

Abstract
Time of flight secondary ion mass spectrometry (ToF-SIMS) is a well-established bioanalytical method for directly imaging the chemical distribution across single-cells. Here we report a protocol for the use of SIMS imaging to comparatively quantify the relative difference in lipid composition between the plasma membranes of two cells. This development enables direct comparison of the chemical effects of different drug treatments and incubation conditions in the plasma membrane at the single-cell level. Relative, quantitative ToF-SIMS imaging has been used here to compare macrophage cells treated to contain elevated levels of cholesterol with respect to control cells. In-situ fluorescence microscopy with two different membrane dyes was used to discriminate morphologically similar but differentially treated cells prior to SIMS analysis. SIMS images of fluorescently identified cells reveal that the two populations of cells have distinct outer leaflet membrane compositions with the membranes of the cholesterol-treated macrophages containing more than twice the amount of cholesterol of control macrophages. Relative quantification with SIMS to compare the chemical composition of single-cells can provide valuable information about normal biological functions, causative agents of diseases, and possible therapies for diseases.
INTRODUCTION
Single-cell experiments promise an enhanced understanding of fundamental biological events, such as intra- and intercellular communication (exocytosis, endocytosis, and signal trafficking), the physiological effect of exogenous agents (drug treatments and the environment), and abnormalities in normal biological function associated with disease.1 Numerous analytical techniques, including electrochemical detection,2–6 separation-based methods,7–9 fluorescence microscopy,10–13 and mass spectrometry14–24 have been used to surmount the inherent difficulties of measurements down to the μm-, pL-, and zmole-scale to study the intricate chemistry that exists within single cells.
Secondary ion mass spectrometry (SIMS) is an emerging technique for single-cell investigations. SIMS operates on the principle that sample bombardment by an electrostatically focused primary ion beam will result in the ejection of secondary species (electrons, neutrals, and ions) from the sample.25 The secondary ions can be electrostatically collected and mass analyzed. Imaging with SIMS can involve scanning the primary ion beam across the sample and collecting mass spectra as a function of the position of the primary ion beam. Alternatively, some instruments image using secondary ion optics that maintain the spatial distribution of the ions.26 Two modes of SIMS are available for imaging, with the first being a dynamic mode that uses high ion doses to quickly erode sample surfaces and obliterate molecular species making it best suited for elemental and isotopic studies, and depth profiling. The second SIMS approach is a static mode that uses low ion doses (less than 1% of the surface molecules are impacted by a primary ion) to generate larger mass fragments.25 Therefore, this mode allows the study of all of the elements as well as molecular and molecular fragment ions. The primary ions can be delivered in pulses which allow the secondary ions to be separated by their time of flight (ToF). Other SIMS analyzer configurations include magnetic sectors and quadropoles.27 Both static and dynamic SIMS have been used successfully to image the distribution of chemicals across individual cells.19–24
SIMS instruments are held under ultra-high vacuum and this requires strict sample preparation considerations for hydrated samples, such as cells. Freeze-fracture is a popular SIMS sample preparation method which cryogenically preserves biological samples.19, 22, 28–30 Freeze-fracture is an attractive protocol because it has been shown to maintain the native distribution of molecules in liposomes and cells.16, 19–23, 30 However, freeze-fracture involves many experimental variables, such as the temperature and pressure conditions during the fracture,30 the portion of the cell exposed to the sample-vacuum interface,21 and the thickness of ice remaining on the sample substrate, that contribute to experiment-to-experiment variations in secondary ion yield and SIMS images. This issue has been insignificant for many SIMS imaging experiments such as the analysis of individual cells,19–22 the comparison of morphologically distinct cells,16 and the comparison of morphologically-defined areas of model systems and cells. 23, 31 These types of experiments were easily accessible with freeze-fracture SIMS because morphology allowed cells to be identified before analysis and therefore could be analyzed within one image which helps to minimize the inherent variability introduced by freeze-fracture; however, the application of comparing morphologically similar cells is challenging because there is no way to know that the cells being compared are from two different populations until the image is acquired.
Quantification of SIMS images requires the use of an internal standard of known concentration, since intensity of the secondary ion current is not solely dependent on the amount of molecule in the sample. Factors influencing signal intensity include the chemical composition of the sample matrix, topography, matrix-primary ion interactions, instrumental transmission and detector response.32 Adding an internal standard to the plasma membrane, however, is problematic. The obvious choice is a lipid molecule that can easily intercalate into the membrane without disrupting it. A challenge with this approach is that it is difficult to know how much lipid has been added to the membrane, also adding amounts of lipid large enough to compare with the native lipids will ultimately affect the lipid composition of the membrane. The solution to this problem is to use a ubiquitous ion signal to normalize the signal of interest, such as the C5H9+ fatty acid fragment. 24 This type of internal standard is useful to obtain comparative data across single24 or conjugated cells.23
Evaluating the molecular content and distribution within treated cells versus control cells is a valuable potential application of SIMS imaging because it may help elucidate cellular function and the effects of exogenously added agents on these cells. One often manipulated compound in cell membrane experiments is cholesterol. Cholesterol is an intriguing membrane component because it controls physical membrane properties, such as fluidity and permeability.33 Cholesterol also is highly concentrated in lipid rafts, and therefore may be a crucial molecule in normal cell function, such as signal transduction.34–36 In addition, better understanding of the role of cholesterol is important because abnormalities in cholesterol content have been observed in numerous diseases, including HIV, Alzheimer’s disease, and atherosclerosis, a major cause of heart disease.37–39 In particular, advanced lesions in atherosclerosis develop from early lesions which consist of aggregated, cholesterol-engorged macrophages.39
One traditional method for manipulating the amount of cholesterol in cells is to incubate the cells in β-cyclodextrin (BCD), a seven-member glucose ring with a cavity for cholesterol binding.40 This treatment depletes cell membranes of cholesterol and has been used to study lipid rafts.10, 34 Alternatively, pre-formed complexes of cholesterol and BCD (chol-BCD) have been used to elevate the native amount of cholesterol in a cell.35
In this paper, SIMS imaging with 2-color in-situ fluorescence is used to identify differentially treated cells, thus permitting simultaneous analysis and comparison of individual cells from each group. Expanding upon previous work, which incorporated fluorescence microscopy into the SIMS analysis chamber,20 a criterion has been developed to address variations that result from freeze-fracture for comparative single-cell studies. These experiments set the groundwork for further single-cell comparative studies with SIMS that can provide valuable information about normal biological functions, causative agents of diseases, drug resistance, and possible therapies for diseases.
EXPERIMENTAL
Secondary Ion Mass Spectrometry
SIMS experiments were performed on a Kratos (Manchester, U.K.) Prism ToF-SIMS spectrometer equipped with a 15-kV FEI (Beaverton, OR) indium liquid metal ion source (LMIS) oriented 45° to the sample. The LMIS was focused to a beam size of 200 nm and delivered 0.5 nA of current to the sample with a 50-ns pulse width. The sample was mounted onto a liquid nitrogen (LN2) cooled analysis stage (Kore Tech. Ltd, Cambridge, U.K.) and biased at +2.5 kV. An extraction lens, biased at −4.7 kV, collected the secondary ions which then traveled along a 4.5-m flight path before being detected at a microchannel plate (MCP) detector (Galileo Co., Sturbridge, MA). SIMS imaging was performed by electrostatically rastering the primary ion beam across the sample and collecting a mass spectrum at every pixel in the 128 × 128 pixel image. All images acquired for this experiment had a field of view of 100 × 100 μm2. From the LMIS scan, software written in-house generated both a “total ion” mass spectrum, which was the summation of the individual spectra for every image pixel, and a “total ion” image, in which the signal of each pixel represented the integrated intensity of all mass spectral peaks for that pixel.
Using these data, three different types of analyses were performed. First, regions of the total ion image were selected and a mass spectrum, representative of the summation of the mass spectra in the selected pixels, was produced. In order to compare two of these mass spectra, each spectrum was normalized to the total signal to account for the different number of selected pixels for each region. In a second type of analysis, mass ranges of interest were chosen from the total ion mass spectrum to create 2D intensity plots of the location of a given analyte in the imaging area. The intensity plots were also called molecule-specific images. The molecule-specific images were then color-coded and overlaid using the SIMS software. Additional colors were generated with Adobe Photoshop. The third type of analysis was a line scan, which used molecule-specific images to graph the summed intensity of a given analyte across the thickness of a line as a function of the lateral distance of the line.
Line Scans and Quantification Methods
The line scans in this experiment used a line width of 15 pixels in order to sample a large percentage of each cell. For a given molecule-specific image, a single line was drawn through each pair of cells. Line scans for both m/z 69 and m/z 366–370 were created for every pair of cells and the same lateral distance was analyzed for both masses for a given cell pair. The raw line scans were then treated with 25 % Savitsky-Golay smoothing in PeakFit software. The smoothed line scan was then used to quantify the average signal intensity for the cells for the two mass ranges. Unlike the raw data, the smoothed line scans allowed plot sections to be selected for quantification based on a change in the slope of the line. The same length range along the line scan was used for every evaluation. Using this approach, raw, average signal intensities were determined for n=8 cell pairs. The average signal intensity for m/z 366–370 was normalized to the average intensity of m/z 69 for each cell to account for the local effects on the ionization efficiency.23, 24 Only the outer leaflet of cell membranes were compared in this experiment.21
Selection Criteria for Semi-quantitative SIMS
Three criteria have determined whether a given pair of cells is included in the subsequent analysis. First, both cells in a pair must be in the same SIMS image thus ensuring the cells being analyzed would be in similar environment and receive the same primary ion dose. Second, each cell must be determined to be intact with optical microscopy. That is, cells which are obviously deformed or ruptured were excluded. The third factor is only cell pairs that have the outer leaflet of their membranes uncovered are analyzed. The exposed cellular surfaces are determined to be from the outer leaflet by molecule-specific images of DiD and DiI. As previously discussed, cholesterol from the chol-BCD treatment will incorporate into different regions of the cell by different amounts, making the identification of the exposed surface crucial.
Microscopy
A vertical illuminator microscope (Olympus, Melville, NY) was previously incorporated into the SIMS instrumental design for brightfield and epifluorescence imaging of samples on the SIMS analysis stage.20 The microscope includes two illuminator columns equipped with a 50-W halogen and a 100-W Hg light source. A Spot RT CCD camera (Diagnostic Instruments, Sterling Heights, MI) was used in monochrome mode to capture fluorescence images. For fluorescence images of DiD, a XF110 filter set (Omega, Battleboro, VT) was used to select excitation wavelengths of 644 nm and emission wavelengths of 663 nm. As in previous experiments, fluorescence images of DiI were possible using a XF101 filter set (Omega, Brattleboro, VT) which selects 549 nm and 565 nm excitation and emission wavelengths, respectively.20, 21 The monochrome DiD and DiI images were colored and combined using the Spot software. The fluorescence images were then rotated, flipped, and cropped in Adobe Photoshop to align them with the SIMS images, as described elsewhere.20
Standards and J774 Cell Preparation
Stock solutions of 10 mg/mL DiD and DiI (Molecular Probes, Eugene, OR) were prepared in ethanol. To obtain a standard spectrum for DiD, 100 μL of the stock solution was drop-dried onto a pirhana etched silicon substrate (Ted Pella, Redding, CA). The standard spectrum was taken using a 300 × 300-μm2 field of view and a dose of 1 million primary ion pulses. J774 cells were the generous gift of R.A. Schlegel and were cultured to confluence in sterile, polystyrene cell culture flasks at 5% CO2 and 37°C. When confluent, the J774 cell flasks were treated for 1 hr with 25 μg of DiD or DiI/mL of serum-free RPMI media (Invitrogen, Carlsbad, CA). Next, 100 μL of a 350 mg/mL aqueous stock solution of chol-BCD (CDT, Inc., High Springs, FL), which corresponded to a 260 μM loading dose of cholesterol, was added to the DiD labeled flask. Both the DiI and chol-BCD-containing DiD flasks were further incubated for 1 hr. After the incubation, both flasks were rinsed 5 times with serum-free RPMI to remove the dye and chol-BCD solution. The cells then were dislodged from the bottom of the flask with gentle tapping and the two cell populations were combined into a culture dish containing sterilized 5 × 5 mm silicon wafers (Ted Pella, Redding, CA). After a short settling period, a top silicon shard was placed on every wafer and the assemblies were cryogenically frozen by plunging them into liquid ethane. Before SIMS analysis, all frozen sample assemblies were stored under LN2. The freeze-fracture sample preparation method used has been described in detail and has been slightly modified.30 Briefly, the sample stub was held in the sample preparation chamber in a cold clamp cooled to −196°C with LN2. Meanwhile, a freeze-fracture sample stage was cooled to −106°C and the pressure inside the vacuum was maintained at 2 × 10−8 torr. Once the fracture stage had reached the desired temperature, the sample was transferred onto it and the top shard was removed with a cryogenically cooled knife. The sample was then cooled back to −196°C and transferred onto a stage in the SIMS analysis chamber that was also cryogenically cooled to −196°C.
RESULTS AND DISCUSSION
Use of DiI and DiD to Classify Cells
To discriminate the quantitative membrane composition of cells that are exposed to different incubation conditions, it is necessary to identify the cell population from which each cell originates. One method for distinguishing morphologically similar cells from two populations is to use two-color fluorescence and two fluorophores with excitation and emission profiles that have suitably different energies. DiI and DiD have been chosen here because they fit this criterion, are lipophillic and are commonly used in fluorescence studies.41 Additionally, these particular dyes adhere only to the outer membrane of a cell, which is advantageous for analysis with a surface-sensitive technique, like SIMS. Earlier SIMS experiments with DiI–stained cells were used to determine chemical signatures for various cellular sections that can be exposed to the vacuum-surface interface during freeze-fracture.21 Detection of DiI signal on a freeze-fractured cell indicates that freeze fracture can be used to uncover the outer leaflet of the cell membrane. In terms of single-cell comparative studies, chemically labeling the outer membrane of the cells also helps classify the exposed cellular plane to identify which cell surfaces are compared.
Because DiI and DiD are present in the cell membranes during SIMS imaging, it is important to collect standard mass spectra in order to identify characteristic dye peaks. The mass spectrum for DiI has formerly been described to contain the molecular ion peak (m/z 834), fragment peaks (m/z 410 and 424), and a hydrolysis product peak (m/z 414).20 The DiD ionization pattern has not been explored with SIMS and its positive ion spectrum is shown in Figure 1. DiD ionizes to form an intense molecular ion peak at m/z 860 (Figure 1). The two conjugated ring systems in the DiD molecule are joined by an unsaturated linker that is 5 carbons in length, allowing it to be symmetrically fragmented across the linker (Figure 2A) to yield a fragment peak at m/z 424 (Figure 1). The symmetrical cleavage accounts for the absence of a peak at m/z 410, which had been identified in the DiI spectrum to result from an asymmetrical fragmentation across the DiI linker which is only 3 carbon atoms in length.20 A DiD hydrolysis peak is observed at m/z 414 (Figure 1 and Figure 2B), similar to the DiI spectrum. Because m/z 414 and m/z 424 are mass spectral peaks for both dye molecules, only m/z 860 can be used to image DiD in the presence of DiI. Similarly, m/z 410 and 834 can be used to distinguish DiI from DiD.
Figure 1.
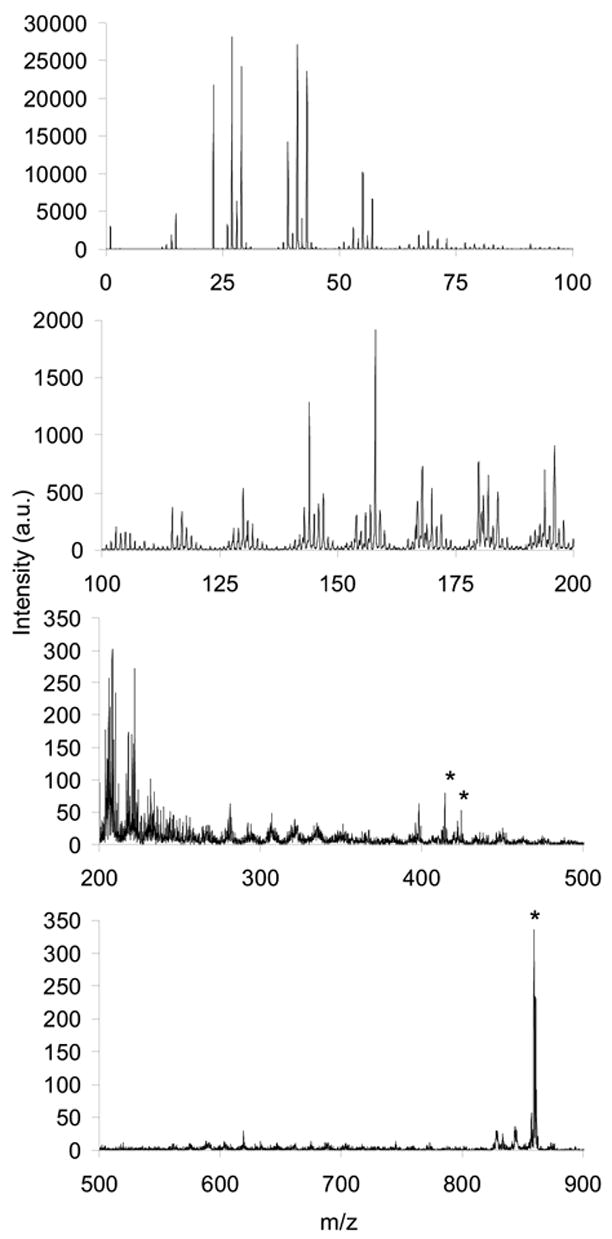
Positive SIMS mass spectrum for a DiD standard on a Si substrate. The peak at m/z 860 corresponds to the DiD molecular ion. The peak at m/z 424 is a fragment of the molecule which cleaved symmetrically about the unsaturated linker. The peak at m/z 414 is a hydrolysis product of the DiD.
Figure 2.
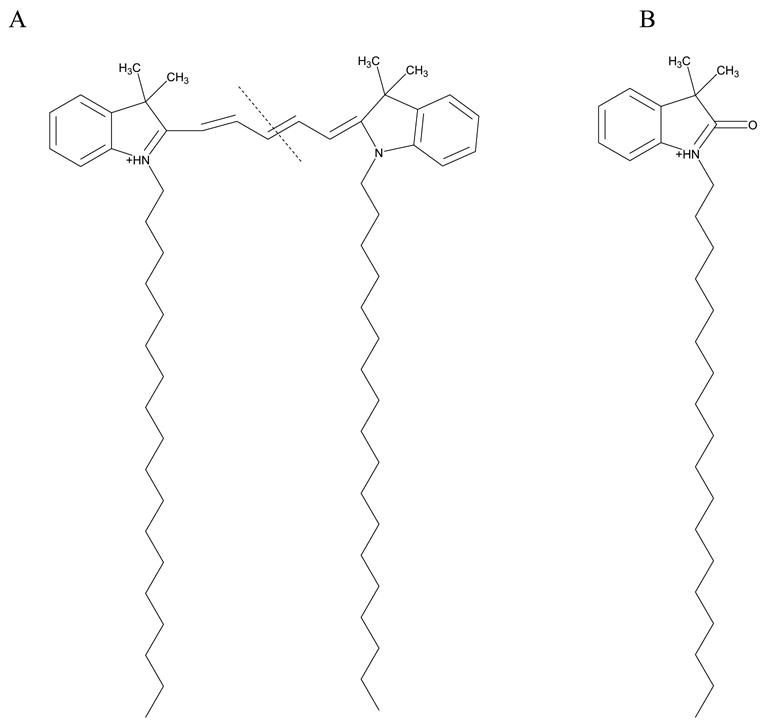
Chemical structures for DiD and DiD fragments. (A) The structure for DiD with a dotted line indicating the position of cleavage for the m/z 424 fragment observed in the standard mass spectrum. (B) Chemical structure for the DiD hydrolysis product observed at m/z 414.
SIMS Spectra and Images Comparing Cholesterol in Cell Membranes
A proof-of-concept experiment has been carried out which uses in-situ fluorescence to demonstrate semi-quantitative SIMS imaging by measuring elevated cholesterol content in treated cells versus control cells where the two cell populations are fluorescently labeled. One population of J774 cells (treated) was incubated with a solution of chol-BCD and labeled with DiD; a second J774 population (control) was incubated in media and labeled with DiI. With this protocol, the treated, DiD-labeled cells should exhibit a higher SIMS cholesterol signal than the DiI-labeled control cells. Because cholesterol is a major analyte of interest for these experiments, its SIMS ionization warrants discussion here. Ionization of cholesterol yields arrays of peaks in the mass range of the molecular ion (m/z 386) and in the mass range of a fragment ion (m/z 369).42 The array of peaks in the mass range of the fragment ion (m/z 366–370) is used for all cholesterol molecule-specific images and quantification in this paper.
From a SIMS image of a fluorescently characterized mixed population of treated and untreated J774 cells, two mass spectra have been generated: one from the integrated intensity across a treated cell (Figure 3A) and the second from the integrated intensity across a control cell (Figure 3B). As expected, the normalized mass spectrum of the treated cell contains higher signal from cholesterol than the control cell. Additionally, the treated cell mass spectrum contains a peak for the molecular ion of the identifying marker, DiD, and the control cell mass spectrum contains signal corresponding to the molecular ion of the identifier, DiI. In addition to supporting the identification of the two cells, the presence of the dye signals indicates that the outer leaflet is uncovered for both cells, as the dyes are present in the outer leaflet and are detected in both cases.
Figure 3.
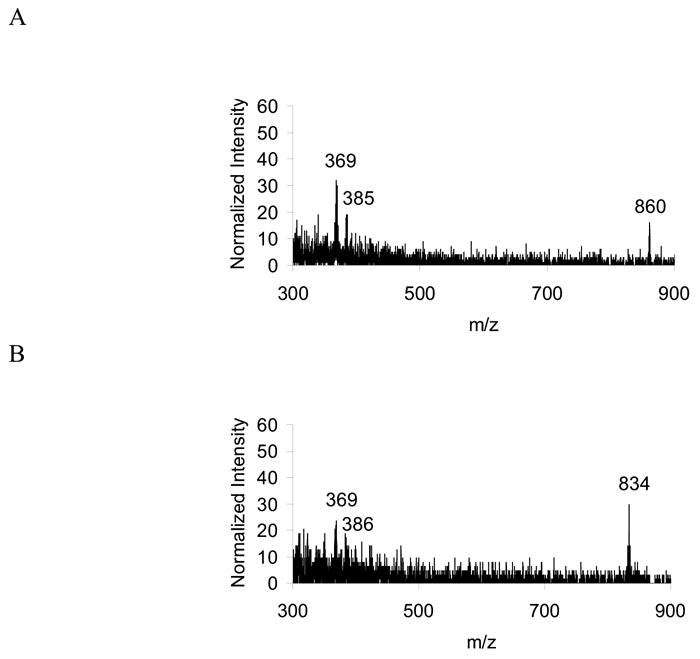
Summed mass spectra from pixels located across two cells. (A) A chol-BCD treated J774 cell and (B) a control J774 cell.
A fluorescence image demonstrates that the excitation and emission profiles of the two dyes do not interfere with one another and that mixing the two populations of cells does not cross-contaminate the dyes (Figure 4A). A composite SIMS image of m/z 834 (DiI; green) and m/z 860 (DiD; red) correlates with the fluorescence image and further shows, by mass spectrometry, that the two dyes have not cross-contaminated on differing cells during sample preparation (Figure 4B). The fluorescence and SIMS images also identify the two cells at the bottom of the image as DiD-treated cells and the two cells at the top as DiI-control cells. Furthermore, the SIMS image indicates again that the outer membranes of the left-most DiD-treated cell and of both DiI-control cells are exposed to the vacuum interface because the signal from the lipophilic dyes, in each case, localizes to the cells (Figure 4B). It is important that the same cellular surface is compared between treated and control cells because different amounts of cholesterol will likely incorporate into the cytoplasm and into the outer leaflet and the inner leaflet of the plasma membrane.33 Since the DiD-treated cell on the right does not appear to be fractured to expose the outer-leaflet, it has not been considered in further analyses. It appears that this cell exhibits less intense fluorescence because part of the DiD-enriched outer membrane was removed during freeze-fracture, resulting in significantly less fluorophore molecules associated with the cell, compared to the outer-leaflet exposed DiD-cell. The brightness and contrast settings in the imaging software that are appropriate for the outer-leaflet exposed cells cause the DiD-cell with less fluorescence to appear smaller than in actuality. The control cell located in the upper right of the images was optically determined to have been damaged during the freeze-fracture process and also has not been further analyzed. Therefore, the remaining left most treated and control cells are the subject for the rest of the discussion of Figures 4 and 5.
Figure 4.
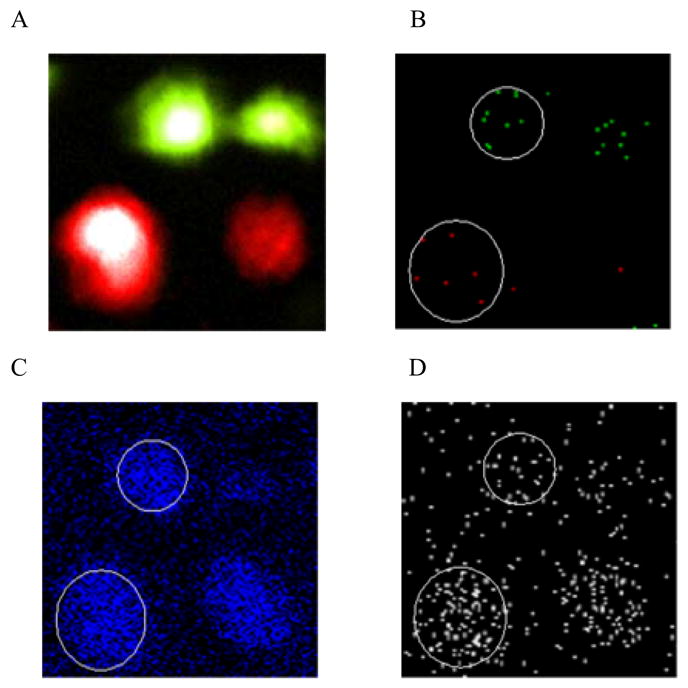
Fluorescence and SIMS images of cholesterol treated and control J774 cells (100 × 100 μm2 field of view). Fluorescence and SIMS images were aligned manually using imaging software.20 (A) In-situ two-color fluorescence image of several freeze-fractured J774 cells. The treated cells were arbitrarily colored red and the control cells are shown in green. (B–D) Positive ion molecule-specific images of J774 cells that correlate with the fluorescence image in (A). In all SIMS images, black pixels indicate an absence of signal. Cells that are discussed in the text and further analyzed in Figure 5 are circled. (B) SIMS composite image for DiD (m/z 860; red) and DiI (m/z 834; green). (C) SIMS image for C5H9+ hydrocarbon (m/z 69; blue). (D) SIMS image for cholesterol (m/z 366–370; white).
Figure 5.
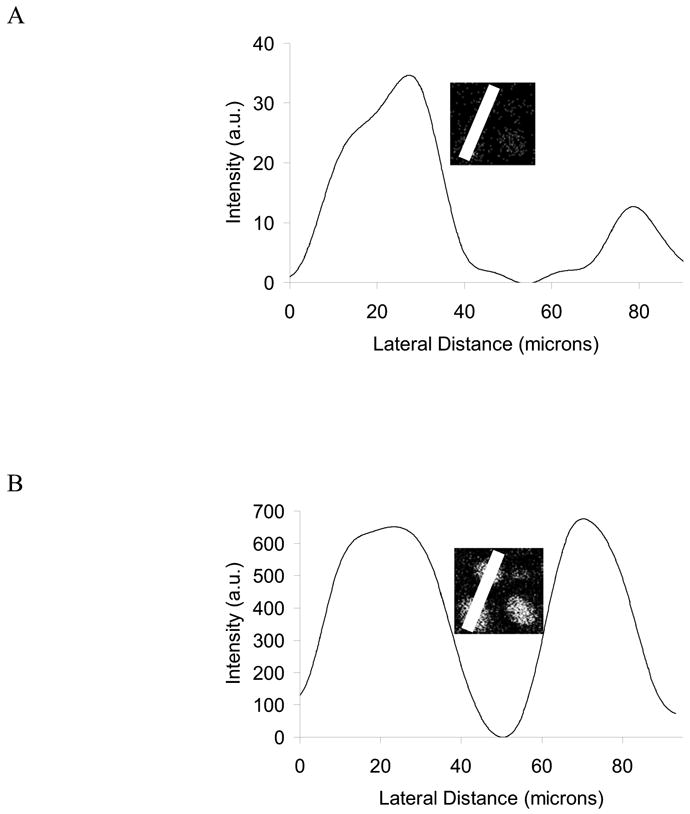
Line scans comparing a cholesterol-treated J774 cell and a control J774 cell. The insets depict the distance selected for the line scan, which was drawn from the lower left to the upper left of the each respective chemical map. (A) Line scan for cholesterol(m/z 366–370). (B) Line scan for C5H9+ hydrocarbon (m/z 69).
Relative quantification in SIMS is possible if the signal intensity is normalized to an internal standard.24(also ref #23 here) In this experiment, the C5H9+ hydrocarbon (m/z 69) serves as an internal standard because it is a generic fragment from lipid fatty acid side chains and should remain constant between the treated and control J774s. The SIMS image for m/z 69 (Figure 4C) shows that both cells contain a similar amount of signal-containing pixels that are of a similar intensity for m/z 69. Therefore, any differences in signal observed for other mass ranges should represent a true dissimilarity in the membrane chemistry between the two cells, and not an artifact of sample topography. In the SIMS image of the cholesterol fragments (m/z 366–370), there are visibly more signal-containing pixels localized to the treated J774 cell, compared to the control cell (Figure 4D). It follows that this chol-BCD treated J774 cell displays higher SIMS cholesterol signal and hence contains elevated amounts of cholesterol, compared to the control J774 cell.
Relative Quantification of Cholesterol Content in Treated versus Control J774 Cells
Line scans for the SIMS images of m/z 69 (Figure 5A) and m/z 366–370 (Figure 5B) provide a more thorough comparison of the cholesterol content in the J774 cell membranes. A line scan is a plot of the summed pixel intensities for a given chemical map as a function of the lateral distance of the line. For the line scans in Figure 5, the line is indicated in the graph insets and was taken from the cholesterol-treated cell located in the lower left through the control cell on the top left of the image. As suggested from Figure 4C, the signal obtained from m/z 69 is very similar for both cells (Figure 5A). In contrast, the cholesterol line scan shows a large discrepancy in cholesterol signal intensity between the treated and control cells (Figure 5B). In this example, there is approximately 180 % more cholesterol signal detected from the treated cell, compared to the control, supporting the conclusion from the SIMS images that the chol-BCD J774 cells contain increased amounts of cholesterol in the outer leaflet of their membranes.
SIMS images have been acquired for numerous pairs of treated and control J774 cells to calculate the average increase in cholesterol in the chol-BCD cells. For each of eight cell pairs that met the selection criteria (Experimental Section) SIMS images have been quantified. Use of a single line scan, obtained for each pair of cells for a given mass of interest (m/z 366–370 or m/z 69) resulted in raw signal intensities detected from every cell. As discussed before, quantification in SIMS requires normalization of the signal of interest to an internal standard, in this case m/z 69. Therefore, the raw signal intensity at m/z 366–370 for a given cell has been normalized to the intensity at m/z 69 from the same cell, prior to calculating the percent increase in cholesterol. The relative difference in cholesterol content between each treated/control pair is displayed as a bar graph in Figure 6. The SIMS data demonstrate that cholesterol in the outer leaflet of J774 cell membranes versus native untreated cells is elevated by an average of 134 % with elevated values between 74 and 366 %. Although the differences are statistically different, some variation in the level of cholesterol elevation is observed and may result from the intrinsic variability in biological systems as well as from environmental factors during the chol-BCD treatment. Both populations of J774 cells were not evenly confluent in their respective culture flasks. A small percentage of chol-BCD cells may have originated from sparsely populated regions of the treated flask, allowing a greater amount of the membrane surface area to interact with the cholesterol complex for a higher amount of cholesterol loading.
Figure 6.

Relative percent of cholesterol in cell membranes treated with chol-BCD. The cholesterol raw peak intensity has been normalized to the intensity for m/z 69 for that cell and each cell is compared to a control cell that was found in the same image.
CONCLUSIONS
A SIMS imaging method has been developed that enables the relative quantification of the chemical content of single cell membranes. Combining treated and control cells on the same sample surface removes the sample-to-sample variability associated with sample preparation and SIMS imaging. Morphologically similar cells from two disparate populations have been identified by use of specific labels and 2-color fluorescence microscopy integrated into the SIMS experimental design. DiD and DiI have been selected to fluorescently label the two populations of cells. Fluorescence images show that the absorption and emission profiles for the two dyes do not overlap. Additionally, fluorescence and molecule-specific SIMS images have demonstrated that mixing the two populations of labeled cells does not cause cross-contamination of the dyes. In a proof-of-concept application, one population of DiD-labeled J774 cells has been chemically altered with chol-BCD and compared to untreated, DiI-labeled control cells. In-situ fluorescence images were first used to discriminate the treated and control J774 cells. Subsequently obtained mass spectra and SIMS images have been correlated with the fluorescence images and demonstrate significant and expected chemical differences between the two cell populations. This quantification method has been used to determine that cholesterol increases by an average of 134 % in the membranes of the treated J774 cells versus control cells in repeated trials.
Acknowledgments
Supported, in part, by a grant from the National Institutes of Health and additional instrumentation funding provided by the National Science Foundation. We thank R.A. Schlegel for providing the J774 cells that were used in these experiments.
References
- 1.Cannon DM, Winograd N, Ewing AG. Annual Review of Biophysics and Biomolecular Structure. 2000;29:239–263. doi: 10.1146/annurev.biophys.29.1.239. [DOI] [PubMed] [Google Scholar]
- 2.Sombers LA, Hanchar HJ, Colliver TL, Wittenberg N, Cans A, Arbault S, Amatore C, Ewing AG. Journal of Neuroscience. 2004;24:303–309. doi: 10.1523/JNEUROSCI.1119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gong LW, Hafez I, de Toledo GA, Lindau M. Journal of Neuroscience. 2003;23:7917–7921. doi: 10.1523/JNEUROSCI.23-21-07917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heien MLAV, Johnson MA, Wightman RM. Analytical Chemistry. 2004;76:5697–5704. doi: 10.1021/ac0491509. [DOI] [PubMed] [Google Scholar]
- 5.Pihel K, Hsieh S, Jorgenson JW, Wightman RM. Analytical Chemistry. 1995;67:4514–4521. doi: 10.1021/ac00120a014. [DOI] [PubMed] [Google Scholar]
- 6.Devadoss A, Burgess JD. Journal of the American Chemical Society. 2004;126:10214–10215. doi: 10.1021/ja047856e. [DOI] [PubMed] [Google Scholar]
- 7.Lee TT, Yeung ES. Analytical Chemistry. 1992;64:3045–3051. doi: 10.1021/ac00047a029. [DOI] [PubMed] [Google Scholar]
- 8.Bergquist J, Josefsson E, Tarkowski A, Ekman R, Ewing A. Electrophoresis. 1997;18:1760–1766. doi: 10.1002/elps.1150181009. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy RT, Oates MD, Cooper BR, Nickerson B, Jorgenson JW. Science. 1989;246:57–63. doi: 10.1126/science.2675314. [DOI] [PubMed] [Google Scholar]
- 10.Hao MM, Mukherjee S, Maxfield FR. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13072–13077. doi: 10.1073/pnas.231377398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmoranzer J, Goulian M, Axelrod D, Simon SM. Journal of Cell Biology. 2000;149:23–31. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang J, Gheber LA, Margolis L, Edidin M. Biophysical Journal. 1998;74:2184–2190. doi: 10.1016/S0006-3495(98)77927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiti S, Shear JB, Williams RM, Zipfel WR, Webb WW. Science. 1997;275:530–532. doi: 10.1126/science.275.5299.530. [DOI] [PubMed] [Google Scholar]
- 14.Hofstadler SA, Severs JC, Smith RD, Swanek FD, Ewing AG. Rapid Communications in Mass Spectrometry. 1996;10:919–922. doi: 10.1002/(SICI)1097-0231(19960610)10:8<919::AID-RCM597>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Garden RW, Shippy SA, Li LJ, Moroz TP, Sweedler JV. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3972–3977. doi: 10.1073/pnas.95.7.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorey DR, Morrison GH, Chandra S. Analytical Chemistry. 2001;73:3947–3953. doi: 10.1021/ac0103266. [DOI] [PubMed] [Google Scholar]
- 17.Strick R, Strissel PL, Gavrilov K, Levi-Setti R. Journal of Cell Biology. 2001;155:899–910. doi: 10.1083/jcb.200105026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinfeld AM, Kampf JP, Lechene C. Journal of the American Society for Mass Spectrometry. 2004;15:1572–1580. doi: 10.1016/j.jasms.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Colliver TL, Brummel CL, Pacholski ML, Swanek FD, Ewing AG, Winograd N. Analytical Chemistry. 1997;69:2225–2231. doi: 10.1021/ac9701748. [DOI] [PubMed] [Google Scholar]
- 20.Roddy TP, Cannon DM, Meserole CA, Winograd N, Ewing AG. Analytical Chemistry. 2002;74:4011–4019. doi: 10.1021/ac0255734. [DOI] [PubMed] [Google Scholar]
- 21.Roddy TP, Cannon DM, Ostrowski SG, Winograd N, Ewing AG. Analytical Chemistry. 2002;74:4020–4026. doi: 10.1021/ac025574w. [DOI] [PubMed] [Google Scholar]
- 22.Cliff B, Lockyer N, Jungnickel H, Stephens G, Vickerman JC. Rapid Communications in Mass Spectrometry. 2003;17:2163–2167. doi: 10.1002/rcm.1169. [DOI] [PubMed] [Google Scholar]
- 23.Ostrowski SG, Van Bell CT, Winograd N, Ewing AG. Science. 2004;305:71–73. doi: 10.1126/science.1099791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monroe EB, Jurchen JC, Lee J, Rubakhin SS, Sweedler JV. Journal of the American Chemical Society. 2005;127:12152–12153. doi: 10.1021/ja051223y. [DOI] [PubMed] [Google Scholar]
- 25.Vickerman JC, Briggs D. ToF-SIMS: Surface Analysis by Mass Spectrometry. IM Publications and SurfaceSpectra Ltd; Chichester, West Sussex: 2001. [Google Scholar]
- 26.Chandra S, Smith DR, Morrison GH. Analytical Chemistry. 2000;72:104a–114a. doi: 10.1021/ac002716i. [DOI] [PubMed] [Google Scholar]
- 27.Benninghoven A, Rüdenauer FG, Werner HW. Secondary Ion Mass Spectrometry: Basic Concepts, Instrumental Aspects, Applications, and Trends. J Wiley; New York: 1987. [Google Scholar]
- 28.Pscheid P, Schudt C, Plattner H. J Microsc. 1981;121:149–167. doi: 10.1111/j.1365-2818.1981.tb01208.x. [DOI] [PubMed] [Google Scholar]
- 29.Chandra S, Morrison GH. Biology of the Cell. 1992;74:31–42. doi: 10.1016/0248-4900(92)90006-m. [DOI] [PubMed] [Google Scholar]
- 30.Cannon DM, Pacholski ML, Winograd N, Ewing AG. Journal of the American Chemical Society. 2000;122:603–610. [Google Scholar]
- 31.Cannon DM, Jr, Pacholski ML, Roddy TP, Winograd N, Ewing AG. Brussels: Elsevier Science B.V.; 1999. pp. 931–934. [Google Scholar]
- 32.Pacholski ML, Winograd N. Chemical Reviews. 1999;99:2977–3005. doi: 10.1021/cr980137w. [DOI] [PubMed] [Google Scholar]
- 33.Yeagle P. The membranes of cells. 2. Academic Press; San Diego: 1993. [Google Scholar]
- 34.Simons K, Toomre D. Nature Reviews Molecular Cell Biology. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen DH, Espinoza JC, Taub DD. Mechanisms of Ageing and Development. 2004;125:641–650. doi: 10.1016/j.mad.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM. Trends in Neurosciences. 2002;25:412–417. doi: 10.1016/s0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- 37.Simons K, Ehehalt R. Journal of Clinical Investigation. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolozin B. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5371–5373. doi: 10.1073/pnas.101123198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lusis AJ. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uekama K, Hirayama F, Irie T. Chemical Reviews. 1998;98:2045–2076. doi: 10.1021/cr970025p. [DOI] [PubMed] [Google Scholar]
- 41.D’Onofrio TG, Hatzor A, Counterman AE, Heetderks JJ, Sandel MJ, Weiss PS. Langmuir. 2003;19:1618–1623. [Google Scholar]
- 42.Ostrowski SG, Szakal C, Kozole J, Roddy TP, Xu J, Ewing AG, Winograd N. Analytical Chemistry. 2005;77:6190–6196. doi: 10.1021/ac0508189. [DOI] [PubMed] [Google Scholar]