

Type 2 diabetes mellitus is an increasingly prevalent disease throughout the world, with dramatic consequences in terms of individual well-being, public health, and economy. It is due to a combination of defective insulin secretion from the pancreatic β-cell and impaired sensitivity of the peripheral tissues to insulin. Because of the key role of β-cell dysfunction in the pathogenesis of type 2 diabetes, the pharmaceutical industry has devoted major efforts in recent years towards developing drugs that can enhance insulin secretion and preserve β-cell function. Such efforts have been somewhat hampered by our incomplete understanding of the mechanisms of insulin secretion. Glucose-stimulated insulin secretion (GSIS) is controlled by a “triggering” pathway, which involves glucose metabolism, membrane depolarization, and insulin exocytosis, as well as an “amplifying” pathway, which potentiates the former (1) (Figure 1). Whereas the nature of the amplifying signals is still debated, ample evidence implies a role for lipid-derived signaling molecules in this process (2). A number of nutrients, hormones, and neurotransmitters can influence GSIS, and most of these act through G-protein coupled receptors (GPCRs). Whereas the majority of GPCRs initially identified in the β-cell inhibit insulin secretion (3), the last few years have seen a multitude of papers ascribing stimulatory functions to newly discovered GPCRs (Figure 1). The number of β-cell GPCRs involved in the control of insulin secretion has increased steadily and to date, there are 6–7 different known G-protein-coupled mechanisms. Except for glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP), which are polypeptides and whose receptors are Class-B GPCRs (4), almost all of the other insulin secretagogue GPCR ligands are lipidic in nature and their cognate receptors belong to the Class-A GPCRs. These include free-fatty acids (FFA) (GPR40) (5–7), 2-arachidonylglycerol / anandamide (CB1/2R) (8), and acetylcholine (M3-muscarinic receptor) (9). In this issue, Chu et al. (10) further demonstrate that the oleoylethanolamide (OEA) / lysophosphatidylcholine (LPC)-activated GPR119 is involved in GSIS (10).
Figure 1. GPCR-mediated amplification of insulin secretion.
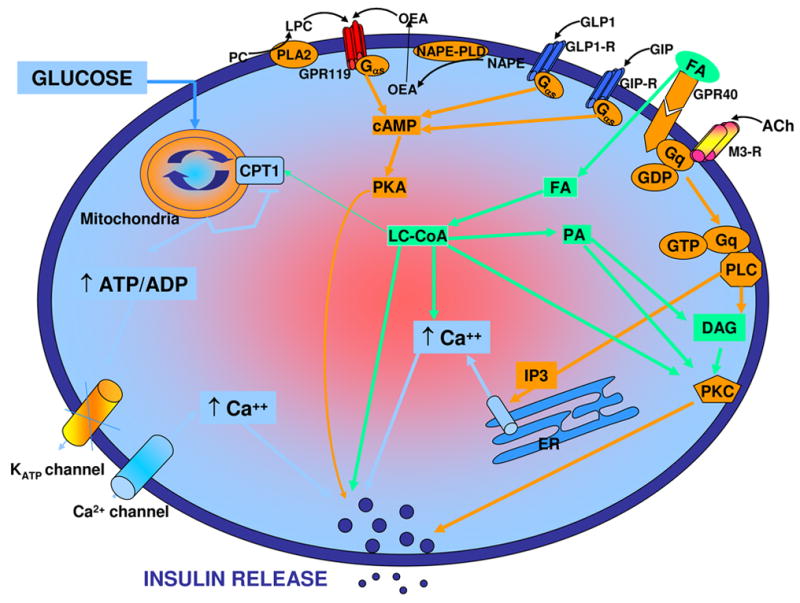
Glucose metabolism raises the ATP/ADP ratio, which closes ATP-sensitive potassium channels (KATP channels), depolarizes the cell membrane, activates voltage-gated calcium channels (Ca2+ channels), and results in calcium influx, which in turn triggers the release of insulin. Several extracellular signals can amplify this process through the activation of GPCRs. These include receptors for GLP1, GIP, FFA, acetylcholine, and the newly described GPR119 activated by LPC / OEA. These receptors couple to various downstream signaling pathways in the cell leading to an increase in cyclic AMP levels and / or intracellular calcium and converge to amplify GSIS. Ach, acetylcholine; CPT1, carnitine-palmitoyl transferase 1; DAG, diacylglycerol; ER, endoplasmic reticulum; FA, fatty acid; GIP, glucose-dependent insulinotropic peptide; GLP-1, glucagon-like peptide-1; IP3, inositol triphosphate; LC-CoA: long-chain coenzyme-A; LPC, lysophosphatidylcholine; M3-R, M3-muscarinic receptor; NAPE, N-acylphosphatidylethanolamine; OEA: oleoylethanolamide; PA, phosphatidic acid; PC, phosphatidylcholine; PKA; cyclic AMP-dependent protein kinase; PLA2, phospholipase A2; PLC, phospholipase C; PLD, phospholipase D. Adapted from (13).
First in class: GPR40 as a model for small molecule drug development
Translation of the knowledge about novel GPCRs that control insulin secretion into potential drug development for type 2 diabetes is only beginning. It has proven difficult to design small molecule agonists or antagonists to Class-B GPCRs, whose ligands are large polypeptides. This is exemplified by the GLP1-R, whose activity could only be enhanced by synthetic long-acting peptides (e.g., exendin-4) or by increasing the biological half-life of GLP1 through inhibition of its degrading enzyme, dipeptidyl peptidase-4 (4). Because of its biological activity and tissue distribution, GPR40 is an attractive drug target for type 2 diabetes, and its druggability has been investigated (11, 12). A GPR40 agonist, GW9508, that activates both GPR40 and GPR120 and stimulates GSIS in insulin-secreting MIN6 cells (but not in isolated islets) and a selective GPR40 antagonist, GW1100, that reverses the effects of GW9508, were described (11). Arylpropionic acid derivatives have also emerged as selective agonists (12). It is somewhat surprising that these compounds are already undergoing pre-clinical development while the mechanisms of action (beyond coupling to Gα/q and increasing intracellular calcium levels) and long-term effects of the receptors are still largely unknown (13). In fact, it is still not clear whether it is the antagonist or the agonist of GPR40 that would be the appropriate therapeutic agent for type 2 diabetes (14). Whereas a study (14) using GPR40-knockout and transgenic mice suggests that the antagonistic approach may be more suitable, evidence indicates that GPR40 mediates part of the insulin response to FFA in vivo (15). Therefore, it remains to be determined whether interfering with this physiological mechanism represents an appropriate approach. In this context, it will be important to resolve whether FFA activation of GPR40 in β-cells, in obesity-associated diabetic conditions, is detrimental to β-cell function or represents a “necessary evil” to enhance insulin release and compensate for insulin resistance. Another aspect to consider is that since the level of the natural ligands of GPR40, FFA, are already elevated in obesity associated diabetes, further addition of agonists may be superfluous.
GPR119: another β-cell specific GPCR
The recently discovered GPR119 (16, 17) stirred up a major interest because its expression is also high in pancreatic β-cells as compared to other tissues, making it an attractive target for drug development. After its initial discovery by Fredriksson et al. (18) as a conserved human rhodopsin GPCR, it was considered an orphan receptor. In 2005, Soga et al. (16) demonstrated that GPR119, like GPR40, is predominantly expressed in pancreatic islets and β-cell lines. These observations were confirmed by others (17, 19), and now more concrete evidence for its near exclusive localization in β-cells is presented by Chu et al. (10). Deletion of GPR119 by siRNA approach in vitro reduces insulin secretion (16), and its genetic ablation in the mouse impairs glucose tolerance (10), indicating a physiologic role in the control of GSIS. LPC and OEA have been described as the physiological ligands for GPR119 (16, 17), even though the ability of LPC to activate GPR119 has been questioned (19). The well-known GSIS-enhancing effect of LPC in β-cells is dependent upon the presence of functional GPR119 (16). It is interesting to note that both ligands can be produced in close vicinity of the receptor on the plasma membrane. LPC is generated by plasma membrane-associated phospholipase A2 activities, which are known to stimulate insulin secretion (20). OEA is produced by N-acylphosphatidylethanolamine-hydrolyzing phospholipase D (21) (Figure 1). Whether or not these metabolites reach sufficiently high concentrations on the outer surface of the plasma membrane to activate the receptor remains to be determined. Little is known about the downstream signaling pathways activated by GPR119, except that it couples to the G-protein alpha subunit Gα/s and increases intracellular cyclic-AMP levels (16).
GPR119: a novel drug target for diabetes ?
Chu et al. (10) described a new agonist for GPR119, AR231453, which was found to enhance GSIS and improve glucose tolerance in both normal and diabetic mice. Interestingly, the drug did not affect feeding behavior at doses that are effective in normalizing glucose tolerance. This is in contrast to what was reported earlier by Overton et al. (17), who showed that another GPR119 agonist, PSN632408, mimicked OEA and could reduce food intake and body weight significantly in animal models. However, these authors did find relatively lower blood glucose levels in treated animals. Chu et al. (10) attributed this difference to possible lower agonist concentrations in the brain and also suggested that the previously seen hypophagic effects might be GPR119-independent. Nevertheless, Chu et al. (10) provided convincing evidence for the dependence of AR231453’s glucose homeostatic effect on GPR119 as this compound was inactive in GPR119 knockout mice. Additional studies in GPR119-overexpressing mice would be helpful in that regard. It may be expected that such transgenic mice will show enhanced glucose tolerance and perhaps feed less and gain less weight on high-fat diet. Such corroborative data using GPR119-KO mice were not provided by Overton et al. (17). Differences in compound screening methodologies and/or chemotypes may also contribute to the apparent discrepancy between the two studies. Systematic drug metabolism - pharmacokinetics studies will need to be done on these compounds to answer these questions. As discussed above for GPR40, the long-term effects of stimulating or inhibiting GPR119 are unknown, and must be addressed before a therapeutic strategy can be envisioned.
In conclusion, the development of a compound which could be administered orally, improve glucose tolerance, and reduce body weight by controlling food intake would represent a major improvement over currently available therapies for type 2 diabetes. Although it is too early to tell whether GPR119 agonists may be able to meet these goals, further investigations into the roles and mechanism of action of this promising receptor are clearly warranted.
Acknowledgments
Work in our laboratories related to GPCRs and insulin secretion was supported by the National Institutes of Health (R21 DK070598), the Canadian Institutes for Health Research, and the Canadian Diabetes Association. V.P. was the recipient of the 2003 Thomas R. Lee Career Development Award from the American Diabetes Association and holds the Canada Research Chair in Diabetes and Pancreatic Beta-cell Function.
References
- 1.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 2.Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M. Fatty Acid Signaling in the {beta}-Cell and Insulin Secretion. Diabetes. 2006;55(Suppl 2):S16–23. doi: 10.2337/db06-s003. [DOI] [PubMed] [Google Scholar]
- 3.Robertson RP, Seaquist ERTFW. G proteins and modulation of insulin secretion. Diabetes. 1991;40:1–6. doi: 10.2337/diab.40.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Covington DK, Briscoe CA, Brown AJ, Jayawickreme CK. The G-protein-coupled receptor 40 family (GPR40-GPR43) and its role in nutrient sensing. Biochem Soc Trans. 2006;34:770–773. doi: 10.1042/BST0340770. [DOI] [PubMed] [Google Scholar]
- 6.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 7.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 8.Sharkey KA. Endocannabinoids: biology, mechanism of action and functions. Int J Obes (Lond) 2006;30(Suppl 1):S4–6. doi: 10.1038/sj.ijo.0803270. [DOI] [PubMed] [Google Scholar]
- 9.Gautam D, Han SJ, Hamdan FF, Jeon J, Li B, Li JH, Cui Y, Mears D, Lu H, Deng C, Heard T, Wess J. A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab. 2006;3:449–461. doi: 10.1016/j.cmet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Chu ZL, Jones RM, He H, Carroll C, Gutierrez V, Lucman A, Moloney M, Gao H, Mondala H, Bagnol D, Unett D, Liang Y, Demarest K, Semple G, Behan DP, Leonard J. A Role for {beta}-Cell-Expressed GPR119 in Glycemic Control by Enhancing Glucose-Dependent Insulin Release. Endocrinology. 2007 doi: 10.1210/en.2006-1608. [DOI] [PubMed] [Google Scholar]
- 11.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, Fornwald JA, Ignar DM, Jenkinson S. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKeown SC, Corbett DF, Goetz AS, Littleton TR, Bigham E, Briscoe CP, Peat AJ, Watson SP, Hickey DM. Solid phase synthesis and SAR of small molecule agonists for the GPR40 receptor. Bioorg Med Chem Lett. 2007;17:1584–1589. doi: 10.1016/j.bmcl.2006.12.084. [DOI] [PubMed] [Google Scholar]
- 13.Poitout V. The ins and outs of fatty acids on the pancreatic beta cell. Trends Endocrinol Metab. 2003;14:201–203. doi: 10.1016/s1043-2760(03)00086-9. [DOI] [PubMed] [Google Scholar]
- 14.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Latour MG, Alquier T, Oseid E, Tremblay C, Jetton TL, Luo J, Lin DCH, Poitout V. GPR40 is Necessary but not Sufficient for Fatty-Acid Stimulation of Insulin Secretion in Vivo. Diabetes. 2007In Press doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, Matsumoto S, Kamohara M, Hiyama H, Yoshida S, Momose K, Ueda Y, Matsushime H, Kobori M, Furuichi K. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. 2005;326:744–751. doi: 10.1016/j.bbrc.2004.11.120. [DOI] [PubMed] [Google Scholar]
- 17.Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Fredriksson R, Hoglund PJ, Gloriam DE, Lagerstrom MC, Schioth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003;554:381–388. doi: 10.1016/s0014-5793(03)01196-7. [DOI] [PubMed] [Google Scholar]
- 19.Sakamoto Y, Inoue H, Kawakami S, Miyawaki K, Miyamoto T, Mizuta K, Itakura M. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem Biophys Res Commun. 2006;351:474–480. doi: 10.1016/j.bbrc.2006.10.076. [DOI] [PubMed] [Google Scholar]
- 20.Ramanadham S, Wolf MJ, Li B, Bohrer A, Turk J. Glucose-responsitivity and expression of an ATP-stimulatable, Ca(2+)-independent phospholipase A2 enzyme in clonal insulinoma cell lines. Biochim Biophys Acta. 1997;1344:153–164. doi: 10.1016/s0005-2760(96)00139-7. [DOI] [PubMed] [Google Scholar]
- 21.Ueda N, Okamoto Y, Morishita J. N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: a novel enzyme of the beta-lactamase fold family releasing anandamide and other N-acylethanolamines. Life Sci. 2005;77:1750–1758. doi: 10.1016/j.lfs.2005.05.018. [DOI] [PubMed] [Google Scholar]