-14), 7.42 (2H, m, H-15, H-16); 13C NMR (100 MHz, CDCl3); δ 15.1 (C-20), 16.4 (C-19), 18.0 (C-7), 30.6 (C-22), 31.8 (C-3), 35.2 (C-9), 38.3 (C-6), 41.9 (C-5), 43.3 (C-11), 49.7 (C-2), 51.3 (C-8), 51.8 (C-23), 52.8 (C-4), 63.8 (C-10), 72.0 (C-12), 108.4 (C-14), 125.5 (C-13), 139.3 (C-16), 143.7 (C-15), 171.2 (C-17), 172.2 (C-18), 191.1 (C-21), 203.8 (C-1); HRESIMS m/z [M+H]+ 449.1687 (calcd for C23H28O7S 448.1556).
- 8.General procedure for synthesis of 2-thiosalvinorin B (5) and 2-epi-2-thiosalvinorin B (8): To a stirred solution of thioacetate (1 mmol) in methanol (10 mL) under argon at −20°C was added sodium thiomethoxide (1 equiv. 1M solution in MeOH). The reaction mixture was stirred for 30 minutes. The solution was then added to aqueous HC1 (0.1M, 20 mL). The aqueous solution was extracted with CH2C12. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated.
- 9.2-epi-2-thiosalvinorin B (8): white solid, m.p. 161–163°C, [α]25D -160 (c 0.05, CHCl3), 1H NMR (400 MHz, MeOH): δ 1.12 (3H, s, H-20), 1.41 (3H, s, H-19), 1.70 (4H, m, 2H-6, H-7b, H-11b, SH), 2.03 (2H, m, H-7a, H-3b), 2.42 (1H, dd, J 3, 12 Hz, H-8), 2.52 (1H, dd, J 5, 13 Hz, H-11a), 2.68 (1H, ddd, J 6, 14, 14, H-3a), 3.08 (1H, dd, J 1, 6, H-4), 3.20 (1H, s, H-10), 3.57 (1H, d, J 6 Hz, H-2), 3.70 (3H, s, OCH3), 5.61 (1H, dd, J 5, 11, H-12), 6.54 (1H, s, H-14), 7.50 (1H, s, H-15), 7.44 (1H, s, H-16); 13C NMR (125 MHz, CDCl3): δ 15.5 (C-20), 16.6 (C-19), 18.4 (C-7), 31.9 (C-3), 35.3 (C-9), 38.3 (C-6), 42.4 (C-5), 43.4 (C-2), 44.2 (C-11), 50.9 (C-4), 51.6 (C-8), 52.0 (C-21), 59.1 (C-10), 72.1 (C-12), 108.4 (C-14), 125.4 (C-13), 139.3 (C-16), 143.5 (C-15), 171.1 (C-17), 172.0 (C-18), 205.9 (C-1); HRESIMS m/z [M+H]+ 407.1597 (calcd for C21H26O6S 406.1450).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.