

Abstract
Non-genomic estrogen signaling pathways involve extranuclear estrogen receptors or function independently from estrogen receptors. These pathways participate in neuroprotection elicited by the hormone. Additional non-genomic neuroprotective effects are attributable to antioxidant and anti-inflammatory actions of estrogens. Numerous chemical modifications to afford neuroprotective compounds from estrogens while eliminating estrogenicity and maintaining or enhancing non-genomic neuroprotection have been described. This review highlights recent structure–activity studies that revealed the importance of antioxidant effects for neuroprotective estrogen analogues and derivatives .
Keywords: Estrogen, neuroprotection, non-genomic mechanism, antioxidant
1. Introduction
Estrogens are ovarian steroid hormones that elicit complex, tissue-specific responses (Turgeon et al., 2004). The abrupt stopping of the large clinical trials under the Women's Health Initiative (WHI) anticipated to reveal benefits and risks of postmenopausal estrogen-only and combined estrogen-progestin therapy has presented the scientific community a challenge to reconcile the results of the studies with evidence from epidemiological and clinical data, well-studied animal models and in vitro experiments that estrogens are potent neuroprotectants (McCullough and Hurn, 2003). There is a growing consensus that the results of the WHI trial should be interpreted within its boundaries given by the population included, drugs of choice, doses and route of administration, age at which hormone exposure occurs or timing of hormone therapy initiation in relation to menopause (Turgeon et al., 2004; Singh and Simpkins, 2005; Henderson, 2006). However, the study has definitely prompted the assessment that additional fundamental and clinical research is required to understand estrogens' neuroprotective action and explore the pharmaceutical potential of estrogen-derived agents as neuroprotectants.
The protective effects of estrogens may take place partly through genomic pathways involving estrogen-receptor (ER) activation. ERs are constitutively expressed in many brain regions and are able to initiate gene transcription after specifically binding to estrogen. In addition, non-genomic (or alternative) signaling pathways, involving extranuclear ERs or functioning independently of ER-binding, respond to estrogens and elicit neuroprotection. Additional neuroprotective effects such as the antioxidant action of the hormone are also independent of ERs. The prevailing mechanism of action commonly depends on the experimental paradigms used for the assessment of protection against neurotoxic insults, and mechanistic interpretation of findings has often been controversial. Recent studies have put into perspectives for pharmaceutical development neuroprotective estrogen analogues or derivatives that reduce or eliminate genomic effects. These analogues and derivatives are expected to curtail endocrine side effects associated with chronic estrogen therapy (Shumaker et al., 2003; Rapp et al., 2003) that impacts ERs in the liver, uterus, breast and other tissues. In addition, they would also allow for treatment of both women and men. The exploration of estrogen analogues and derivatives for neuroprotective therapy depends on an understanding of the mechanism of action and structure–activity relationships, specifically regarding non-genomic effects.
2. Estrogens and neuroprotection
Estrogens are effective neuroprotectants in vivo and in vitro. Recent reviews (Behl, 2002; Marin et al., 2005; Amantea et al., 2005) offer a comprehensive summary of numerous investigations. Consistent observations of potent neuroprotection by estrogens makes this ovarian steroid a prime candidate for protection of a variety of cell types during chronic neurodegenerative diseases and acute brain cell-compromising conditions, such as stroke and brain trauma (McCullough and Hurn, 2003; Sribnick et al., 2004). Before the discussion of structure–activity relationships, a brief overview of potential neuroprotective mechanisms is presented in this section.
2.1. Classical genomic pathways
The genomic pathway involving the binding of estrogen to the cognate nuclear ERs (which are expressed in ERα or ERβ isoforms) followed by gene transcription (Hall et al., 2001) has been implicated in the promotion of neuronal survival by the hormone. Estrogens are believed to distribute freely across biological membranes because their low molecular weight and lipophilic nature (Aloisi, 2003; Martin and Behbehani, 2006). As shown schematically in Fig. 1, the ligated ERs can form homo- (ERα/ERα or ERβ/ERβ) or heterodimers (ERα/ERβ) that can bind to the estrogen-response element (ERE) of the nuclear DNA. The dimers can associate with a multitude of coregulator proteins (Tremblay and Giguere, 2002). These proteins participate in and recruit many enzymatic and structural proteins that allow modulation of chromatin structure to facilitate stimulation or repression of gene expression.
Fig. 1.

A simplified model for the genomic action of estrogen. Estrogen, ER, co-regulator (co-r) and the nuclear DNA are symbolized by the filled circles, rounded rectangles and elongated rectangle showing an ERE (shaded area), respectively.
The differential expression of distinct ER subtypes has been implicated in the pleiotropic and tissue-specific effects of estrogens (Moggs and Orphanides, 2001). In the brain, there is a large overlap in the expression of ERα or ERβ, the two ER isoforms that are produced from different genes located on separate chromosomes (Enmark et al., 1997) yet sharing common structural features (Sand et al., 2002). ER density is higher in the hypothalamus than in extrahypothalamic areas (Shughrue et al., 1997; Shughrue et al., 2000), and ERα or ERβ may be co-localized in cells of some brain regions (Shughrue et al., 1998). However, ERβ is highly expressed in the cortex (Zhang et al., 2002) and hippocampus (Milner et al., 2005). Consequently, estrogen impacts the function of extrahypothalamic areas that are not involved in sex maturation and reproduction (McEwen, 2001).
Potential neuroprotective target genes induced by estrogen include neurotrophic factors, such as nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophins 3 and 4, and insulin-like growth factor 1 (IGF-1) (Sorabji et al., 1995; Jezierski and Sorabji, 2000), as well as their receptors (Blurton-Jones et al., 1999). These proteins directly support vital functions of neurons. Additional target genes implicated in estrogen-mediated neuroprotection are associated with apoptosis. Apoptosis is the regulated form of cell death utilized by multicellular organisms (metazoans) to remove unneeded, damaged, or potentially deleterious cells (Hail et al., 2006) and plays a central role in development and homeostasis (Yan and Shi, 2005). By the genomic mechanism associated with apoptosis, estrogen may rescue neurons through the induction of anti-apoptotic proteins such as Bcl-2 (Singer et al., 1998; Alkayed et al., 2001) and Bcl-x (Stolzner et al., 2001), or down-regulation of apoptotic proteins such as Bcl-2-associated X protein (BAX) (Sribnick et al., 2004), or by the induction of inhibitors against caspases (Zhang et al., 2001). Estrogens may also induce several gene products that maintain cellular architecture such as neurofilament (Scoville et al., 1997) and microtubulin-associated proteins (Shah et al, 2003). Many additional genomic pathways potentially associated with estrogen neuroprotection have been revealed (see review by Behl, 2002).
2.2. Modulation of intracellular signaling
Estrogen rapidly induces a variety of cellular responses that cannot be reconciled with the delayed genomic activity of the hormone. However, it is important to recognize that, in addition to nuclear ERs, the existence of membrane/cytoplasmic pools of ERα and ERβ have also been demonstrated (reviewed by Levine, 2001) and an estrogen-ER complex can function as a cytoplasmic signaling molecule involved in neuroprotection (Manavathi and Kumar, 2006). Specifically, cellular responses may arise through ERs that interact with intracellular pathways promoting the expression of neuroprotective genes (Behl, 2002). A typical example is shown schematically in Fig. 2. ERα has been shown to bind in a ligand-dependent manner to the p85 alpha regulatory subunit of phosphatidylinositol 3-kinase (PI3K) (Simoncini et al., 2000). Therefore, stimulation with estrogen increases ERα-associated PI3K activity, leading to the activation of protein kinase B/Akt and endothelial nitric oxide synthase (eNOS).
Fig. 2.

Indirect action of estrogen via interaction with intracellular signaling pathways. As in Fig. 1, estrogen, ER and the nuclear DNA are represented by the filled circles, rounded rectangles and elongated rectangle indicating a regulated gene promoter (shaded area), respectively. An ER-interacting protein (↔ PI3K) is also shown by a rounded rectangle.
The mitogen-activated protein kinase (MAPK) cascade (Singer et al., 1999; Bryant et al., 2005) and the cyclic-AMP-responsive element-binding (CREB) protein signaling pathway (Carlstrom et al., 2001) also respond rapidly to estrogen and have been implicated in its neuroprotective effects such as expression of anti-apoptotic proteins. Upstream mechanisms that initiate the signaling cascade leading to estrogen-inducible neuroprotection may be associated with rapid Ca2+-influx, which activates Src/extracellular signal-regulated kinase (ERK) and cAMP signaling pathways and induces Bcl-2 protein expression (Wu et al., 2005). Modulation of intracellular pathways may occur though the binding of estrogen to ERs (Simoncini et al., 2000), or independently of ligand binding (Coleman and Smith, 2001; Yu et al., 2004).
In addition to the classical intranuclear estrogen receptors (ERα and ERβ), a novel, plasma membrane-associated, putative ER (ER-X) that activates the MAPK cascade has been implicated in estrogen neuroprotection (Toran-Allerand, 2004). It has also been well established that estrogen can directly influence neurotransmission by binding to various transmembrane ion channels (Rupprecht et al., 2001; Peinado et al., 2005). Very recently, localization of ERβ in the mitochondria has been shown (Yang et al., 2004), and neuroprotective mechanisms implicating estrogen in preventing cell death largely by maintaining functionally intact mitochondria has been proposed (Simpkins et al., 2005). In addition, GPR30 (a G protein-coupled receptor uniquely localized to the endoplasmic reticulum) has been found to bind estrogen and fluorescent estrogen derivatives and its activation by estrogen resulted in the intracellular Ca2+-mobilization and synthesis of phosphatidylinositol 3,4,5-triphosphate in the nucleus (Revankar et al., 2005). However, continued research is necessary to understand molecular details of the interactions between ERs and signaling pathways involved in the mechanisms discussed briefly in this section.
2.3. Antioxidant effect
Free-radical damage of neurons has been implicated in neurodegenerative diseases (Gotz et al., 1994; Varadarajan et al., 2000) and aging (Beckman and Ames, 1998; Sohal, 2002). Estrogen's ER-independent antioxidant effects (Mooradian, 1993; Gomez-Zubeldia et al., 2001) is mainly due to its direct free-radical scavenging ability (Prokai et al., 2005), although indirect mechanisms such as upregulation of antioxidant enzymes (Borras et al., 2005), and chelation of redox-active metal ions (Ruiz-Larrea et al., 1995) may also be involved. Involvement of antioxidant mechanisms in the neuroprotective effect of the hormone has been recognized (Green et al., 1997a; Moosman and Behl, 1999, Vedder et al., 1999), in part by structure–activity relationship studies (Behl et al., 1997; Green et al., 1997b).
A simplified mechanism of direct oxyradical-scavenging by estrogen functioning as a phenolic antioxidant is shown in Fig. 3. The process involves H-atom transfer and is thought to cause an interruption of free-radical chain reactions, such as lipid peroxidation (R = LO, where L represents a lipid; Prokai et al., 2005). Estrogen, indeed, reduces lipid peroxidation in cells and tissues of the central nervous system (Vedder et al., 1999). However, the chain-braking reaction leaves behind a radical product (phenoxyl radical) whose fate has to be explained for a complete understanding of the antioxidant action. In vivo, phenolic antioxidants may be regenerated from the corresponding phenoxyl radicals by a reaction with ascorbic acid (vitamin C; Packer et al., 1979), or through glutathione (GSH) dependent free-radical reductase (FRR) (McCay et al., 1989). Therefore, a continuous cycle (Fig. 3) is maintained during the antioxidant of the hormone. Ascorbic acid has been shown to influence estrogen levels in postmenopausal women receiving hormone therapy (Vihtamaki et al., 2002). Estrogen also interacts synergistically with the water-soluble antioxidant glutathione (Green et al., 1998; Gridley et al., 1998). Therefore, both ascorbic acid and the GSH-dependent FRR may be involved into the reduction of estrogen-derived phenoxyl radical back to the parent hormone. An alternative neuroprotective antioxidant cycle involving a para-quinol intermediate and NAD(P)H-mediated reductive aromatization to regenerate the parent estrogen (Fig. 4) has also been discovered recently (Prokai et al., 2003).
Fig. 3.

Free-radical scavenging effect of estrogens according to the classical phenolic antioxidant mechanism. The scheme only shows the part of the molecule that participates in this mode of action. The solid arrows represent the chain-braking H-atom transfer, while the dashed arrows indicate the conversion of the phenoxyl radical back to the phenolic compound by an endogenous reductant (AH).
Fig. 4.

Hydroxyl-radical scavenging effect of estrogens according to a para-quinol-based antioxidant mechanism. The scheme only shows the part of the molecule that participates in this mode of action.
Because of its lipophilicity, estrogens concentrate in lipid-rich regions of the cell such as cellular membranes (Simpkins et al., 2005) affecting thereby membrane fluidity (Liang et al., 2001). In addition to binding to ERs in the cellular membrane, cytosol and nuclear compartments, they may also interact with lipid-transport proteins (Tikkanen et al., 2002). Therefore, it is likely that estrogens act in vivo as a highly localized antioxidant (Prokai et al., 2005).
2.4. Anti-inflammatory action
The previous sections dealt with direct effects on neurons as a key component of estrogen neuroprotection. However, the hormone has been shown to mediate inflammatory events. Estrogen has been shown to suppress the induction of the cyclooxygenase-2 pathway mediated by interleukin-1β (IL-1β) in cerebral blood vessels and prevents migration of microglia into the CNS following inflammatory challenge (Ospina et al., 2004). Brain macrophages, i.e. microglia cells, have also been implicated in estrogen's action on the brain. The hormone prevents the activation of microglia and the recruitment of peripheral monocytes induced by intraventricular injection of lipopolysaccharide. This effect occurs through ERα activation and reduces the expression of neuroinflammatory mediators such as the inducible form of NO synthase (iNOS), prostaglandin-E2 , the matrix metalloproteinase 9, lysosomal enzymes and complement C3 receptor, as well as by preventing morphological changes occurring in microglia during the inflammatory response (Vegeto et al., 2001; Vegeto et al., 2004). Estrogen also decreases microglial superoxide production and phagocytic activity by both an ER- and MAPK-dependent pathway (Bruce-Keller et al., 2000), and the hormone inhibits pro-inflammatory gene expression by controlling intracellular localization of nuclear factor-κ B (NF-κ B, Ghisletti et al., 2005).
3. Structure–activity relationships
The neuroprotective effects of estrogen have been widely documented in animal models of neurological disorders, such as Alzheimer's and Parkinson's diseases, as well as cerebral ischemia. The hormone protects cultured neurons against a variety of insults, including amyloid-β peptide (Aβ), the excitatory neurotransmitter L-glutamate, deprivation of nutritional and trophic factors, and a wide range of oxidative stressors (as summarized by Behl, 2002). Although many investigations have attempted to confine the neuroprotective action of estrogens to a single predominant mechanism, various processes discussed in the previous sections may be involved. The most potent activator of the classical genomic pathway is 17β-estradiol (17β-E2, Fig. 5), one of the primary export products from the human ovary (Turgeon et al., 2004). Even subtle changes in the structure of this endogenous ligand are often associated with a significant decrease in affinity to ERα and ERβ and, therefore, in propensity to invoke neuroprotective effects through binding to these receptors. For example, 17α-estradiol (17α-E2, a stereoisomer of 17β-E2 as shown in Fig. 5) binds to these two ER-isoforms with an approximately 40-fold lower affinity (Perez et al., 2006). [On the other hand, 17α-E2 has been proposed as the preferred ligand of ER-X (Toran-Allerand, 2004).] Therefore, structure–genomic neuroprotection relationship based on steroid scaffold (Fig. 6) should be considered in the framework of ligand structure–estrogen-receptor binding affinity relationship. This assessment holds true for non-genomic effects mediated by an ER (see section 2.2). The ligand structure–ER-binding affinity relationship has been thoroughly reviewed by Anstead et al. (1997) based on affinity-labeling studies of 17β-E2 and its analogues. In general, the ER is intolerant of polar substituents. Small hydrophobic substituents enhance binding affinity at positions 4, 12β, 14, and 16α, whereas, larger hydrophobic substituents are tolerated at positions 7α, 11β, and 17α. On the other hand, the ligand-binding pockets ERα and ERβ appear to have subpockets of different size and flexibility, which have resulted in “eclectic” structure–ligand-binding affinity relationships for estrogens (Katzenellenbogen et al., 2003) and presented challenges in developing computational methods for the prediction of ER-binding affinity. Nevertheless, comparative quantitative structure–activity relationship (QSAR) analysis of ER-ligands has been available (Gao et al., 1999a), and a new QSAR-like methodology that can correlate less quantitative assay data (i.e., “active” versus “inactive”) has also been introduced for potential use in high-throughput screening (Gao et al., 1999b). The examination of the findings is outside the scope of our review; for details, interested readers should consult publications cited above (Anstead et al. 1997; Gao et al., 1999a; Gao et al., 1999b; Katzenellenbogen et al., 2003).
Fig. 5.

17β-Estradiol [estra-1,3,5(10)-triene-3,17β-diol] and 17α-estradiol [estra-1,3,5(10)-triene-3,17α-diol].
Fig. 6.
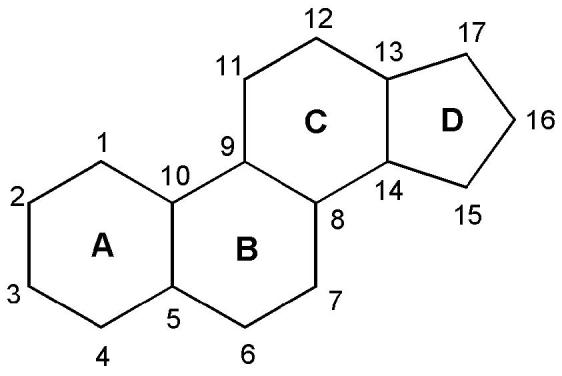
Chemical nomenclature of the steroid scaffold that shows rings A–D and positions 1–17 referred to in the text.
Structure–activity relationships have been explored for ER-independent modes of action, immediately after these types of action had been recognized (Behl et al., 1997; Green et al., 1997b). Our review below will focus on this subject, particularly considering neuroprotection as target.
3.1. Protection of neuronal cells by non-genomic mechanisms
Deciphering structure–non-genomic neuroprotection relationship for estrogen-derived compounds and analogues may be hindered by the multitude of mechanisms by which these molecules exert their effect. Different modes of action (genomic versus non-genomic) have often been reported for identical cell lines (Mize et al., 2003, and Deecher et al., 2005 versus Behl et al., 1995). However, many researchers have been able to distinguish non-genomic and genomic actions by appropriate selection of models and/or experimental conditions, and studies employing cultured rat primary cortical neurons (Behl et al., 1997), mouse hippocampal HT22 neuronal cells (Behl et al., 1997; Perez et al., 2006) and human SK-N-SH neuroblastoma cells (Green et al., 1997b) with neurotoxic insults involving an Aβ fragment, hydrogen peroxide, buthionine sulfoximine (Behl et al., 1997), glutamate (Behl et al., 1997; Perez et al., 2006), serum deprivation (Green et al., 1997b) and iodoacetic acid (Perez et al., 2006) have revealed meaningful structure–activity relationships for pathways not associated with ER-binding.
The seminal finding that the ER-α/β ligand 17β-E2 (Fig. 4) and its enantiomer 17αE2 (Fig. 5), which binds to these ER-isoforms with an approximately 40-fold lower affinity (Perez et al., 2006), were equally potent in protecting neuronal cells and the effects were not blocked by an estrogen antagonist (Green et al., 1997a) prompted a closer look at the structural requirements for neuroprotection among steroids (Green et al., 1997b; Behl et al., 1997). All estrogens tested with an intact phenolic A-ring were neuroprotective including 17β-E2 and 17α-E2 (Fig. 5), as well as estrone (E1), estriol (E3), the potent synthetic estrogen ethinylestradiol (EE) and the catechol estrogens (Mcluskey et al., 1981) representing the principal cytochrome P450 metabolites of the hormone (Fig. 7). However, 3-O-methyl (Green et al., 1997b) and 3-O-butyl ether congeners (Prokai et al., 2001), 3-O-cycloalkyl ether and 3-O-acetyl derivatives of neuroprotective estrogens (Fig. 8) were found inactive indicating the importance of a phenolic A-ring. Steroids that lack a phenolic A ring (testosterone, dihydrotestosterone, progesterone, corticosterone, prednisolone, 6α-methylprednisolone, aldosterone, and cholesterol), phenol, lipophilic phenols and tetrahydronapthol also were inactive (Green et al., 1997b). Therefore, a phenolic A-ring and at least three rings of the steroid nucleus were found necessary for the neuroprotective activity. This requirement was suggested as a basis for the design and synthesis of estrogen derivatives in which the structural modification would render the compound non-estrogenic (Behl et al., 1997). One recommendation was the synthesis of estrogens with bulky alkyl substituents in both the 2- and 4-position on the A-ring that diminishes ER-binding but, nevertheless, retains an antioxidant activity (Miller et al., 1996).
Fig. 7.

Estrone (E1), estriol (E3), ethinylestradiol (EE) and catechol estrogens (2-OH-17β-E2 and 4-OH-17β-E2).
Fig. 8.
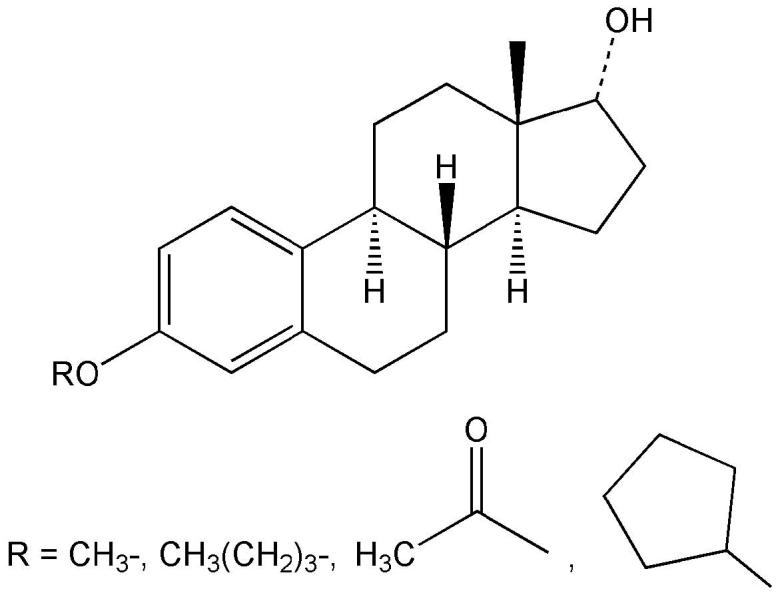
3-O-substituted estrogens.
In the most comprehensive structure–activity study to date, estratrienes (over 70 compounds) were tested in HT-22 (murine hippocampal) cells for their ability to inhibit cell toxicity against glutamate and iodoacetic acid and determined EC50 values to ascertain potency comparisons with 17β-E2 (Perez et al., 2006). Again, no correlation between ER-α/β binding and neuroprotection was found. The rank order of unsubstituted estratrienes for neuroprotection against glutamate and iodoacetic acid was an enantiomer of 17β-E2 (ent-E2, Fig. 9)>17β-E2>E1>17α-E2 and E1>17β-E2>17α-E2>ent-E2, respectively. Consistent with the reduced ER binding in vitro, 17α-E2 did not show feminizing effects, yet were effective neuroprotectants in an animal model for cerebral ischemia in vivo (Simpkins et al., 2004).
Fig. 9.
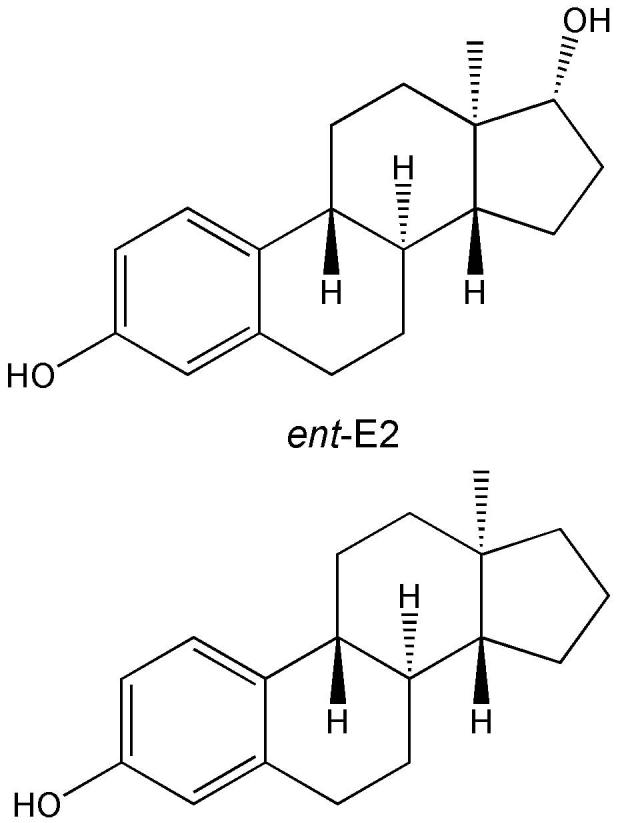
An enantiomer of 17β-E2 (ent-E2, top) and its 17-desoxy analogue (bottom).
A-ring derivatives with electron donating (alkyl, alkenyl, cycloalkyl, arylalkyl, etc.) substituents in the 2- and/or 4-position were more potent than 17β-E2 in protecting HT22 cells from neurotoxic insults. 2,4-Disubstituted compounds were more potent than the corresponding monosubstituted compounds. Electron donating substituents may enhance the antioxidant effect by weakening the phenolic O–H bond and providing stability to the phenoxyl radical formed (see Fig. 3). On the other hand, the electron-withdrawing iodo (I) group at position 2 of the A-ring diminished neuroprotection, whereas I-substitution at position 4 was less detrimental in this regard.
There was no apparent relationship between neuroprotective potency of estratriene and the size of the bulky substituent (methyl, sec-butyl, 1-methyl-allyl, tert-butyl, 3,3-dimethyl-butyl, 3,3-dimethyl-1-butynyl, adamantyl, 4-hydroxyphenylethyl and 4-methoxyphenylethyl) added. However, A-ring derivatives with the bulky adamantyl group (Fig. 10) were among the most potent compounds in vitro. 2-(1-Adamantyl)-3-hydroxyestra-1, 3, 5(10)-trien-17-one, 2-(1-adamantyl)-4-methyl-3-hydroxyestra-1, 3, 5(10)-trien-17-one and 2-(1-adamantyl)-4-methyl-estra-1,3,5(10)-triene-3,17β-diol (Fig. 11), which did not bind to either ERα or ERβ, have also been evaluated in vivo using ischemia-reperfusion injury induced by temporary middle cerebral artery occlusion in rats (Liu et al., 2002; Simpkins et al., 2004; Perez et al., 2005). Infarct volume was significantly reduced, while cerebral blood flow was increased compared to ovariectomized control animals. The magnitude of these beneficial effects was similar to that of the endogenous estrogens. Altogether, these compounds possessed both neuroprotective and vasoactive effects, which offered the attractive possibility of clinical application for stroke without the side effects of estrogen.
Fig. 10.
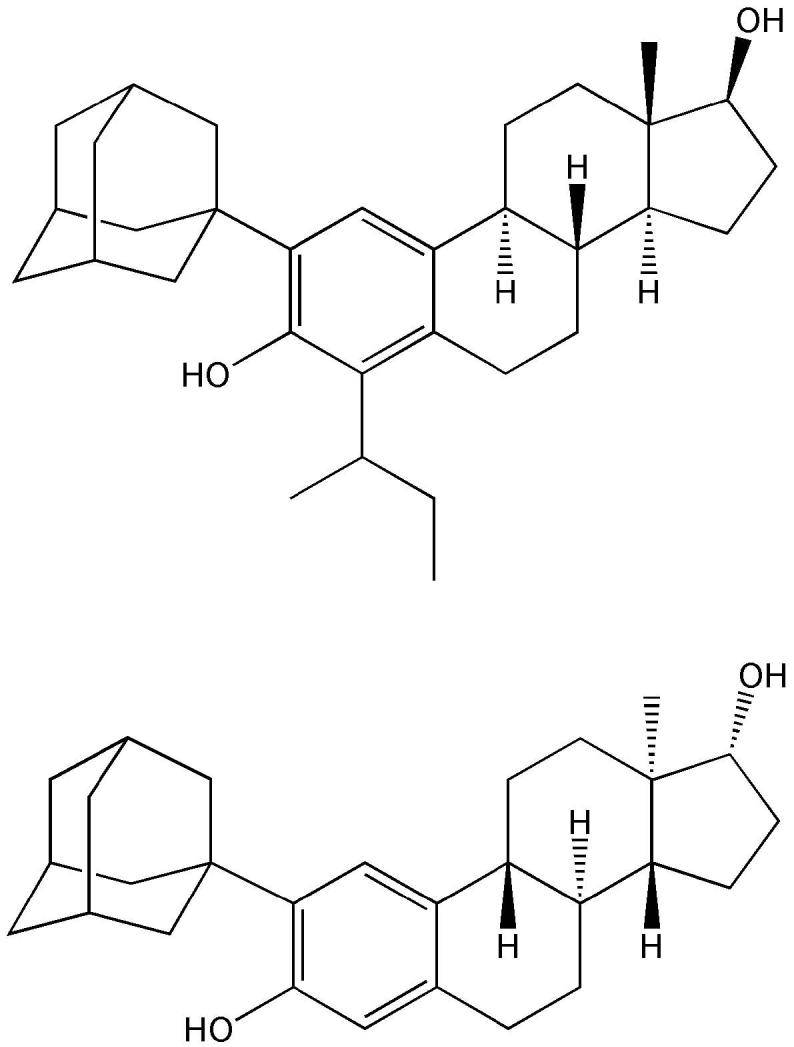
A-ring substituted estratrienes with high potency to protect mouse hippocampal HT22 cells in vitro.
Fig. 11.
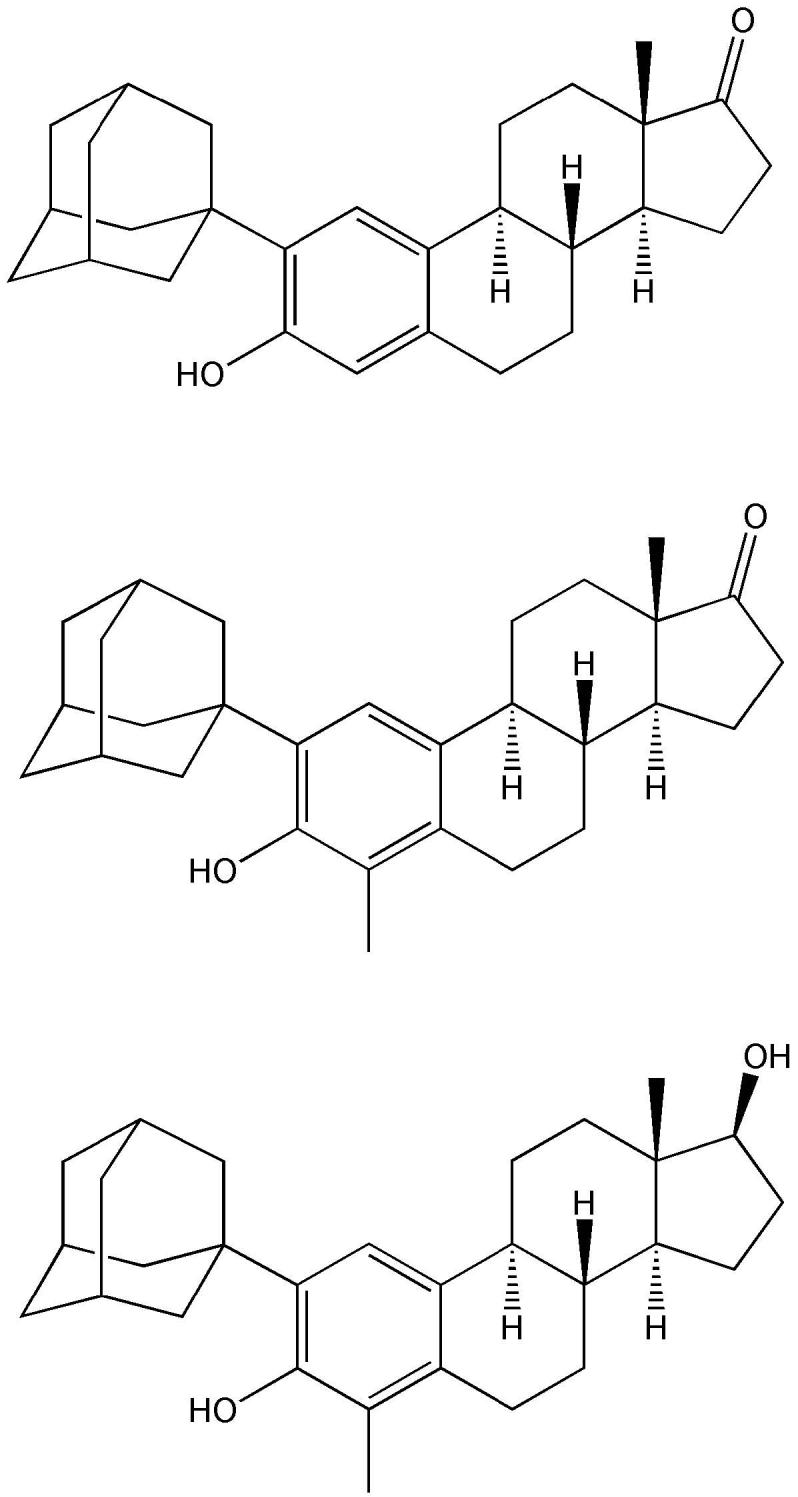
2-(1-Adamantyl)-3-hydroxyestra-1, 3, 5(10)-trien-17-one (top), 2-(1-adamantyl)-4-methyl-3-hydroxyestra-1, 3, 5(10)-trien-17-one (middle) and 2-(1-adamantyl)-4-methyl-estra-1, 3, 5(10)-triene-3,17β-diol (bottom), which do not bind to either ERα or ERβ, are effective in vivo in rat against ischemia-reperfusion injury as a model for stroke.
Addition of a hydroxyl group to the B-and C-rings completely abolished neuroprotection against toxic insults by both glutamate and iodoacetic acid (Perez et al., 2006). These structural changes may reduce antioxidant effect by affecting the streghth ot the phenolic O–H bond and the stability of the phenoxyl radical formed (see Fig. 3); however, additional molecular properties such as lipophilicity may also be affected. Introduction of double bonds into the B- and C-rings markedly increased potency to protect against insults by both glutamate and iodoacetic acid. However, an aromatic B-ring only improved neuroprotection against iodoacetic acid, but did not increase protection against glutamate toxicity when compared to 17β-E2. Esterification of the 17β-OH or replacing the 17α-H with a side chain did not enhance potency. However, straight-chain 17β-O-alkyl ethers had increased protection of HT22 cells compared to 17β-E2 against glutamate toxicity (iodoacetic acid was not tested), when the number of carbons in alkyl chain reached three or more (Fig. 12, Prokai et al., 2001). Additionally, increased lipophilicity (usually correlated with the logarithm of the n-octanol/water partition coefficient, logP) and planarity was also proposed to increase the ability of estratrienes with a phenolic hydroxyl group in their A-ring to protect cells from the stressors employed (Perez et al., 2006). These parameters have not been evaluated quantitatively. Collectively, these observations indicate that the impact of introducing substituents into the B, C and D rings may not be explained simply by substituent effects (on electronic transmission, π delocalization, etc.), although electron-donating substituents generally increase and electron-withdrawing substituents decrease neuroprotection by the resultant estrogen derivatives.
Fig. 12.

17β-O-alkyl ethers with improved potency to protect HT22 cells against glutamate neurotoxicity in vitro.
3.2. Antioxidant effect
The brain is prone to oxidative stress, and is inadequately equipped with antioxidant defense systems to prevent ‘ongoing’ oxidative damage, let alone the extra oxidative damage imposed by neurodegenerative diseases (Halliwell, 2006). Neuronal membrane lipids are rich in highly polyunsaturated fatty acid components and, thus, subject to peroxidation. The overall effects of lipid peroxidation are to decrease membrane fluidity, make it easier for phospholipids to exchange between the two halves of the bilayer, increase the ‘leakiness’ of the membrane to substances that do not normally cross it other than through specific channels (e.g., K+ and Ca2+) and damage membrane proteins, inactivating receptors, enzymes and ion channels (reviewed by Halliwell and Gutteridge, 2006). In addition, products of lipid peroxidation can injure neurons (Halliwell, 2006). Therefore, therapeutic interventions based on reducing oxidative stress and, thereby, attenuating lipid peroxidation have been implicated for at least some of the neurodegenerative diseases (Barnham et al., 2004). For estrogens, their analogues and derivatives, there was a positive correlation between inhibition of iron-induced lipid peroxidation and in vitro neuroprotective potency in cell death models that utilized glutamate and iodoacetic acid as insults (Perez et al., 2006). However, higher concentrations of estratrienes were required to inhibit peroxidation than were needed for neuroprotection. This latter observation indicates that inhibition of lipid peroxidation is not the only mechanism involved in the neuroprotective activity of these compounds. Nevertheless, structure–antioxidant potency relationship based on inhibition of thiobarbituric acid reactive substance (TBARS) in brain tissue was found to mirror the trend discussed in the previous section.
Although not directly related to neuroprotection, a recent study exploring structural determinants for the in vitro antioxidant activity of estrogen derivatives in aqueous lipoprotein solutions (Badeau et al., 2005) is worthy of discussion. The methodical framework of this study, which monitored the suppression of conjugated diene formation (using copper ions as pro-oxidants) by the steroids, may be considered an appropriate model for antioxidant mechanisms operative in any lipid–aqueous system. The results confirmed the phenolic A-ring requirement for activity. Additionally, compounds with one or two adjacent methoxy groups to the phenolic OH provided the strongest antioxidant protection (Fig. 13). Again, the electron donating methoxy group(s) may enhance the antioxidant effect by weakening the phenolic O–H bond and providing stability to the phenoxyl radical formed (see Fig. 3). The incorporation of an aromatic B-ring was also found to have increased potency to inhibit lipid peroxidation, which was in agreement with an earlier report on the strong antioxidant effect with an unsaturated B- or D-ring (Ravi Subbiah et al., 1993). On the other hand, the most hydrophilic “tetrol” compound with three unsubstituted A-ring hydroxyl groups was inactive, and additional hydroxyl groups in the B-, C- or D-ring also seemed to weaken the antioxidant effect. Accordingly, both the presence of unsubstituted hydroxyl groups and adjacent substituents, as well as the lipophilicity evaluated by calculated logP values was found to control the antioxidant activity of estrogen derivatives (Badeau et al., 2005). Distribution into lipid membrane increases with increasing log P; hence, lipophilic estrogen analogues and derivatives can more readily exert their inhibitory effect on lipid peroxidation than those bearing polar, hydrophilic substituents.
Fig. 13.
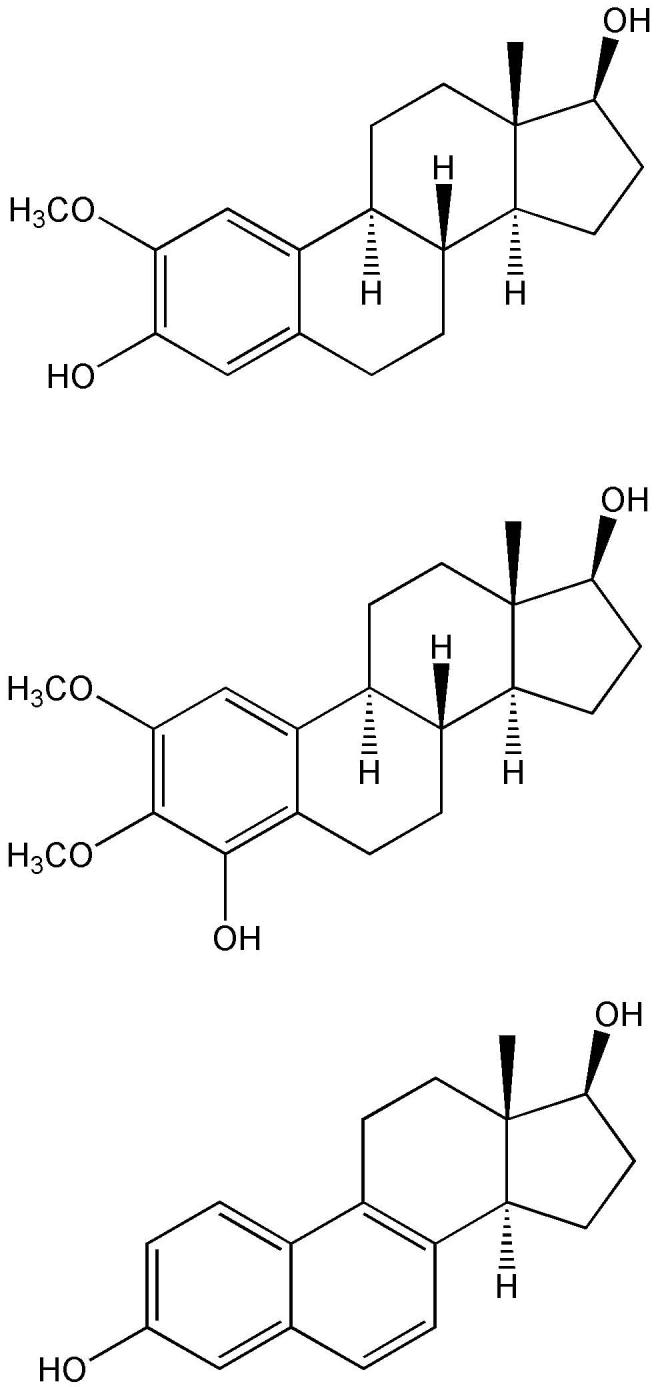
Examples of steroids with antioxidant effect stronger than that of 17β-E2.
4. Conclusion
Numerous chemical modifications that afford neuroprotective estrogen analogues or derivatives to reduce or eliminate estrogenicity (Table 1) while maintaining or enhancing neuroprotection have been described. Knowledge on structure–activity relationships is invaluable, when further molecular optimization and eventual pharmaceutical development of these modified compounds are pursued. Although in vitro paradigms employed to screen these compounds for in vivo evaluation are subject to many competing mechanisms invoked by estrogen on neurons and they should always be evaluated critically, recent studies that apply this approach to address non-genomic neuroprotection have offered insights into structural determinants of neuroprotective activity by estrogen analogues and derivatives. Essentially, several studies suggest that antioxidant effect is important. Therefore, neuroprotective action can often be interpreted in terms of electronic effect of the substituents and based on lipophilicity. These findings hold the promise for meaningful quantitative non-genomic neuroprotection relationship studies for estrogens and estrogen-derived compounds in the near future.
Table 1.
Relative binding affinities (RBA) of selected estrogens and neuroprotective estrogen analogues/derivatives.a
| Compound | RBA: ERα | RBA: ERβ |
|---|---|---|
| 17β-E2 b | 100 c | 100 d |
| 17α-E2 b | 18 | 5 |
| E1 e | 18 f | 10 f |
| Ent-E2 g | 12 | 80 |
| 2-(1-Adamantyl)-3-hydroxyestra-1, 3, 5(10)-trien-17-one h | ∼0 i | ∼0 i |
| 2-(1-Adamantyl)-4-methyl-3-hydroxyestra-1, 3, 5(10)-trien-17-one j | ∼0 i | ∼0 i |
| 2-(1-Adamantyl)-4-methyl-estra-1,3,5(10)-triene-3,17β-diolk | ∼0 i | ∼0 i |
| Estra-1,3,5(10),6,8(9)-pentaene-3,17β-diol l | 51 | 190 |
| 17β-Butoxyestra-1,3,5(10)-triene-3-ol m | 11 | 5 |
RBA=IC50(17β-E2)/ IC50(compound)·100, compiled from Perez et al., 2006, and Prokai et al., 2003
Based on IC50 of 3.0 nM (Perez et al., 2006) or 1.3 nM (Prokai et al., 2003) was considered 100
Based on IC50 of 4.5 nM (Perez et al., 2006) or 0.7 nM (Prokai et al., 2003) as 100
Fig. 7 (first structure)
Average value based on the IC50's reported
Fig. 11 (first structure)
RBA<0.1
Fig. 11 (second structure)
Fig. 11 (third structure)
Fig. 13 (third structure)
Fig. 12 (R=n-C4H9).
Acknowledgements
The authors were supported in part by grants from the National Institutes of Health (NS04765, AG10485 and AG022550). Laszlo Prokai is the Robert A. Welch Professor at the University of North Texas Health Science Center.
Abbreviations
- 17α-E2
estra-1,3,5(10)-triene-3,17α-diol
- 17β-E2
estra-1,3,5(10)-triene-3,17β-diol (estradiol)
- Aβ
amyloid-β peptide
- BAX
Bcl-2-associated X protein
- Bcl
B-cell lymphoma/leukemia
- BDNF
brain-derived neurotrophic factor
- CREB
cyclic AMP-responsive-element binding protein
- E1
3-hydroxyestra-1,3,5(10)-trien-17-one (estrone)
- E3
estra-1,3,5( l0)-triene-3,16α,l7β-triol (estriol)
- EC50
concentration to reach 50% of maximal effect
- EE
17α-ethynyl-estra-1,3,5(10)-triene-3,17β-diol (ethinylestradiol)
- ent-E2
enantiomer of estradiol
- ER
estrogen receptor
- ERE
estrogen-response element
- ERK
extracellular signal-regulated kinase
- FRR
free-radical reductase
- GSH
glutathione
- IGF-1
insulin-like growth factor 1
- IL-1β
interleukin-1β
- IGF-1
insulin-like growth factor 1
- IP3K
phosphatidylinositol 3-kinase
- logP
logarithm of the n-octanol/water partition coefficient
- MAPK
mitogen-activated protein kinase
- NAD(P)H
nicotinamide-adenine dinucleotide (phosphate), reduced form
- NF-κ
nuclear factor kappa
- NMDA
N-methyl-D-aspartate
- NOS
nitric oxide synthase
- PI3K
phosphatidylinositol-3-kinase
- RBA
relative binding affinity
- ROS
reactive oxygen species
- TBARS
thiobarbituric acid reactive substance
- WHI
Women's Health Initiative
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: Gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloisi AM. Gonadal hormones and sex differences in pain reactivity. Clin J Pain. 2003;19:168–174. doi: 10.1097/00002508-200305000-00004. [DOI] [PubMed] [Google Scholar]
- Amantea D, Russo R, Bagetta G, Corasaniti MT. From clinical evidence to molecular mechanisms underlying neuroprotection afforded by estrogens. Pharmacol Res. 2005;52:119–132. doi: 10.1016/j.phrs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- Badeau M, Adlercreutz H, Kaihovaara P, Tikkanen M. Estrogen A-ring structure and antioxidative effect on lipoproteins. J Steroid Biochem Mol Biol. 2005;96:271–278. doi: 10.1016/j.jsbmb.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- Behl C. Oestrogen as a neuroprotective hormone. Nat Rev Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- Behl C, Widmann M, Trapp T, Hoelsboer F. 17-Beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc'h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Blurton-Jones MM, Roberts JA, Tuszynski MH. Estrogen receptor immunoreactivity in the adult primate brain: Neuronal distribution and association with p75, trkA, and choline acetyltransferase. J Comp Neurol. 1999;405:529–542. [PubMed] [Google Scholar]
- Borras C, Gambini J, Gomez-Cabrera MC, Sastre J, Pallardo FV, Mann GE, Vina J. 17β-Oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2[MAPK]/NFκB cascade. Aging Cell. 2005;4:113–118. doi: 10.1111/j.1474-9726.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141:3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- Bryant DN, Bosch MA, Ronnekleiv OK, Dorsa DM. 17-beta estradiol rapidly enhances extracellular signal-regulated kinase 2 phosphorylation in the rat brain. Neuroscience. 2005;133:343–352. doi: 10.1016/j.neuroscience.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Carlstrom L, Ke ZJ, Unnerstall JR, Cohen RS, Pandey SC. Estrogen modulation of the cyclic AMP response element-binding protein pathway – effects of long-term and acute treatments. Neuroendocrinology. 2001;74:227–243. doi: 10.1159/000054690. [DOI] [PubMed] [Google Scholar]
- Coleman KM, Smith CL. Intracellular signaling pathways: Nongenomic actions of estrogens and ligand-independent activation of estrogen receptors. Front Biosci. 2001;6:D1379–D1391. doi: 10.2741/coleman. [DOI] [PubMed] [Google Scholar]
- Deecher DC, Daoud P, Bhat RA, O'Connor LT. Endogenously expressed estrogen receptors mediate neuroprotection in hippocampal cells (HT22) J Cell Biochem. 2005;95:302–312. doi: 10.1002/jcb.20413. [DOI] [PubMed] [Google Scholar]
- Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjold M, Gustafsson JA. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metabol. 1997;82:4258–4265. doi: 10.1210/jcem.82.12.4470. [DOI] [PubMed] [Google Scholar]
- Gao H, Katzenellenbogen JA, Garg R, Hansch C. Comparative QSAR analysis of estrogen receptor ligands. Chem Rev. 1999a;99:723–744. doi: 10.1021/cr980018g. [DOI] [PubMed] [Google Scholar]
- Gao H, Williams C, Labute P, Bajorath J. Binary quantitative structure-activity relationship (QSAR) analysis of estrogen receptor ligands. J Chem Inf Comp Sci. 1999b;39:164–168. doi: 10.1021/ci980140g. [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Meda C, Maggi A, Vegeto E. 17 beta-estradiol inhibits inflammatory gene expression by controlling NF-kappa B intracellular localization. Mol Cell Biol. 2005;25:2957–2968. doi: 10.1128/MCB.25.8.2957-2968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Zubeldia MA, Arbués JJ, Hinchando G, Nogales AG, Millan LC. Influence of estrogen replacement therapy on plasma lipid peroxidation. Menopause. 2001;8:274–280. doi: 10.1097/00042192-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Gotz ME, Kunig G, Riederer P, Youdim MBH. Oxidative stress: Free radical production in neural degeneration. Pharmacol Ther. 1994;63:37–122. doi: 10.1016/0163-7258(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Green PS, Bishop J, Simpkins JW. 17α-Estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997a;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Gordon K, Simpkins JW. Phenolic A ring requirement for neuroprotective effects of steroids. J Steroid Biochem Mol Biol. 1997b;63:229–235. doi: 10.1016/s0960-0760(97)00124-6. [DOI] [PubMed] [Google Scholar]
- Green PS, Gridley KE, Simpkins JW. Nuclear estrogen receptor-independent neuroprotection by estratrienes: A novel interaction with glutathione. Neuroscience. 1998;84:7–10. doi: 10.1016/s0306-4522(97)00595-2. [DOI] [PubMed] [Google Scholar]
- Gridley KE, Green PS, Simpkins JW. A novel, synergistic interaction between 17β-estradiol and glutathione in the protection of neurons against beta-amyloid 25-35-induced toxicity in vitro. Mol Pharmacol. 1998;54:874–880. doi: 10.1124/mol.54.5.874. [DOI] [PubMed] [Google Scholar]
- Hail N, Carter BZ, Konopleva M, Andreeff M. Apoptosis effector mechanisms: A requiem performed in different keys. Apoptosis. 2006;11:889–904. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- Hall JM, Couse JF, Korach KF. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th Ed. Oxford University Press; Oxford: 2006. [Google Scholar]
- Henderson VW. Estrogen-containing hormone therapy and Alzheimer's disease risk: Understanding discrepant interferences from observational and experimental research. Neuroscience. 2006;138:1031–1039. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Jezierski MK, Sohrabji F. Region- and peptide-specific regulation of the neurotrophins by estrogen. Mol Brain Res. 2000;85:75–84. doi: 10.1016/s0169-328x(00)00244-8. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. Nature of the ligand-binding pocket of estrogen receptor alpha and beta: The search for subtype-selective ligands and implications for the prediction of estrogenic activity. Pure Appl Chem. 2003;75:2397–2403. [Google Scholar]
- Levin ER. Cell Localization, physiology and nongenomic actions of estrogen receptors. J. Applied Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- Liang Y, Belford S, Tang F, Prokai L, Simpkins JW, Hughes JA. Membrane fluidity effects of estratrienes. Brain Res Bull. 2001;54:661–668. doi: 10.1016/s0361-9230(01)00483-x. [DOI] [PubMed] [Google Scholar]
- Liu R, Yang S-H, Perez E, Yi KD, Wu SS, Eberst K, Prokai L, Prokai-Tatrai K, Cai ZY, Covey DF, Day AL, Simpkins JW. Neuroprotective effects of a novel non-receptor binding estrogen analogue: In vitro and in vivo analysis. Stroke. 2002;33:2485–2491. doi: 10.1161/01.str.0000030317.43597.c8. [DOI] [PubMed] [Google Scholar]
- Maclusky NJ, Naftolin F, Krey LC, Franks S. The catechol estrogens. J Steroid Biochem. 1981;15:111–124. doi: 10.1016/0022-4731(81)90265-x. [DOI] [PubMed] [Google Scholar]
- Manavathi B, Kumar R. Steering estrogen signals from the plasma membrane to the nucleus: Two sides of the coin. J Cell Physiol. 2006;207:594–604. doi: 10.1002/jcp.20551. [DOI] [PubMed] [Google Scholar]
- Marin R, Guerra B, Alonso R, Ramirez CM, Diaz M. Estrogen activates classical and alternative mechanisms to orchestrate neuroprotection. Curr Neurovasc Res. 2005;2:287–301. doi: 10.2174/156720205774322629. [DOI] [PubMed] [Google Scholar]
- Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—Part I. Headache. 2006;46:3–23. doi: 10.1111/j.1526-4610.2006.00309.x. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrin Metab. 2003;14:228–235. doi: 10.1016/s1043-2760(03)00076-6. [DOI] [PubMed] [Google Scholar]
- McCay PB, Brueggemann G, Lai EK, Powell SR. Evidence that α-tocopherol functions cyclically to quench free radicals in hepatic microsomes. Ann N Y Acad Sci. 1989;570:32–45. doi: 10.1111/j.1749-6632.1989.tb14906.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Estrogenic effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–2801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- Miller CP, Jirkovsky I, Hayhurst DA, Adelman SJ. In vitro antioxidant effects of estrogens with a hindered 3-OH function on the copper-induced oxidation of low density lipoprotein. Steroids. 1996;61:305–308. doi: 10.1016/0039-128x(95)00234-h. [DOI] [PubMed] [Google Scholar]
- Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- Mize AL, Shapiro RA, Dorsa DM. Estrogen receptor-mediated neuroprotection from oxidative stress requires activation of the mitogen-activated protein kinase pathway. Endocrinology. 2003;144:306–312. doi: 10.1210/en.2002-220698. [DOI] [PubMed] [Google Scholar]
- Moggs JG, Orphanides G. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep. 2001;2:775–781. doi: 10.1093/embo-reports/kve185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooradian AD. Antioxidant properties of steroids. J Steroid Biochem Mol Biol. 1993;45:509–511. doi: 10.1016/0960-0760(93)90166-t. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ospina JA, Brevig HN, Krause DN, Duckles SP. Estrogen suppresses IL-1 beta-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am J Physiol. 2004;286:H2010–H2019. doi: 10.1152/ajpheart.00481.2003. [DOI] [PubMed] [Google Scholar]
- Packer JE, Slater TF, Wilson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1979;278:737–738. doi: 10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- Peinado V, Gonzalez JC, Leret ML. Effect of 17-beta-estradiol on dopamine, serotonine and GABA striatal levels in 6-hydroxydopamine-treated rats. Toxicology. 2004;204:155–160. doi: 10.1016/j.tox.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Perez EJ, Cai YC, Covey DF, Simpkins JW. Neuroprotective effects of estratriene analogs: structure-activity relationships and molecular optimization. Drug Dev Res. 2006;66:78–92. [Google Scholar]
- Perez E, Liu R, Yang SH, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005;1038:216–222. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Prokai L, Oon S-M, Prokai-Tatrai K, Abboud K, Simpkins JW. Synthesis and biological evaluation of 17β-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J Med Chem. 2001;44:110–114. doi: 10.1021/jm000280t. [DOI] [PubMed] [Google Scholar]
- Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez EJ, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci. 2003;100:11741–11746. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai L, Prokai-Tatrai K, Perjesi P, Simpkins JW. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev Res. 2006;66:118–125. [Google Scholar]
- Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. WHIMS Investigators. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. J Am Med Assoc. 2003;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- Ravi Subbiah MT, Kessel B, Agrawal M, Rajan R, Abplanalp W, Rymaszewski Z. Antioxidant potential of specific estrogens on lipid peroxidation. J. Clin. Endocrinol Metab. 1993;77:1095–1097. doi: 10.1210/jcem.77.4.8408459. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Ruiz-Larrea B, Leal A, Martin C, Martinez R, Lacort M. Effects of estrogens on the redox chemistry of iron: a possible mechanism of the antioxidant action of estrogens. Steroids. 1995;60:780–783. doi: 10.1016/0039-128x(95)00119-b. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, di Michele F, Herman B, Strohle A, Lancel M, Romeo E, Holsboer F. Neuroactive steroids: molecular mechanisms of action and implications for neuropsychopharmacology. Brain Res Rev. 2001;37:59–67. doi: 10.1016/s0165-0173(01)00123-0. [DOI] [PubMed] [Google Scholar]
- Sand P, Luckhaus C, Schlurmann K, Gotz M, Deckert J. Untangling the human estrogen receptor gene structure. J Neural Transm. 2002;109:567–583. doi: 10.1007/s007020200047. [DOI] [PubMed] [Google Scholar]
- Scoville SA, Bufton SM, Liuzzi FJ. Estrogen regulates neurofilament gene expression in adult female rat dorsal root ganglion neurons. Exp Neurol. 1997;146:596–599. doi: 10.1006/exnr.1997.6565. [DOI] [PubMed] [Google Scholar]
- Shah RD, Anderson KL, Rapoport M, Ferreira A. Estrogen-induced changes in the microtubular system correlate with a decreased susceptibility of aging neurons to beta amyloid neurotoxicity. Mol Cell Neurosci. 2003;24:503–516. doi: 10.1016/s1044-7431(03)00166-0. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Scrimo PJ, Merchenthaler I. Evidence for the colocalization of estrogen receptor-beta mRNA and estrogen receptor-alpha immunoreactivity in neurons of the rat forebrain. Endocrinology. 1998;139:5267–5270. doi: 10.1210/endo.139.12.6525. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Scrimo PJ, Merchenthaler I. Estrogen binding and estrogen receptor characterization (ER alpha and ER beta) in the cholinergic neurons of the rat basal forebrain. Neuroscience. 2000;96:41–49. doi: 10.1016/s0306-4522(99)00520-5. [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, III, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. J Am Med Assoc. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezl-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Wang J, Wang X, Perez EJ, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr Drug Targets. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Wen Y, Perez EJ, Yang S, Wang X. Role of nonfeminizing estrogens in brain protection from cerebral ischemia. An animal model of Alzheimer's disease neuropathology. Ann N Y Acad Sci. 2005;1052:233–242. doi: 10.1196/annals.1347.019. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Yang S-H, Liu R, Perez E, Cai ZY, Covey DF, Green PS. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004;35:2648. doi: 10.1161/01.STR.0000143734.59507.88. [DOI] [PubMed] [Google Scholar]
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen protection in NT2 neurons. Neuroreport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- Singh M, Simpkins JW, editors. The Future of Hormone Therapy: What Basic Science and Clinical Studies Teach Us. Annals of the New York Academy of Sciences. Vol. 1052. The New York Academy of Sciences; New York, NY: 2005. pp. ix–260. [Google Scholar]
- Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med. 2002;33:37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- Sohrabji F, Miranda RCG, Toran-Allerand CD. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sribnick EA, Ray SK, Nowak MW, Li L, Banik NL. 17 beta-estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. J Neurosci Res. 2004;76:688–696. doi: 10.1002/jnr.20124. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Berchtold NC, Cotman CW, Pike CJ. Estrogen regulates bcl-x expression in rat hippocampus. Neuroreport. 2001;12:2797–2800. doi: 10.1097/00001756-200109170-00009. [DOI] [PubMed] [Google Scholar]
- Tikkanen MJ, Vihma V, Jauhiainen M, Hockerstedt A, Helisten H, Kaamanen M. Lipoprotein-associated estrogens. Cardiovasc Res. 2002;56:184–188. doi: 10.1016/s0008-6363(02)00535-7. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD. Estrogen and the brain: beyond ER-alpha and ER-beta. Exp Gerontol. 2004;39:1579–1586. doi: 10.1016/j.exger.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Tremblay GB, Giguere V. Coregulators of estrogen receptor action. Crit Rev Eukar Gene Expr. 2002;12:1–22. doi: 10.1615/critreveukaryotgeneexpr.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- Turgeon JL, McDonnell DP, Martin KA, Wise PM. Hormone therapy: physiological complexity belies therapeutic simplicity. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer's amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- Vedder H, Anthens N, Stumm G, Wurz C, Behl C, Krieg JC. Estrogen hormones reduce lipid peroxidation in cells and tissues of the central nervous system. J Neurochem. 1999;72:2531–2538. doi: 10.1046/j.1471-4159.1999.0722531.x. [DOI] [PubMed] [Google Scholar]
- Vegeto E, Belcredito S, Etteri S, Ghisletti S, Brusadelli A, Meda C, Krust A, Dupont S, Ciana P, Chambon P, Maggi A. Estrogen receptor-alpha mediates the brain anti-inflammatory activity of estradiol. Proc Natl Acad Sci USA. 2004;101:9614–9619. doi: 10.1073/pnas.1531957100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtamaki T, Parantainen J, Kolvisto AM, Metsa-Ketela T, Táimala R. Oral ascorbic acid increases plasma oestradiol during postmenopausal hormone replacement therapy. Maturitas. 2002;42:129–135. doi: 10.1016/s0378-5122(02)00005-1. [DOI] [PubMed] [Google Scholar]
- Wu TW, Wang JM, Chen S, Brinton RD. 17β-Estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-amp response element binding protein signal pathway and Bcl-2 expression in rat hippocampal neurons: A potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Yan N, Shi YG. Mechanisms of apoptosis through structural biology. Ann Rev Cell Dev Biol. 2005;21:35–56. doi: 10.1146/annurev.cellbio.21.012704.131040. [DOI] [PubMed] [Google Scholar]
- Yang S, Liu R, Perez EJ, Wen Y, Stevens SM, Jr., Valencia T, Brun-Zinkernagel A-M, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XR, Rajala RVS, McGinnis JF, Li F, Anderson RE, Yan XR, Li S, Elias RV, Knapp RR, Zhou XH, Cao W. Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17 beta-estradiol-mediated neuroprotection. J Biol Chem. 2004;279:13086–13094. doi: 10.1074/jbc.M313283200. [DOI] [PubMed] [Google Scholar]
- Zhang JQ, Cai WQ, Zhou DS, Su BY. Distribution and differences of estrogen receptor beta immunoreactivity in the brain of adult male and female rats. Brain Res. 2002;935:73–80. doi: 10.1016/s0006-8993(02)02460-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tounekti O, Akerman B, Goodyer CG, LeBlanc AA. 17-beta-estradiol induces an inhibitor of active caspases. J Neurosci. 2001;21:RC176. doi: 10.1523/JNEUROSCI.21-20-j0007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]