

Abstract
Urokinase plasminogen activator (uPA) and its receptor (uPAR) play a major role in invasion and proliferation. A growing body of evidence has suggested that the uPA system promotes tumor metastasis by several different mechanisms, and not just solely by breaking down the ECM. In this study we have used RNAi mediated simultaneous downregulation of uPAR and uPA to determine the signaling pathway molecules and caspase mediated apoptosis. From our in vitro experiments, we have observed that plasmid based RNAi mediated downregulation of uPAR and uPA in SNB19 human glioma cells caused a decrease in the levels of uPAR protein and uPA enzyme activities. In addition, we observed a decrease in the phosphorylation of the Ras-activated pathway molecules such as FAK, p38MAPK, JNK and ERK1/2, as well as the MEK-activated PI3k pathway, and also retarded the dephosphorylation of p-AKTser473 and p-mTORser2448, indicative of a feedback signaling mechanism of the uPAR-uPA system. Activation of Caspase 8 accompanied by the release of cytochrome c and cleavage of PARP was also observed and indicative of Fas-mediated apoptosis. The use of FMK-VAD-FAK peptides coupled with FITC indicated activation of poly-caseases, which was accompanied by the presence of fragmented nuclei. Our studies provide evidence for the presence of a feedback response of the uPAR-uPA system indicative of the multifaceted role of uPAR, and also the therapeutic potential of simultaneously targeting uPAR and uPA in cancer patients.
Keywords: uPAR, uPA, apoptosis, Pi3k, AKT
INTRODUCTION
Malignant tumors have the capacity to degrade the extracellular matrix (ECM) by controlled proteolysis. One proteolytic system involved in these processes is the urokinase-type plasminogen activator (uPA) system, which consists of uPA, uPA receptor (uPAR) and uPA inhibitors 1 and 2 (PAI-1 and PAI-2). A growing body of evidence suggests that the uPA system promotes tumor metastasis by several different mechanisms, and not solely through the breakdown of the ECM (1). Initially, urokinase receptors (uPAR) were thought to function simply as a mechanism to concentrate the urokinase/plasmin system near the cell surface. However, extensive evidence has since shown that this glycolipid-anchored receptor also functions in both the adhesive and signaling pathways of many migratory cells. One mechanism through which uPAR directs these actions is by complexing with other membrane proteins (e.g., integrins) for signal transduction. There are three basic steps involved in invasion and intracellular signaling: (1) uPA/uPAR promotes extracellular proteolysis by regulating plasminogen activation; (2) uPA/uPAR regulates cell/ECM interactions as an adhesion receptor for vitronectin (Vn) and through its capacity to modulate integrin function; and (3) uPA/uPAR regulates cell migration as a signal transduction molecule and by its intrinsic chemotactic activity (2). Recent studies point to important structural features of uPAR:integrin interactions, indicating uPAR to be a cis-acting integrin ligand (3). Although uPAR transcripts are induced by adhesion, rapid synthesis of the protein uses constitutive mRNA without a requirement for new transcription and is regulated by mammalian target of rapamycin, demonstrating new biologic roles for the signal-dependent translation pathway controlled by this intracellular kinase (4). Integrins are known to activate the PI3k and the MEK pathway and recent studies have shown that the higher activation of Akt is associated with increased phosphorylation of its downstream targets glycogen synthase kinase 3beta (GSK3beta), FKHR, and mTOR (5) indicating a feedback mechanism. Studies with non-small cell lung carcinoma cells found that fibronectin stimulated the phosphorylation of Akt, an upstream inducer of mTOR, and induced the phosphorylation of p70S6K1 and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), which are both downstream targets of mTOR. In contrast, fibronectin inhibited the phosphatase and tensin homologue deleted on chromosome 10 (PTEN), a tumor suppressor protein that antagonizes the phosphatidylinositol 3-kinase/Akt signal. In addition, treatment with fibronectin inhibited the mRNA and protein expression of LKB1 as well as the phosphorylation of AMP-activated protein kinase (AMPKalpha), both known to downregulate mTOR. Rapamycin, an inhibitor of mTOR, blocked the fibronectin-induced phosphorylation of p70S6K and 4E-BP1. Akt small interfering RNA (siRNA) and an antibody against the fibronectin-binding integrin alpha5beta1 also blocked the p70S6K phosphorylation in response to fibronectin, and the combination of rapamycin and siRNA for Akt blocked fibronectin-induced cell proliferation (6). In this study, we have attempted to further delineate the complex role of uPAR and its associated molecules.
MATERIALS AND METHODS
Construction of hpRNA expressing plasmid
A pcDNA 3 plasmid with a CMV promoter was used in the construction of the hpRNA-expressing vector. We used the following siRNA sequences
The uPA inverted repeat sequence, agcttGagagccctgctggcgcgccatatataatggcgcgccagcagggctctca; and
The uPAR inverted repeat sequence, gatccTacagcagtggagagcgattatatataataatcgctctccactgctgtag.
Both were synthesized for both uPA and uPAR. The inverted repeats were laterally symmetrical, making them self-complimentary, with a five-base pair mismatch in the loop region. These five-base pair mismatches aids in the loop formation of the hpRNA. Oligos were heated in a boiling water bath in 6X SSC for 5 min and allowed to self-anneale by slow cooling to room temperature. The resulting annealed oligos were sequentially ligated to pcDNA 3 at the Hind III site for uPA and BamHI site for uPAR. The resulting plasmid was named pU2. Two single constructs were also made: puPAR targeting uPAR alone, and puPA targeting only uPA. An inverted repeat sequence targeting GFP mRNA was also synthesized and cloned into the pcDNA 3 Hind III site as described above, which was used as a negative control.
Cell culture and transfection
An SNB19 (or SNB19-GFP) cell line established from a high-grade human glioma was used for this study. Cells were grown in Dulbecco’s modified Eagle medium/F12 media (1:1, v/v) supplemented with 10% fetal calf serum in a humidified atmosphere containing 5% CO2 at 37°C. SNB19 cells at 60% confluency in a 100mm tissue culture plate were transfected with 10 μg of siRNA expressing plasmid constructs (empty vector, scrambled vector, puPAR, puPA or pU2) using lipofectamine as per manufacturer’s instructions (Life Technologies, Rockville, MD). Following transfection, conditioned media and cell lysates were used to determine the expression levels of uPAR and uPA by western blot analysis and fibrin zymography as per standard protocols. Experiments were performed by transfection of 5×105 SNB19 cells with 10 μg of pU2 in 100mm tissue culture plates.
Western blot analysis
SNB19 cells were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR, or pU2. After 48 h, cells were collected and total cell lysates were prepared in standard RIPA extraction buffer containing aprotinin, and phenylmethylsulfonyl fluoride. The extracts were incubated at 37°C for 5 min and then centrifuged to separate the lower (detergent) phase that contains mainly hydrophobic membrane proteins, including the glycosylphosphatidylinositol-anchored uPAR. Subsequently, 20 μg of protein from these samples were separated under non-reducing conditions by 12% SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The membranes were probed for 2 h with antibodies against uPAR, FAK, pFAKser843, p38, p-p38tyr182, JNK, pJNKtyr183/tyr185, ERK1/2, pERK1/2, RAPTOR, mTOR, phospho-mTORser2448, Caspase 8, Ki67 and cleaved PARP as per standard protocols, GAPDH levels served as loading controls. The membranes were subsequently washed three times with PBS to remove excess primary antibodies, incubated with appropriate HRP conjugated secondary antibodies as required, and then developed according to enhanced chemiluminescence protocol (Amersham, Arlington Heights, IL). For loading control, the membranes were stripped and probed with monoclonal antibodies for GAPDH, as per standard protocol.
Fibrin zymography
The enzymatic activity and molecular weight of electrophoretically separated forms of uPA in the conditioned media of SNB19 cells transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR, or pU2 were determined by SDS-PAGE as described previously (7,8). As stated, the acrylamide gels were enriched with purified plasminogen and fibrinogen before polymerization. Equal amounts of sample proteins were electrophoresed and the gel was washed and stained to determine enzymatic activity as per standard protocols.
In situ caspase activity assay
Caspase activation was detected using the polycaspase detection kit (Immunochemistry Technologies, Bloomington, IL) as per manufacturer’s instructions. Briefly, SNB19 cells cultured on chamber slides were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR, or pU2. The detection of polycaspase was performed as previously described (9). This assay used a cell-permeable, non-cytotoxic Fluorochrome Inhibitor of Caspases (FLICA) that binds covalently to a reactive cysteine residue on the large subunit of the active caspase heterodimer. This kit uses a carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of many caspases (caspase 1, -3, -4, -5, -6, -7, -8 and -9; FAM-VAD-FMK), which is a generic probe for the detection of most caspases and emits green fluorescence. The green fluorescent signal is a direct measure of the amount of active caspase in the cell at the time the reagent was added. After 48 h of transfection, caspase activation was detected by staining the cells with the FAM-VAD-FMK dye (in situ marker). The bound marker was localized by fluorescence detection and observed with a confocal microscope. DAPI was used for nuclear staining. The results were quantified as percent polycaspase activity and graphically represented.
Visualization of apoptotic cells by nuclear staining
SNB19 cells were cultured on 6-well chamber slides and transfected with mock, EV, puPAR, puPA, or pU2. After 48 h of transfection, glass coverslips were mounted using DAPI-containing mounting media (Vector Laboratories, Burlingame, CA). The coverslips were allowed to set for 20 min in the dark. Cells were examined for nuclear fragmentation with a fluorescent microscope and photographed using a high resolution CCD camera. Ten fields were photographed and the percentage of apoptotic cells was determined compared to the average number of cells with apoptotic bodies per field.
Isolation of mitochondrial and cytosolic cell fractions
SNB19 cells cultured on chamber slides were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR, or pU2 and cultured for 48 h. Cells were then harvested, washed twice in ice-cold distilled water and resuspended in 0.5 g/mL osmotic buffer (10 mM NaPO4, 1.35 M sorbitol, 1 mM EDTA, 2.5 mM dithiothreitol, pH 7.5) with Zymolyase 20T (Seikagaku Corp., E. Falmouth, MA) added to a final concentration of 100μg/ml. The cells were then incubated at 30°C with gentle shaking for 5 min, after which they were resuspended in lysis buffer (0.6 M mannitol, 2 mM EDTA, pH 7.0). Protease inhibitors, PMSF, and aprotinin were added to the lysis buffer immediately prior to cell lysis. Cells were lysed by vigorous vortexing, and cellular debris was removed by centrifugation (1,900xg for 5 min). The supernatant containing both the mitochondrial and cytosolic fractions was then centrifuged (12,100xg, 10 min.) to pellet the mitochondria. The resulting supernatant, designated as the cytosolic fraction, was frozen at –80 °C or used immediately for western blotting as cytosolic fraction. The mitochondrial pellet was washed once in 0.6 M mannitol, 2 mM EDTA, pH 7.0, and centrifuged at 1,651xg for 5 min to pellet any remaining debris. The supernatant containing the mitochondria was then pelleted by centrifugation (23,000xg, for 10 min), resuspended in 10 mM NaPO4, pH 7.0, to a final concentration of 5–10 μg/μL, and frozen at –80°C or used immediately. Intact mitochondria were lysed in lysis buffer (150 mM NaCl, 50 mM Tris-Hcl, 2 mMEDTA, 1% NP-40, PH 7.4) and western blotted. Western blotting for cytochrome c was performed as per standard protocol for the cytosolic and mitochondrial fractions.
RESULTS
RNAi-mediated downregulation of uPAR and uPA causes the reduction of uPAR mRNA and protein expression and a reduction in uPA enzymatic activity
SNB19 glioma cells were transfected with siRNA expressing plasmids targeting uPAR (puPAR), uPA (puPA) and uPAR-uPA simultaneously (pU2). Cell lysates were used to determine the expression level of uPAR and the conditioned media was used to determine uPA activity by fibrin zymography. Figure 1A shows the decrease in the expression level of uPAR in puPAR and pU2-, transfected cells when compared to control EV/SV. From the fibrin zymography results, we observed a decrease in activity of uPA in puPAR- and puPA-transfected cells compared to control and EV/SV. uPAR protein expression in puPAR-transfected cells showed an 8-fold decrease, whereas pU2- transfected cells showed 25-fold decreases when compared to the controls (Fig 1B). Cells transfected with puPA showed a decrease in uPA activity with no significant decrease in uPAR expression level, whereas the downregulation of uPAR alone did show a significant decrease in uPA level.
Figure 1.
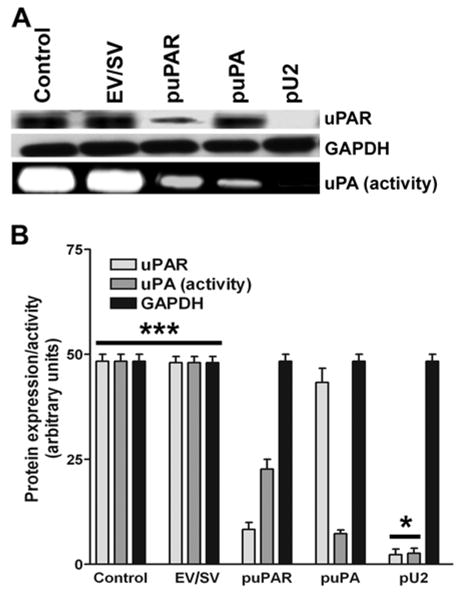
SNB19 cells were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 plasmid constructs. After 48 h, cells were collected, total cell lysates were prepared, fractionated by SDS-PAGE, and western blotted as per standard protocols. The blots were immunoprobed for uPAR and GAPDH (A). To determine the enzyme activity of uPA, conditioned media from SNB19 cells transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 were collected and fractionated on a fibrin gel as described earlier (A). Quantitative analysis of uPAR protein levels and uPA enzyme activity per 10×106 cells was normalized to GAPDH expression levels (B). The values shown are mean ±SD from four different experiments (p<0.002)
Simultaneous downregulation of uPAR and uPA retards the phosphorylation of pro-survival proteins FAK, p38MAPK, JNK and ERK1/2
Figure 2 shows the expression levels of FAK and pFAKser843. Total FAK expression remained more or less unchanged whereas the levels of pFAKser843 decreased in pU2-transfected cells. Similar results were obtained for the expression of ERK1/2 and pERK1/2. In the case of p38MAPK, total p38MAPK showed a decrease in pU2-transfected cells and p-p38MAPKtyr182 levels were almost undetectable. The levels of p38MAPK and p-p38MAPKtyr182 in control, EV/SV-, puPAR- and puPA-transfected cells essentially remained unchanged. In the case of JNK, levels of total JNK remained essentially unchanged in puPAR-, puPA- and pU2-transfected cells when compared to controls, whereas the levels of p-JNKtyr183/tyr185 decreased in puPAR- and puPA-transfected cells. Further, very low levels of p-JNKtyr183/tyr185 were observed in pU2-transfected cells.
Figure 2.
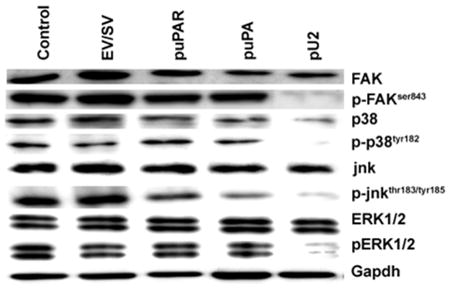
SNB19 cells were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 plasmid constructs. After 48 h, cells were collected, total cell lysates were prepared, fractionated by SDS PAGE, and western blotted as per standard protocols. The blots were probed for normal and phosphorylated forms of FAK, p38, JNK and ERK. GAPDH level served as a loading control.
RNAi-mediated simultaneous downregulation of uPAR and uPA induces the activation polycaspase and causes nuclear fragmentation in SNB19 cells
Figure 3A shows an increase in green fluorescence of SNB19 cells indicating caspase activation. Control, EV- and SV-transfected cells did not show activation of caspases, whereas puPAR-, puPA- and pU2-transfected cells showed caspase activation. Quantitative analysis of FMA-VAD-FAK (seen as green florescence), as determined by fluoroscopic measurement of the RGB values using a confocal microscope, indicated that approximately 80% of pU2-transfected cells exhibited caspase activation, whereas only 25% and 20% of puPAR- and puPA-transfected cells, respectively, exhibited caspase activation (Fig. 3B). Figure 3C shows the nuclear morphology of SNB19 cells in control, EV-, SV-, puPAR-, puPA- and pU2-transfected cells. Quantitative analysis of the percentage of apoptotic cells showed that pU2-transfected cells showed the highest number of apoptotic cells (80%), whereas puPAR (25%) and puPA (20%) transfected cells showed a lower percentage of apoptotic cells (Fig. 3D). The percentage of apoptotic cells correlated well with the polycaspase activity as seen in FMA-VAD-FAK-stained cells.
Figure 3.
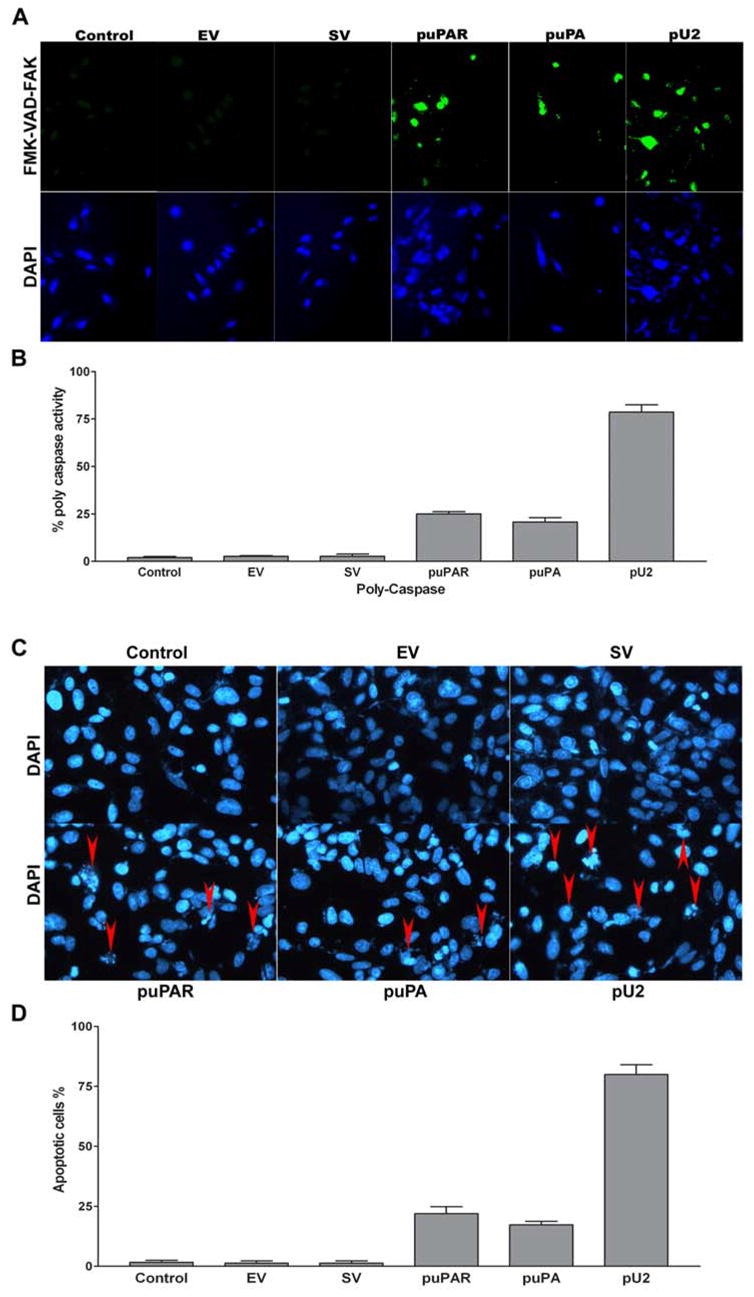
SNB19 cells in chamber slides were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 plasmid constructs. After 48 h, cells were processed as per manufacturer’s instructions (see Materials and Methods). The green fluorescent signal is a direct measure of the amount of active caspase in the cell. Caspase activation was detected by staining the cells with the FAM-VAD-FMK dye (A). Caspase activity was quantified as number of cells showing caspase activity and expressed as percent polycaspase activity (B). The values shown are mean ±SD from three different experiments (p<0.005). Initiation of nuclear degradation was determined by staining the cells with DAPI nuclear dye (C) and quantified as a percentage of apoptotic cells as determined by nuclear fragmentation (D). The values shown are mean ±SD from four different experiments (p<0.001)
Simultaneous downregulation of uPAR and uPA causes the activation of Caspase 8, cleavage of PARP, dephosphorylation of p-AKTser473, dephosphorylation of p-mTORser2448 and also induces the downregulation of PI3-k, RAPTOR and Ki67
Figure 4 shows that SNB19 cells transfected with pU2 showed an increase in Caspase 8 expression with almost uniform expression in control, EV/SV-, puPAR- and puPA-transfected cells. Cleavage of PARP was also observed in cells transfected with puPAR, puPA and pU2, with pU2-transfected cells showing the highest level of PARP cleavage. No detectable levels of PARP cleavage were observed in controls or in EV/SV-transfected cells. Phosphorylation of AKT at ser 473 was also reduced in puPAR- and puPA-transfected cells. In pU2-transfected cells, almost undetectable levels of p-AKTser473 were observed. The levels of PI3K were also downregulated in correlation with the levels of p-AKTser473. The mammalian target of rapamycin mTOR showed a decrease in of phosphorylation at ser 2448, with no appreciable change in the levels of unphosphorylated protein. Levels of RAPTOR (regulatory associated protein of mTOR) were almost undetectable in puPAR- and pU2-transfected cells. Ki67, an indicator of proliferation, showed a decrease in pU2-transfected SNB19 cells.
Figure 4.
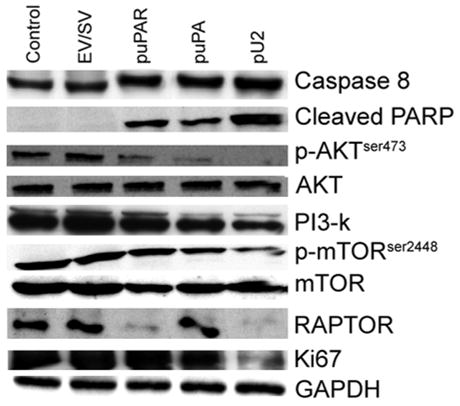
SNB19 cells were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 plasmid constructs. After 48 h, cells were collected, total cell lysates were prepared, fractionated by SDS-PAGE, and western blotted as per standard protocols. The blots were immunoprobed for activated Caspase 8, cleaved PARP, AKT, pAKT, PI3K, mTOR, pmTOR, RAPTOR and Ki67. GAPDH level served as a loading control.
Simultaneous downregulation of uPAR and uPA induces a release of cytochrome c from the mitochondria into the cytoplasm
Figure 5A shows no change in the mitochondrial cytochrome c levels, whereas in cells transfected with puPAR, puPA and pU2, the cytosolic fraction showed the presence of cytochrome c. Quantification of the levels of cytosolic cytochrome c indicated detectable levels of cytochrome c in puPAR-, puPA- and pU2-transfected cells, with pU2-transfected cells showing the highest levels of cytosolic cytochrome c when compared to control and EV/SV (Fig. 5B).
Figure 5.
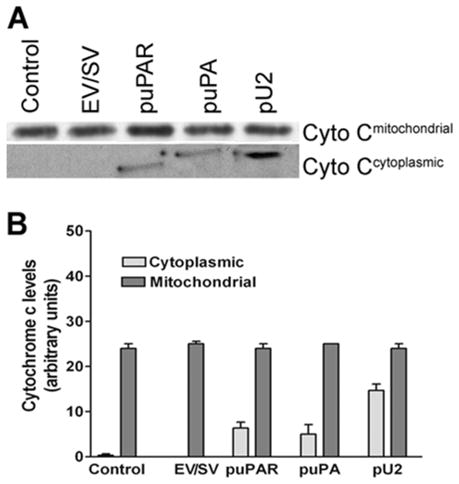
SNB19 cells were transfected with mock, empty vector (EV)/scrambled vector (SV), puPA, puPAR and pU2 plasmid constructs. After 48 h, cells were collected and mitochondrial and cytoplasmic fractions separated as described in Materials and Methods. The mitochondrial fraction was lysed and loaded onto SDS-PAGE gels and after electrophoresis, western blotted and immunoprobed for cytochrome c levels. Cytoplasmic cytochrome c levels, which indicate mitochondrial membrane permeability, were determined (A). Cytoplasmic and mitochondrial cytochrome c levels were quantified and expressed as arbitrary units (B). The values shown are mean ±SD from four different experiments (p<0.01)
DISCUSSION
Recent advances in RNAi technology have revolutionized the way gene expression is targeted, and even the most difficult genes are now relatively easy to silence. In this study, we have used RNAi to downregulate uPAR and uPA simultaneously. As described earlier (9), we have used bicistronic constructs to simultaneously target uPAR and uPA in prostate cancer cell lines. It is well documented that RNAi phenomenon is catalytic when compared to antisense, is highly specific, and works in the nanomolar range (10). Further, other studies have reported that RNAi is three times more active in the absence of translation (11). In our study, we have demonstrated that hairpin dsRNA molecules having a poly-A tail can induce RNAi in mammalian cells (9). uPAR and uPA function at the leading edge of the tumor to facilitate invasion of the surrounding extracellular matrix (ECM) by activating plasmin to plasminogen (1,12). Also, uPAR has been shown to be associated with vitronectin and integrins (13), which may account for its possible intra cellular signaling role. UPA has been shown to regulate the expression of its receptor both at the transcriptional and post-transcriptional levels, and to upregulate uPAR expression in MDA-MB-231 breast carcinoma cells by increasing SP1 binding activity and in lung carcinoma cells by increasing uPAR mRNA stability (14,15). Also, uPA has been shown to positively regulate vitronectin binding to uPAR (16). The role of uPA as a signaling initiator is probably via uPAR mediated by integrins. Integrins represent one set of molecules that are able to interact with uPAR on the cell surface. Fluorescence resonance energy transfer analysis (FRET), immunolocalization and co-immunoprecipitation have identified uPAR in complex with several integrin families, such as β1, β2, β3 and β5 (17–20). Our results demonstrated that the simultaneous downregulation of uPAR and uPA causes the dephosphorylation of p-FAK, p-p38 and pERK1/2, indicating a signaling feedback event associated with uPAR and integrins probably mediated via uPA. As has been demonstrated by others, uPA is capable of regulating the expression of uPAR. In MDA-MB-231 breast cancer cells, uPA-induced uPAR- α3 β1 association and enhanced the spreading and focal adhesion kinase (FAK) phosphorylation also on FN or collagen type I (21) indicating its FN and collagen interactions. Similar results were obtained with the simultaneous downregulation of uPAR and cathepsin B (22) where we demonstrated a decrease in pERK and pFAK levels. It is known that uPAR and cathepsin B are in close proximity and since uPAR is associated with integrins (23), internal Ras-mediated signaling via integrin cytoplasmic tails is facilitated. Here, we have demonstrated that the interference of two or more components associated with integrins can influence the intracellular signaling cascade (24). FAK is known to be directly associated with the cytoplasmic tail of integrins and is also associated with Paxillin (25–27). In turn, Paxillin, a direct substrate of FAK, has a physical association with filamentous actin cytoskeleton and is capable of influencing the migration ability of cells. In a previous study, we demonstrated that the antisense-mediated downregulation of uPAR and uPA in SNB19 cells using antisense adenovirus-mediated constructs results in decreased migration, invasion and retards intracranial tumor growth in nude mice (28), demonstrating that the targeting of this receptor ligand system has significant therapeutic potential. We have also shown that similar results were obtained with the simultaneous downregulation of uPAR and cathepsin B (22), and that cathepsin B is associated with β-integrin complex and integrin heterodimers are in association with uPAR making the uPAR –uPA and cathepsin B complex with integrins. Our previous results and the present study demonstrated that the molecules associated with uPAR and integrins play crucial roles in the survival and proliferation of cancer cells. Other researchers have shown that an antisense oligonucleotide significantly retards the metastasis of prostate cancer to the bone and that this effect can be attributed to the downregulation of uPAR alone (29). Since pro-uPA can be activated via tPA as well, downregulation of uPAR alone would not completely inhibit uPA mediated activation of plasmin. From our in vitro studies, we observed that SNB19 cells transfected with puPAR, puPA and pU2 induced an elevation in caspase activity, providing evidence that highly proliferative cells require uPAR-uPA for survival. Chemotaxis has been shown to be induced through an uPA-dependent conformational change in uPAR, which uncover potent chemotactic epitope acting through a pertussis-toxin sensitive step and activating intracellular tyrosine kinases as demonstrated using uPAR deficient mice (30). Similarly, in our results we demonstrated that uPAR levels are necessary in maintaining tyrosine kinase activation indicative of an invasive phenotype. We previously demonstrated that uPAR downregulation caused the induction of Caspase 9, an important component of the apoptosome complex, in SNB19 cells (31). It has also been suggested that uPA may be capable of mediating intracellular signals through uPAR via integrins and that the uPA-uPAR system is required for the maintenance of a invasive and metastatic phenotype (32). From our western blot analysis, it was observed that there was a significant increase in Caspase 8 activity with a corresponding increase in the cleavage of PARP. The Caspase 8-mediated pathway is well documented and the activation of Caspase 8 from its pro form via Fas and FasL has been established and has been extensively reviewed (33,34). It is, therefore, probable that the presence of uPAR and uPA may induce a shielding effect on Fas receptor inhibiting the activation of caspase 8. It has also been reported that the inhibition of Akt causes an upregulation of FasL, thereby activating the Caspase 8-mediated apoptotic pathway (35). Because Akt is a downstream molecule to FAK, and PI3K cascades from the cytoplasmic tail of cell surface integrins, it probable that any signaling event at the cell surface involving integrins would initiate a cascade of signaling events up to Akt (36). We observed similar results when the downregulation of uPAR and uPA caused the dephosphorylation of pAKT, the activation of Caspase 8, and a collapse in the mitochondrial membrane potential as determined by the release of cytochrome c into the surrounding cytoplasm. Earlier studies demonstrated that the inhibition of Caspase 8 activation retarded the loss of the mitochondrial membrane potential, indicating that activated Caspase 8 is the initiator for the mitochondrial membrane potential (37). PARP cleavage was also observed in uPAR and uPA downregulated cells indicating activation of DNA degradation and initiation of apoptosis (38). PARP normally recognizes DNA damage and is involved in DNA repair. At the onset of apoptosis, PARP is cleaved, thereby inhibiting the DNA repair function and enabling DNA damage to continue, which ultimately leads to cell death. The actual mediators and mechanisms of PARP cleavage are not clearly understood, but a detailed review of the possible role of PARP and PARP cleaved products is discussed by C. Soldani and A.I. Scovassi (39). In the present investigation, we also observed that the levels of p-mTOR were reduced in pU2-transfected cells. Mammalian target of rapamycin (mTOR), a downstream molecule of PI3K and Akt, is considered as a nutrient response protein and is involved in the cap-dependent translation of mRNA to proteins (40). As indicated by our results, total mTOR levels did not change, but the activated form of mTOR decreased significantly in pU2-transfected cells. As a downstream molecule of PI3K, mTOR would show a decrease in the phosphorylated form. Here, PI3K seemed to play a central role in the phosphorylation of mTOR. Earlier researchers have shown that upon inhibition of PI3K phosphorylation, mTOR activation is restored and continues at ser2448 mediated by p70S6 (41). Mounting evidence also suggests the presence of membrane-associated mTOR kinases (MA-mTORk) with possible regulatory functions (42). Whether these mTOR kinases respond to or are regulated by integrin cytoplasmic tails is not yet understood. Our results indicated that integrins may play a larger role in signaling than previously assumed. Our results also indicated that the downregulation of uPAR alone was sufficient to downregulate RAPTOR (regulatory associated protein of mTOR), but the downregulation of uPA alone did not have any effect on the levels of RAPTOR. RAPTOR is known to be a nutrient sensitive complex that signals cell growth machinery activation (43). The downregulation of RAPTOR in response to uPAR downregulation is not understood, but it is probable that cell adhesion regulates gene expression at translational checkpoints. uPAR transcripts are known to be induced by adhesion. Moreover, rapid synthesis of the protein uses constitutive mRNA without a requirement for new transcription and is regulated by mTOR (4) with a possible feed back mechanism to regulate mTOR levels. Since mTOR regulates the translation of other mRNAs, as well the downregulation by feedback, uPAR signaling would be unlikely. Ki67 levels were determined to gauge the overall viability of the cells and as an index for proliferation. From our mitochondrial studies, we observed that the simultaneous downregulation of uPAR and uPA results in the collapse of the mitochondrial membrane potential, releasing cytochrome c into the cytoplasm and initiating the apoptosome complex formation. As we have shown in earlier studies, antisense-mediated downregulation of uPAR causes elevated levels of Caspase 9 in SNB19 cells (31), and since cytochrome c and Caspase 9 are important components of the apoptosome complex (44), the formation of this complex is essential for the proper apoptotic process as demonstrated in our results. The downregulation of uPA with uPAR only seems to enhance the apoptotic process and the downregulation of uPA with uPAR seems more synergistic than additive. Scheme 1 represents the probable mechanisms involved in cellular signaling at the membrane level indicating the shielding effect of uPAR-uPA-Cathepsin B-integrin complexes on Fas receptor.
Scheme 1.
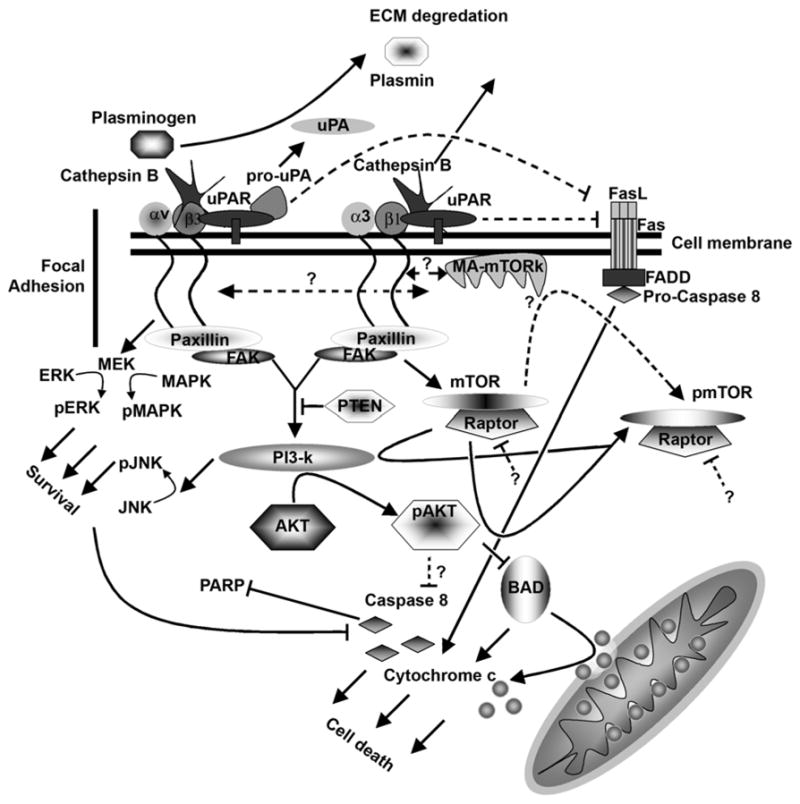
Schematic representation showing the plausible signaling events from uPAR integrin complex.
Acknowledgments
We thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for manuscript review.
Abbreviations used
- uPA[R]
urokinase-type plasminogen activator [receptor]
- CMV
cytomegalovirus
- BGH
bovine growth hormone
- PCR
polymerase chain reaction
- PBS
phosphate-buffered saline
- FITC
fluoresceine-5-isothiocyanate
- GFP
green fluorescent protein
- ECM
extracellular matrix
Footnotes
This research was supported by National Cancer Institute Grant CA 75557, CA 92393, CA 95058, CA 116708, N.I.N.D.S. NS47699 and NS057529, and Caterpillar, Inc., OSF Saint Francis, Inc., Peoria, IL (to J.S.R.).
Reference List
- 1.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 2.Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22:205–222. doi: 10.1023/a:1023099415940. [DOI] [PubMed] [Google Scholar]
- 3.Kugler MC, Wei Y, Chapman HA. Urokinase receptor and integrin interactions. Curr Pharm Des. 2003;9:1565–1574. doi: 10.2174/1381612033454658. [DOI] [PubMed] [Google Scholar]
- 4.Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in human myeloid leukocytes. Proc Natl Acad Sci USA. 2001;98:10284–10289. doi: 10.1073/pnas.181201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dogra C, Changotra H, Wergedal JE, Kumar A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol. 2006 doi: 10.1002/jcp.20696. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 6.Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006;66:315–323. doi: 10.1158/0008-5472.CAN-05-2367. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K, Rao VH, Tomonaga M, Nicolson GL, Rao JS. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res. 1994;54:3656–3661. [PubMed] [Google Scholar]
- 8.Mohanam S, Chintala SK, Go Y, Bhattacharya A, Venkaiah B, Boyd D, Gokaslan ZL, Sawaya R, Rao JS. In vitro inhibition of human glioblastoma cell line invasiveness by antisense uPA receptor. Oncogene. 1997;14:1351–1359. doi: 10.1038/sj.onc.1200963. [DOI] [PubMed] [Google Scholar]
- 9.Pulukuri SM, Gondi CS, Lakka SS, Jutla A, Estes N, Gujrati M, Rao JS. RNA Interference-directed Knockdown of Urokinase Plasminogen Activator and Urokinase Plasminogen Activator Receptor Inhibits Prostate Cancer Cell Invasion, Survival, and Tumorigenicity in Vivo. J Biol Chem. 2005;280:36529–36540. doi: 10.1074/jbc.M503111200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Putral LN, Gu W, McMillan NA. RNA interference for the treatment of cancer. Drug News Perspect. 2006;19:317–324. doi: 10.1358/dnp.2006.19.6.985937. [DOI] [PubMed] [Google Scholar]
- 11.Gu S, Rossi JJ. Uncoupling of RNAi from active translation in mammalian cells. RNA. 2005;11:38–44. doi: 10.1261/rna.7158605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohanam S, Sawaya R, McCutcheon I, Ali-Osman F, Boyd D, Rao JS. Modulation of in vitro invasion of human glioblastoma cells by urokinase-type plasminogen activator receptor antibody. Cancer Res. 1993;53:4143–4147. [PubMed] [Google Scholar]
- 13.Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63:1028–1037. doi: 10.1007/s00018-005-5428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montuori N, Mattiello A, Mancini A, Taglialatela P, Caputi M, Rossi G, Ragno P. Urokinase-mediated posttranscriptional regulation of urokinase-receptor expression in non small cell lung carcinoma. Int J Cancer. 2003;105:353–360. doi: 10.1002/ijc.11091. [DOI] [PubMed] [Google Scholar]
- 15.Zannetti A, Del VS, Carriero MV, Fonti R, Franco P, Botti G, D'Aiuto G, Stoppelli MP, Salvatore M. Coordinate up-regulation of Sp1 DNA-binding activity and urokinase receptor expression in breast carcinoma. Cancer Res. 2000;60:1546–1551. [PubMed] [Google Scholar]
- 16.Sidenius N, Andolfo A, Fesce R, Blasi F. Urokinase regulates vitronectin binding by controlling urokinase receptor oligomerization. J Biol Chem. 2002;277:27982–27990. doi: 10.1074/jbc.M111736200. [DOI] [PubMed] [Google Scholar]
- 17.Reinartz J, Schafer B, Batrla R, Klein CE, Kramer MD. Plasmin abrogates alpha v beta 5-mediated adhesion of a human keratinocyte cell line (HaCaT) to vitronectin. Exp Cell Res. 1995;220:274–282. doi: 10.1006/excr.1995.1316. [DOI] [PubMed] [Google Scholar]
- 18.Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 19.Xue W, Kindzelskii AL, Todd RF, III, Petty HR. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J Immunol. 1994;152:4630–4640. [PubMed] [Google Scholar]
- 20.Xue W, Mizukami I, Todd RF, III, Petty HR. Urokinase-type plasminogen activator receptors associate with beta1 and beta3 integrins of fibrosarcoma cells: dependence on extracellular matrix components. Cancer Res. 1997;57:1682–1689. [PubMed] [Google Scholar]
- 21.Wei Y, Eble JA, Wang Z, Kreidberg JA, Chapman HA. Urokinase receptors promote beta1 integrin function through interactions with integrin alpha3beta1. Mol Biol Cell. 2001;12:2975–2986. doi: 10.1091/mbc.12.10.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. RNAi-mediated inhibition of cathepsin B and uPAR leads to decreased cell invasion, angiogenesis and tumor growth in gliomas. Oncogene. 2004;23:8486–8496. doi: 10.1038/sj.onc.1207879. [DOI] [PubMed] [Google Scholar]
- 23.Degryse B, Resnati M, Czekay RP, Loskutoff DJ, Blasi F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J Biol Chem. 2005;280:24792–24803. doi: 10.1074/jbc.M413954200. [DOI] [PubMed] [Google Scholar]
- 24.Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513–2524. doi: 10.1038/sj.onc.1205342. [DOI] [PubMed] [Google Scholar]
- 25.Mukhopadhyay NK, Gordon GJ, Chen CJ, Bueno R, Sugarbaker DJ, Jaklitsch MT. Activation of focal adhesion kinase in human lung cancer cells involves multiple and potentially parallel signaling events. J Cell Mol Med. 2005;9:387–397. doi: 10.1111/j.1582-4934.2005.tb00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 27.Schaller MD. FAK and paxillin: regulators of N-cadherin adhesion and inhibitors of cell migration? J Cell Biol % 2004;19(166):157–159. doi: 10.1083/jcb.200406151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gondi CS, Lakka SS, Yanamandra N, Siddique K, Dinh DH, Olivero WC, Gujrati M, Rao JS. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene. 2003;22:5967–5975. doi: 10.1038/sj.onc.1206535. [DOI] [PubMed] [Google Scholar]
- 29.Margheri F, D'Alessio S, Serrati S, Pucci M, Annunziato F, Cosmi L, Liotta F, Angeli R, Angelucci A, Gravina GL, Rucci N, Bologna M, Teti A, Monia B, Fibbi G, Del Rosso M. Effects of blocking urokinase receptor signaling by antisense oligonucleotides in a mouse model of experimental prostate cancer bone metastases. Gene Ther. 2005;12:702–714. doi: 10.1038/sj.gt.3302456. [DOI] [PubMed] [Google Scholar]
- 30.Blasi F. The urokinase receptor. A cell surface, regulated chemokine. APMIS. 1999;107:96–101. doi: 10.1111/j.1699-0463.1999.tb01531.x. [DOI] [PubMed] [Google Scholar]
- 31.Yanamandra N, Konduri SD, Mohanam S, Dinh DH, Olivero WC, Gujrati M, Nicolson GL, Obeyeseke M, Rao JS. Downregulation of urokinase-type plasminogen activator receptor (uPAR) induces caspase-mediated cell death in human glioblastoma cells. Clin Exp Metastasis. 2001;18:611–615. doi: 10.1023/a:1011941114862. [DOI] [PubMed] [Google Scholar]
- 32.Ma Z, Webb DJ, Jo M, Gonias SL. Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J Cell Sci. 2001;114:3387–3396. doi: 10.1242/jcs.114.18.3387. [DOI] [PubMed] [Google Scholar]
- 33.Curtin JF, Cotter TG. Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003;15:983–992. doi: 10.1016/s0898-6568(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 34.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Semin Immunol. 2003;15:185–193. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 35.Uriarte SM, Joshi-Barve S, Song Z, Sahoo R, Gobejishvili L, Jala VR, Haribabu B, McClain C, Barve S. Akt inhibition upregulates FasL, downregulates c-FLIPs and induces caspase-8-dependent cell death in Jurkat T lymphocytes. Cell Death Differ. 2005;12:233–242. doi: 10.1038/sj.cdd.4401549. [DOI] [PubMed] [Google Scholar]
- 36.Gondi CS, Kandhukuri N, Kondraganti S, Gujrati M, Olivero WC, Dinh DH, Rao JS. Down-regulation of uPAR and cathepsin B retards cofilin dephosphorylation. Int J Oncol. 2006;28:633–639. [PMC free article] [PubMed] [Google Scholar]
- 37.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 38.Hermisson M, Wagenknecht B, Wolburg H, Glaser T, Dichgans J, Weller M. Sensitization to CD95 ligand-induced apoptosis in human glioma cells by hyperthermia involves enhanced cytochrome c release. Oncogene. 2000;19:2338–2345. doi: 10.1038/sj.onc.1203554. [DOI] [PubMed] [Google Scholar]
- 39.Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- 40.Proud CG. Role of mTOR signalling in the control of translation initiation and elongation by nutrients. Curr Top Microbiol Immunol. 2004;279:215–244. doi: 10.1007/978-3-642-18930-2_13. [DOI] [PubMed] [Google Scholar]
- 41.Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 42.Wu S, Mikhailov A, Kallo-Hosein H, Hara K, Yonezawa K, Avruch J. Characterization of ubiquilin 1, an mTOR-interacting protein. Biochim Biophys Acta. 2002;1542:41–56. doi: 10.1016/s0167-4889(01)00164-1. [DOI] [PubMed] [Google Scholar]
- 43.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 44.Adams JM, Cory S. Apoptosomes: engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–720. doi: 10.1016/s0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]