

Abstract
Purpose
The induction of apoptotic pathways in cancer cells offers a novel and potentially useful approach to improve patient responses to conventional chemotherapy. Tissue factor pathway inhibitor-2 (TFPI-2) is a protease inhibitor that is abundant in the extracellular matrix (ECM) and highly expressed in non-invasive cells, but absent or undetectable in highly invasive human glioblastoma cells.
Experimental Design
Using a recombinant adeno-associated viral vector carrying human TFPI-2 cDNA (rAAV-TFPI-2), we stably expressed TFPI-2 in U-251 cells, a highly invasive human glioblastoma cell line. Our previous studies demonstrated that restoration of TFPI-2 in glioblastomas effectively prevents cell proliferation, angiogenesis and tumor invasion. In this study, we determined whether TFPI-2 restoration could induce apoptosis through the caspase mediated signaling pathway.
Results
The results from nuclear chromatin staining, TUNEL assay, and FACS analysis demonstrated increased apoptosis in U251 cells after restoration of TFPI-2. Caspase 9 and caspase 3 activity assays showed increased activity, indicating enhanced apoptosis. Immunofluorescence for cleaved caspase 9 and 3 depicted increased expression and co-localization of both molecules. Western blot analysis demonstrated increased transcriptional activities of FasL, TNF-α, BAX, FADD and TRADD as well as elevated levels of cleaved caspases and PARP. Semiquantitative RT-PCR depicted increased expression of TNF-α and FasL and the related death domains TRADD and FADD.
Conclusions
Taken together, these results demonstrate that restoration of TFPI-2 activates both intrinsic and extrinsic caspase-mediated, pro-apoptotic signaling pathways and induces apoptosis in U251 cells. Furthermore, our study suggests that rAAV-mediated gene expression offers a novel tool for cancer gene therapy.
Keywords: Tissue factor pathway inhibitor-2, Apoptosis, Caspases, Glioblastoma, U-251 cells, Gene therapy
INTRODUCTION
Glioblastomas are highly invasive and aggressive primary brain tumors associated with a dismal prognosis (1). Glioblastomas comprise 23% of primary brain tumors in the United States and are the most commonly diagnosed brain tumor in adults (2). The median survival of patients with glioblastoma treated with surgery, radiotherapy and chemotherapy is from 10 to 22 months (3). Although the understanding of the pathophysiology of gliomas has increased significantly over the past few years, an effective treatment has not been developed for this type of cancer. Limits in the efficacy of current treatment modalities call for the development of novel therapeutic approaches targeting the specific biological features of glioblastomas.
Human tissue factor pathway inhibitor-2 (TFPI-2) is a Kunitz-type proteinase inhibitor that acts against a wide range of serine proteases through their non-productive interaction with a P1 residue in its first Kunitz-type domain (4). A wide variety of cells, including keratinocytes (5), dermal fibroblasts (5), smooth muscle cells (6), synoviocytes (7) and endothelial cells (8), synthesize and secrete TFPI-2, primarily into the extracellular matrix (ECM). Three isoforms of TFPI-2 are synthesized by these cells and migrate with an apparent molecular weight of 33, 31 and 27 kDa due to differential glycosylation (9). TFPI-2 exhibits strong inhibitory activity towards a broad spectrum of proteinases, including trypsin, plasmin, chymotrypsin, cathepsin G, plasma kallikrein and the factor VIIa-tissue factor complex. In contrast, TFPI-2 exhibits little or no inhibitory activity towards urokinase plasminogen activator (uPA), tissue-type plasminogen activator (tPA) and α-thrombin (10). Recent studies have shown that TFPI-2 expression plays a significant role in inhibiting tumor invasion and metastasis by a mechanism that involves its inhibitory activity (11–13). However, little is known about the role of TFPI-2 in the induction of apoptotic pathways in glioblastomas.
Apoptosis, the programmed cell death, is critical for the development and maintenance of healthy tissues. There are two alternative pathways that initiate apoptosis: one is mediated by death receptors on the cell surface and the other is mediated by mitochondria (14, 15). Fas ligand (FasL) and tumor necrosis factor-α (TNF-α) play important roles by rapidly inducing apoptosis under numerous physiological and pathological conditions (16). Upon ligand activation to their receptors, Fas and TNF-R1 associate with death domain containing adaptor proteins FADD (Fas-associated death domain) and TRADD (TNF-R1-associated death domain), which contain a death domain homologous region (17, 18). FADD has a C-terminal death domain interacting with the cytoplasmic tail of the membrane receptor and an N-terminal death effector domain interacting with the caspase-8 (19). Clustering of the receptors upon stimulation causes FADD to oligomerize caspase-8 and triggers a caspase mediated signaling pathway. Caspase-8 subsequently activates downstream effector caspases, such as caspase-3, resulting in the cleavage of proteins involved in the execution of apoptosis. Alternatively, apoptosis driven by TNF-R1 binding to TRADD involves association of TRADD and FADD, which then leads to activation of caspase-8 (20). Further, overexpression of TRADD leads to NF-κB activation and apoptosis in the absence of TNF-R1 (21).
Pro-apoptotic stimuli, such as DNA damage and oxidative stress, require a mitochondrial-dependent step that involves the release of cytochrome c, which is normally located in the intermembrane space (22, 23). Liberated cytochrome c then initiates formation of an apoptosome along with apoptotic protease-activating factor-1 (Apaf-1) in the presence of adenosine nucleotides (22, 24). The apoptosome processes procaspase-9 into a large active fragment and a small fragment by self-cleavage at Asp315, and then initiates a sequential pathway that ends with DNA fragmentation and eventual apoptosis (25–27). In contrast, apoptosis-inducing factor (AIF) translocates from mitochondria via the cytoplasm to the nucleus where it interacts with DNA and causes nuclear condensation and DNA fragmentation (23). Thus, the mitochondrial apoptotic pathway involves both caspase-dependent and caspase-independent processes in apoptotic cell death. The pathophysiological roles of the apoptotic signaling pathway have recently been identified in several human tumors including glioblastomas (28–31). In the present investigation, we selected an established glioblastoma cell line, U251 where TFPI-2 expression is totally absent due to the aberrant hypermethylation of TFPI-2 promoter CpG islands. We restored TFPI-2 protein levels in U251 cells through an adeno-associated viral vector carrying TFPI-2 gene and evaluated the effect of restored TFPI-2 on the signaling of cell surface death receptors as well as mitochondrial-mediated, pro-apoptotic pathways.
MATERIALS AND METHODS
Cloning of human TFPI-2 cDNA
The procedures for the cloning and purification of the recombinant adeno-associated viral vector carrying human TFPI-2 cDNA used in the present study have been described previously (13). Briefly, the entire coding region (a 0.8-kb fragment) of the human TFPI-2 gene was cloned into the adeno-associated viral vector (pCMV-MCS) at the BamH1 site. The plasmids were propagated in JM109 (Promega Corporation, Inc., Madison, WI) competent Escherichia coli strain. The viral vector particles were propagated in the AAV-293 cell line (Stratagene, La Jolla, CA), which produces higher viral titers. The viral preparation was purified by ultracentrifugation using an iodixanol density gradient system.
Cell culture conditions
The highly invasive human glioblastoma cell line U251 was procured from National Cancer Institute, Maryland. The cells were propagated in RPMI-1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere containing 5% CO2 at 37°C. The human adeno-associated viral vector carrying the TFPI-2 cDNA (rAAV-TFPI-2) was suitably diluted in serum-free medium at concentrations of 25, 50 and 100 MOI (multiplicity of infection). The virus particles were reconstituted in a minimum volume of serum-free medium and added to the cell monolayers. Cells were then incubated at 37°C for 1 to 2 hrs to complete the transduction of virus particles into the cells. The serum-free medium was replaced with serum-containing medium and cells were incubated for desired time periods.
Western blot analysis for TFPI-2
Extracellular matrix (ECM) proteins were extracted from U251 parental cells, cells transfected with an empty viral vector or with 25, 50 and 100 MOI of rAAV-TFPI-2. ECM proteins were also extracted from Hs 683 cells, a low-grade, non-invasive astrocytoma (procured from ATCC, #HTB-138); the Hs 683 cells were used as a standard for TFPI-2 as these cells highly express TFPI-2. All the cell cultures were treated with 200 nmol of 4-α-phorbol 12-myristate 13-acetate (PMA) (Cell Signaling Technology, Danvers, MA) per ml of the culture medium overnight. The cultures were washed with phosphate buffered saline (PBS) three times and then lysed with PBS containing 0.5% Triton X-100 (v/v) for 20 min at room temperature. After aspirating the Triton X-100 along with the lysed cells, the remaining ECM was washed three times with PBS and then twice with 20 mM Tris-HCl (pH 7.4) containing 100 mM NaCl and 0.1% (v/v) Tween 20. The culture dishes were then examined under a light microscope and ensured that no visible cells were present in the dishes. To collect ECM proteins, 200 μl of 1X SDS-PAGE sample buffer was added to each dish and agitated for 20 min at room temperature. About 30 μl of these extracts were resolved on 12% SDS-PAGE and the proteins were subsequently transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked with 10% nonfat dry milk and incubated overnight at 4°C with anti-TFPI-2 antibody diluted at 1:3000 (kindly provided by Dr. Walter Kisiel, University of New Mexico). After 3 washes, the membranes were incubated with a 1:5000 diluted peroxidase-conjugated secondary antibody (Biomeda, Foster City, CA). TFPI-2 proteins were developed using an enhanced chemiluminescence (ECL) method according to the manufacturer’s instructions (Amersham Biosciences, Buckinghamshire, UK). The Western blots were quantified using Image-Pro Discovery software (Media Cybernetics, Silver Spring, MD, USA) to evaluate the percentage restoration of TFPI-2 protein levels in U-251 cells.
Nuclear chromatin staining for apoptosis
Double nuclear staining was carried out to study the induction of apoptosis in U251 cells after restoration of TFPI-2. U251 parental cells were cultured on 12-well culture dishes at a density of 2×104 cells per well. After 24 h, the cells were transfected with rAAV-TFPI-2 at concentrations of 25, 50 and100 MOI in serum-free medium for 2 h. We used an empty vector transfected at 100 MOI for the vector control. The serum-free medium was replaced with serum-containing medium and the cultures were incubated for 48 h. The cells were removed from the incubator and 2 μl (2 μg) of Hoechst 33342 (#H3570, Invitrogen, Carlsbad, CA) was added per ml of culture media. The cells were mixed gently and incubated for 15 min. Afterwards, 10 μl (conc. 100 mg/ml) of membrane impermeable propidium iodide (#537059, Calbiochem, EMD Biosciences, San Diego, CA) was added per ml of culture media, mixed gently and allowed to stand for 1 min. The cells were then observed under a fluorescent microscope (Olympus BX61, Tokyo, Japan) using the RGB filter and photographed.
TUNEL assay
The TUNEL (Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End labeling) assay was performed to detect apoptotic cells after transfection with rAAV-TFPI-2. U251 parental cells were cultured on 8-well chamber slides at a density of 2×103 cells per well. 24 h later, the cells were transfected with rAAV-TFPI-2 at concentrations of 50 and100 MOI in serum-free medium. We used an empty vector transfected at 100 MOI for the vector control. The cultures were terminated at 48 h after transfection and the cells were fixed in 10% phosphate buffered formalin for 15 min. TUNEL staining for detection of apoptotic cells was performed using the TUNEL apoptotic detection kit (Upstate Cell Signaling Solutions, Lake Placid, NY) as per manufacturer’s instructions. Briefly, the fixed cells were washed in PBS three times (5 min/wash). The cells were then incubated with 0.05% Tween-20 in PBS containing 0.2% BSA for 15 min at room temperature (RT). The cells were washed twice in PBS and incubated with 50 μl of TdT end-labeling cocktail for 60 min at RT. The reaction was terminated, slides washed 3 times in PBS and blocked with blocking buffer for 20 min at RT. Then, the slides were incubated with 50 μl of avidin-FITC in the dark for 30 min at RT, washed three times in PBS and mounted with anti-fading gel mount (Biomeda, Foster City, CA). Slides were allowed to dry in the dark, observed under a fluorescent microscope (Olympus BX61, Tokyo, Japan) and photographed. Fluorescent apoptotic cells were quantitatively evaluated (10 randomly selected microscopic fields per sample) using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD).
FACS analysis
Fluorescence-activated cell sorting (FACS) based on DNA fragmentation was performed to determine the percentage of apoptotic cells after TFPI-2 restoration. U251 parental cells were cultured on 6-well plates (Corning Life Sciences, Corning, NY) at a density of 1×105 cells per well. After 24 h, the cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI in serum-free medium. An empty viral vector transfected at a concentration of 100 MOI was used as a vector control. The cells were harvested at 48 h after transfection using TrypLE Express (Invitrogen, Carlsbad, CA) and washed once with PBS. The cells were then dispersed in 1 ml of membrane permeable propidium iodide (50 μg/ml) (Biosure, Grass Valley, CA) and incubated for 20 to 30 min in the dark at 4°C. The cells were sorted on a FACS machine (FACSCalibur, Becton and Dickinson, Franklin Lakes, NJ) and the DNA content was analyzed based on the red fluorescence of propidium iodide at 488 nm. Data are represented in a dot plot graph and FACS histogram.
Fluorometric protease activity assay
Protease activity assay was carried out to measure the increased enzymatic activity of caspase-9 after TFPI-2 restoration in U251 cells. The assay is based on detection of cleavage of the fluorescent-tagged peptide LEHD-AFC (AFC: 7-amino-4-trifluoromethyl coumarin) by caspase-9. U251 parental cells were cultured in 100 mm plates (Corning Life Sciences, Corning, NY) at a density of 1×106 cells per plate. After 24 h, the cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI in serum-free medium. A simultaneous transfection was carried out with an empty viral vector at a concentration of 100 MOI to use as a vector control. The cells were harvested at 48 h after transfection and counted. The caspase-9 activity assay was performed using a fluorometric protease activity assay kit (BioVision Research Products, Mountain view, CA) as per manufacturer’s instructions. Briefly, approximately 2×106 cells were suspended in chilled lysis buffer and incubated with 5 μl of 1 mM LEHD-AFC and incubated at 37°C for 2 h. The final fluorescent product was measured on a fluorometer (Fluoroskan Ascent FL, Thermo Labsystems, Milford, MA) with a 390 nm excitation filter and 510 nm emission filter. The assay was carried out in 4 independent samples in duplicate and the data was quantitatively represented as percentage activity of caspase-9.
Activity assay for Caspase-3
The activity assay for caspase-3 (colorimetric) was performed using a kit (Sigma, St. Louis, MO) as per manufacturer’s instructions. The cells were cultured and transfected with rAAV-TFPI-2 as described above. The harvested cells were transferred to 96-well plates and treated with the caspase-3 peptide substrate conjugated with p-nitroaniline (Ac-DEVD-pNA) and the release of the p-nitroaniline by caspase-3 was measured on a microplate reader at 405 nm. The assay was carried out in 4 independent samples in triplicate. Data are quantitatively represented as percentage activity of caspase-3.
Double immunofluorescent staining
U251 parental cells were cultured on 8-well chamber slides at a density of 2×103 cells per well. After 24 h, the cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI in serum-free medium. We used an empty viral vector transfected at a concentration of 100 MOI for the vector control. The cultures were terminated at 48 h after transfection and cells were fixed in cold acetone for 10 min. Cells were then washed twice in PBS and blocked with 1% BSA in PBS for 30 min. The cells were then treated with 1:50 diluted (1% BSA in PBS) goat polyclonal human specific cleaved caspase-9 (Santa Cruz Biotechnology, Santa Cruz, CA) and 1:200 diluted rabbit monoclonal anti-human cleaved caspase-3 (Cell Signaling Technology, Danvers, MA) primary antibodies simultaneously and incubated overnight at 4°C. The cells were then washed three times in PBS and incubated with Texas red conjugated anti-goat (Biomeda, Foster City, CA) and FITC conjugated anti-rabbit (Biomeda, Foster City, CA) secondary antibodies at room temperature for 1 h. The cells were further washed and treated with 1:100 diluted Hoechst 33342 (Invitrogen, Carlsbad, CA) for 5 min at room temperature for nuclear counterstaining. Cells were mounted with anti-fading gel mount (Biomeda, Foster City, CA, USA) and examined under a fluorescent microscope (Olympus BX61, Tokyo, Japan) using both dsRed and GFP filters for the expression of cleaved caspase-9 and cleaved caspase-3, respectively. The pictures were then merged electronically with Hoechst 33342 staining using Spot advanced software (Meyer Instruments, Houston, TX).
Western blot analysis for molecules involved in the caspase-mediated apoptotic pathway
(1) Preparation of cell lysate
U251 parental cells were cultured in 100 mm plates at a density of 1×106 cells per plate. After 24 h, the cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI in serum-free medium as described above. An empty vector was transfected at a concentration of 100 MOI and used as a vector control. All the cultures were terminated after at 48 h. The cells were washed twice with ice-cold PBS and scraped with 1 ml of PBS. Cells were centrifuged at 12,000 rpm for 10 min at 4°C in an Eppendorf centrifuge. The supernatant was discarded and the cell pellet was suspended in freshly prepared RIPA (Radio-immunoprecipitation assay) buffer with protease inhibitors (Tris-HCl, 50 mM, pH 7.4 containing 1% Nonidet P-40, 150 mM NaCl, 1 mM activated sodium orthovanadate, 1 mM sodium fluoride, 1mM PMSF, 1 mM EDTA, 5μg/mL aprotinin and 5μg/mL pepstatin) and sonicated gently in an ultra-cell disruptor (Sonic Dismembrator, Fisher Scientific, Pittsburg, PA). Cells were centrifuged at 14,000 rpm for 10 min at 4°C and the supernatants were collected. The protein concentration in the supernatant was determined using bicinchoninic acid (BCA) (Pierce Biotechnology, Rockford, IL) protein assay as described by Smith et al.,(32). The samples were stored at −20°C until assayed. To measure released cytochrome c levels, mitochondria were isolated from the total cell lysate using the mitochondria isolation kit for cultured cells (Pierce Biotechnology, Rockford, IL) and the cytosol fraction was used to determine cytochrome c levels.
(2) Western blot analysis
Protein concentrations ranging from 5 to 100 μg were used for analysis of various molecules involved in the caspase-mediated apoptotic pathway after prior standardization of each molecule. The protein samples were mixed with 6X loading buffer containing 600 mM dithiothreitol (DTT). The samples were kept in a boiling water bath for 3 min and instantly loaded on SDS-polyacrylamide gel ranging from 7–12% depending on the molecular weight of the protein. The separated proteins were electrophoretically transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). The membranes were treated with 10% nonfat dry milk for 30 min at room temperature to block the nonspecific sites. The membranes were then incubated overnight at 4°C with either polyclonal or monoclonal human specific antibodies for various protein molecules. All antibodies were diluted at either 1:1000 or per manufacturer’s instructions in 5% nonfat dry milk. The antibodies for caspases 9, 8, 7, 6 & 3, lamin A, Bcl-2, Apaf-1, Bax, FasL were purchased from Cell Signaling Technology (Danvers, MA); antibodies for TRADD and FADD were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); antibodies for caspase-10 and TNF-α were purchased from Abcam (Cambridge, MA); the DFF40 antibody was purchased from Chemicon International (Temecula, CA); the PARP antibody was purchased from Oncogene-Calbiochem (EMD Biosciences, San Diego, CA); and the cytochrome c antibody was from BD Pharmingen (San Jose, CA). After an overnight incubation, the membranes were washed three times in 0.05% Tween-20 and treated with 1:2000 diluted respective anti-rabbit or anti-mouse HRP conjugated secondary antibodies (Biomeda, Foster City, CA) at room temperature for 1 h. The membranes were then washed three times in 0.05% Tween-20 and treated with enhanced chemiluminescence (ECL) reagent (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Finally, the membranes were exposed to autoradiography hyperfilm (Amersham Biosciences). The thickness of the band on the film was adjusted appropriately with different exposure times. The membranes were reprobed using western re-probe buffer (Gbiosciences, St. Louis, MO) and analyzed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content to demonstrate that similar amounts of protein were loaded in each lane. Mouse monoclonal GAPDH antibody (Novus Biologicals, Littleton, CO) was used.
Semiquantitative RT-PCR
U251 parental cells were cultured in 6-well chambers (Corning Life Sciences, Corning, NY) up to 80% confluence and transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI. We used an empty viral vector transfected at a concentration of 100 MOI for the vector control. Total cellular RNA was isolated using RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Gene-specific primers were designed using Beacon Designer software (Premier Biosoft International, Palo Alto, CA). We used the following primer sequences: TNF-α (forward 5’ CAC CAC GCT CTT CTG CCT GCT G 3’, reverse 5’ TCT GGT AGG AGA CGG CGA TGC G 3’); FasL (forward 5’ AGC AAA TAG GCC ACC CCA GTC C 3’, reverse 5’ TGG CTC AGG GGC AGG TTG TTG 3’); TRADD (forward 5’ CGC TCT GTG GGT CTC AAA TGG C 3’, reverse 5’ AGT CCT CTG CCA GGC TGG TGA G 3’); FADD (forward 5’ TTG GAG AAG GCT GGC TCG TCA G 3’, reverse 5’ ACA TGG CCC CAC TCC TGT TCT G 3’); and GAPDH (forward 5’ AAG GCT GTG GGC AAG GTC ATC C 3’ reverse 5’ GGA GGA GTG GGT GTC GCT GTT G 3’). All the primers were transcribed with approximately 100 ng of isolated total RNA using a one-step RT-PCR kit (Invitrogen, Carlsbad, CA) in a thermocycler (GeneAMP PCR Systems 9700, Applied Biosystems, Foster City, CA) under the following reaction conditions: cDNA synthesis, 50°C for 30 min; inactivation, 94°C for 2 min; PCR amplification of 35 cycles, denature at 94°C for 20 sec, annealing at 58°C for 30 sec, chain extension at 72°C for 45 sec and a final chain extension at 72°C for 10 min. The amplified products were separated on 1% agarose gels with ethidium bromide and visualized using a transilluminating UV (Alpha Innotech Corporation, San Leandro, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene.
We used the same primer sequences for quantitative real-time RT-PCR analysis of TNF-α, FasL, TRADD and FADD mRNA expression. Real-time RT-PCR was carried out using a one step RT-PCR kit with SYBR green (Bio-Rad Laboratories, Hercules, CA) on a Real-time PCR machine (iCycler, Bio-Rad Laboratories, Hercules, CA) under the following reaction conditions: cDNA synthesis at 10 min at 50°C; reverse transcriptase inactivation at 95°C for 5 min; thermal cycling and detection (up to 35 cycles) at 95°C for 10 sec; and data collection at 56°C for 30 sec. Approximately 100 ng of total isolated RNA was transcribed. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene.
Statistical Analysis
Arithmetic mean and standard deviation were calculated for all quantitative data. The results were statistically evaluated using one-way analysis of variance (ANOVA). Mean control values were compared with empty vector and rAAV-TFPI-2 treated cultures at 50 and 100 MOI using least significant difference method. A value of p<0.05 was considered as statistically significant.
RESULTS
Restoration of TFPI-2 in U251 cells
Previous studies from our laboratory have demonstrated that TFPI-2 plays an important role in the regulation of cell migration, invasion and angiogenesis (12, 13). The results of the present study proved that TFPI-2 restoration in U251 cells triggers caspase-mediated signaling pathway and apoptosis. Western blot analysis (Fig. 1) demonstrates that U251 cells transfected with the recombinant adeno-associated virus carrying human TFPI-2 cDNA successfully restored TFPI-2 protein levels in U251 cells in a dose-dependant manner. The three characteristic bands of TFPI-2 at 33, 31 and 27 kDa were distinctly present in the protein extracts of the U251 cells transfected with rAAV-TFPI-2. In contrast, TFPI-2 protein was completely absent in U251 parental cells and cells transfected with the empty vector. Figure 1B demonstrates the percentage restoration of TFPI-2 expression in U-251 cells when correlated with TFPI-2 expression in a non-malignant tumor cell line Hs-683.
Figure 1.
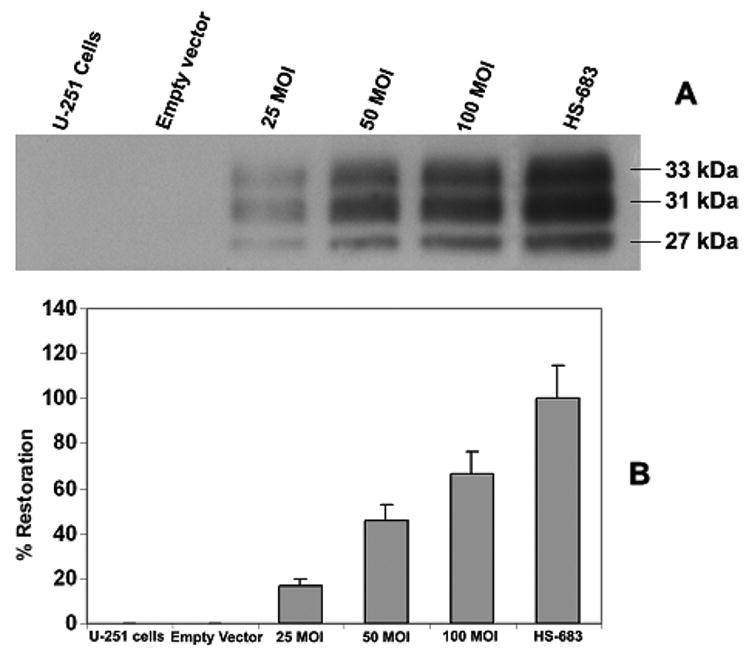
(A). Western blot analysis for the expression of tissue factor pathway inhibitor-2 (TFPI-2) in the extracellular matrix (ECM) of U251 glioblastoma cells. U251 cells were transfected with recombinant adeno-associated viral vectors engineered to express human TFPI-2 at concentrations of 25, 50 and 100 MOI and the levels of TFPI-2 in the ECM of U-251 cells were determined by western blotting. A 0.8-kb fragment of human TFPI-2 was cloned into a human adeno-associated viral vector (pCMV-MCS) at the BamH1 site. TFPI-2 protein from the ECM extracts of Hs 683 cells was used as a positive control. (B). Quantitative evaluation of the percentage restoration of TFPI-2 protein levels in U-251 cells. The Western blots were quantified using Image-Pro Discovery software to assess the percentage restoration of TFPI-2 protein levels in U-251 cells (Mean ± SD; N=6).
Increased apoptosis in nuclear chromatin staining
We examined the rate of apoptosis, necrosis and senescence in U251 cells after restoration of TFPI-2 using a double nuclear staining technique employing Hoechst 33342 (bis-benzimidazole) and membrane impermeable propidium iodide (MIPI). Hoechst 33342 is capable of penetrating the nuclear membrane and stains the chromatin blue, thereby allowing for the visual differentiation of the apoptotic nuclei. In contrast, MIPI is unable to penetrate the cells undergoing apoptosis and can stain only the necrotic cells. The results of double nuclear staining are shown in Figure 2A. As evident from the Figure, the staining indicates a dose-dependant increase of apoptosis and necrosis, as well as senescent cells at 48 h after TFPI-2 restoration. We observed a marked increase in the number of apoptotic cells (where the nuclei is shrunk and stained in a zigzag manner) at 100 MOI.
Figure 2.
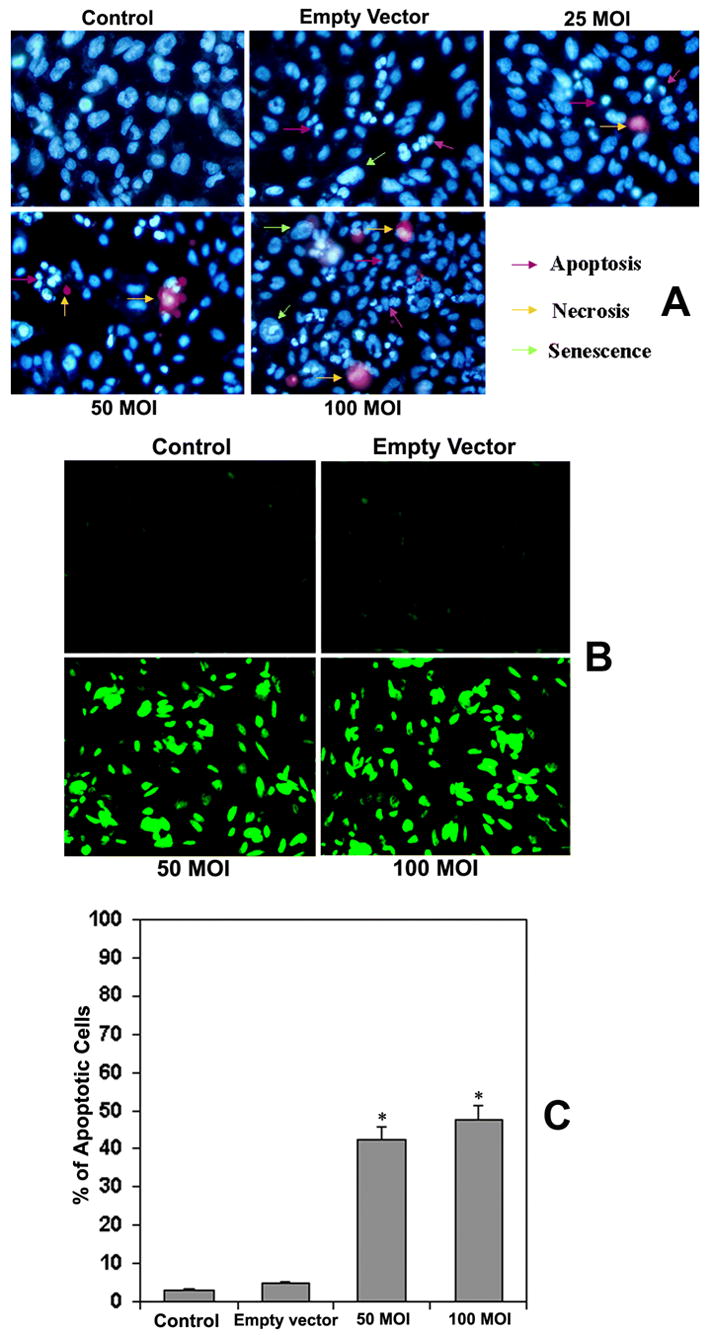
(A) Nuclear chromatin staining of U251 cells with Hoechst 33342 (bis-benzimidazole) and membrane impermeable propidium iodide. U251 cells were transfected with rAAV-TFPI-2 at concentrations of 25, 50 and 100 MOI in serum free media. An empty vector transfected at a concentration of 100 MOI was used as a vector control. (B) Fluorescent TUNEL (Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling) assay of rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2 in serum free media. An empty vector transfected at 100 MOI was used as a vector control. (C) Quantitative evaluation of TUNEL assay. We used Image Pro Discovery software to quantify the results of the TUNEL assay. Data are representative of 4 independent experiments performed in duplicate (*p<0.001).
Increased apoptosis as indicated by TUNEL assay after TFPI-2 restoration
To confirm the apoptosis observed in the results from the nuclear chromatin staining, we next performed the TUNEL assay, which is a highly specific technique that demonstrates the apoptotic cells both in cell cultures and paraffin sections. The TUNEL assay indicated a large number of apoptotic cells in the U251 cell cultures transfected with rAAV-TFPI-2 both at 50 and 100 MOI (Fig. 2B). However, staining was almost absent or insignificant in both U251 parental cells and cells transfected with the empty vector. Quantitative evaluation of the TUNEL assays using computer-assisted Imagepro plus software revealed 43% and 48% apoptotic cells after treatment with rAAV-TFPI-2 at 50 and 100 MOI, respectively (Fig. 2C).
Increased apoptosis as indicated by flow cytometry & DNA fragmentation analyses
Flow cytometry is an excellent tool for accurate detection of apoptotic cells. We performed flow cytometry for DNA fragmentation in U251 cells after TFPI-2 restoration via transfection of rAAV-TFPI-2. Figure 3A represents the dot plot of U251 cells after transfection with rAAV-TFPI-2 at 50 and 100 MOI. A marked increase in cell population was observed in the column R1 area, which represents apoptotic cells. However, there was no significant difference in the cell population in the column R1 area between U251 parental cells and cells transfected with the empty vector. Figure 3B denotes the FACS histogram of DNA fragmentation analysis after restoration of TFPI-2 in U251 cells. As indicated in the Figure, the R1 area represents the apoptotic cell population. It is clearly evident that the apoptotic cell population is remarkably increased after transfection of U251 cells with rAAV-TFPI-2. The data also demonstrate a strong positive correlation with the results of the fluorescent TUNEL assay for the detection of apoptotic cells. Figure 3C represents the quantitative evaluation of FACS data for DNA fragmentation analysis using Imagepro plus software. Quantitative analysis demonstrated 41% and 46% apoptotic cells after treatment with rAAV-TFPI-2 at 50 and 100 MOI, respectively.
Figure 3.
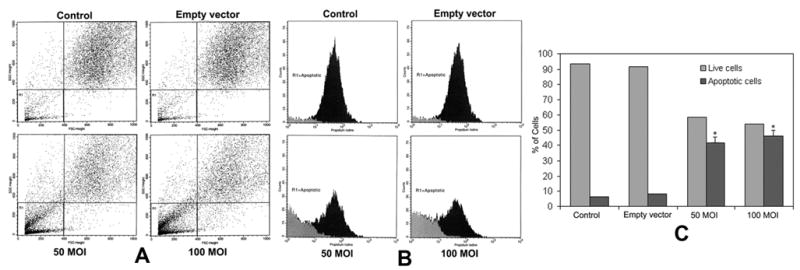
(A) Flow cytometry dot plot of rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2 in serum free media. An empty vector transfected at a concentration of 100 MOI was used as a vector control. The cells were treated with 50 μg/mL propidium iodide for 30 min at 4°C in the dark. Column R1 represents the apoptotic cell population. (B) FACS histogram of rAAV-TFPI-2-transfected U251 cells. (C) Quantitative representation of FACS data for live and apoptotic cells. Data are representative of 4 independent experiments (*p<0.001).
TFPI-2 restoration enhances activity of caspases 9 and 3
We have measured the activity of mature caspase-9 using a specific and sensitive fluorometric assay after TFPI-2 restoration in U251 cells. Figure 4A shows caspase-9 activity in U251 parental cells, cells transfected with the empty vector or rAAV-TFPI-2 at 50 and 100 MOI. A significant increase (p<0.001) was observed in the activity of mature caspase-9 after transfection of U251 parental cells with rAAV-TFPI-2 at both 50 and 100 MOI. The increase was more than 2.5 fold at 100 MOI when compared with the caspase-9 activity in U251 parental cells. We did not detect a significant difference in the activity of caspase-9 between U251 parental cells and cells transfected with the empty vector.
Figure 4.
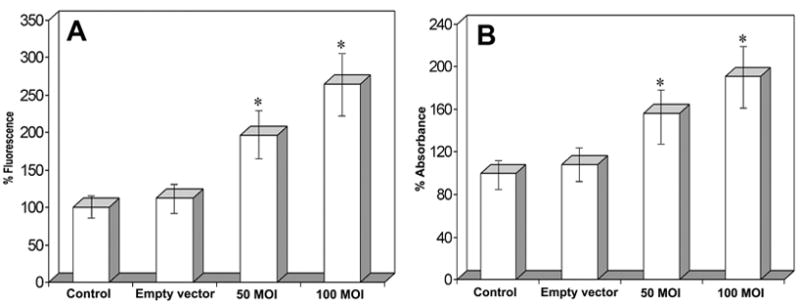
(A) Caspase 9 fluorometric protease activity assay of rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector transfected at a concentration of 100 MOI was used as a vector control. Data represents 4 independent experiments (*p<0.001). (B) Caspase 3 colorimetric protease activity assay of rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector at a concentration of 100 MOI was used as a vector control. A standard curve was prepared with 100 μl of 10–200 μM p-nitroaniline solution. Data are representative of 4 independent experiments (*p<0.001).
Caspase-3 is partially or totally responsible for the proteolytic cleavage of many key proteins including the nuclear enzyme PARP and DFF 45. In the current study, we observed a significant increase (p<0.001) in caspase-3 activity after transfection of U251 cells with rAAV-TFPI-2 at both 50 and 100 MOI (Fig. 4B). A 2-fold increase was observed in the activity of mature caspse-3 in U251 cells transfected with rAAV-TFPI-2 at 100 MOI. Similar to the caspase-9 results, we detected no significant difference in caspase-3 activity between U251 parental cells and cells transfected with the empty vector.
Double immunofluorescent staining reveals increased activity of caspases 9 and 3
To confirm the increased activity of cleaved caspase-9 and cleaved caspase-3, we used double immunofluorescent staining and an electronic merging technique. We used cleaved caspase-9 and cleaved caspase-3 antibodies to avoid any cross-reaction with procaspases 9 and 3. The staining results clearly demonstrated marked increases in the activity of both caspases after TFPI-2 restoration (Fig. 5B and C). In contrast, we detected no significant difference in the staining pattern between U251 parental cells and the cells transfected with the empty vector; staining for both caspases was absent or insignificant. The double immunofluorescent staining for the active fragments of caspases 9 and 3 illustrates the activity of both caspases in rAAV-TFPI-2-transfected cells. Further, nuclear counter staining with Hoechst 33342 reveals several apoptotic cells after transfection with rAAV-TFPI-2 at both 50 and 100 MOI (Fig. 5A). The apoptotic cells were very prominent at concentrations of 50 and 100 MOI (Fig. 5D) after electronic merging of the Hoechst 33342 with caspases 9 and 3 staining using Spot advanced software (Meyer Instruments, Houston, TX).
Figure 5. Double immunofluorescent staining for caspase 9 and caspase 3 expression.
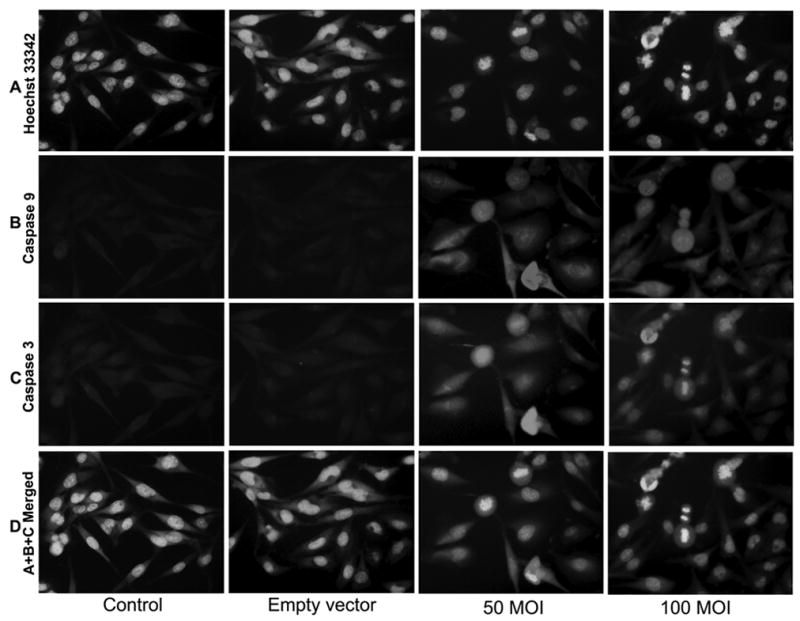
U251 parental cells were cultured on 8-well chamber slides at 2×103 cells per well and transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI. An empty vector transfected at a concentration of 100 MOI was used as a vector control. Cells were blocked with 1% BSA in PBS and incubated overnight at 4°C with mouse monoclonal caspase 9 and rabbit polyclonal caspase 3 primary antibodies simultaneously. Cells were washed and incubated with Texas red conjugated anti-mouse and FITC-conjugated anti-rabbit secondary antibodies at room temperature for 1 h. The cells were then washed and treated with 1:100 diluted Hoechst 33342 for nuclear staining. (A) Hoechst 33342; (B) caspase 9; (C) caspase 3; and (D) A+B+C merged.
TFPI-2 restoration increased the expression of death factors and death domains
Since TNF-α, Fas ligand and their respective death domains TRADD and FADD play key roles in apoptosis, we analyzed the protein levels of these important molecules after TFPI-2 restoration in U251 cells. Apoptosis mediated by death factors such as Fas ligand and TNF-α involves the formation of death-inducing signaling compound (DISC) to their respective receptors (33). Western blot analysis revealed a significant increase in the protein levels of TNF-α, FasL, TRADD and FADD after restoration of TFPI-2 in U251 cells (Fig. 6A). There was no significant difference in the levels of all these molecules in the samples treated with the empty vector when compared with the parental cells. Reprobing and analysis of the nitrocellulose membrane for GAPDH clearly demonstrated equal loading of protein in all the samples analyzed.
Figure 6.
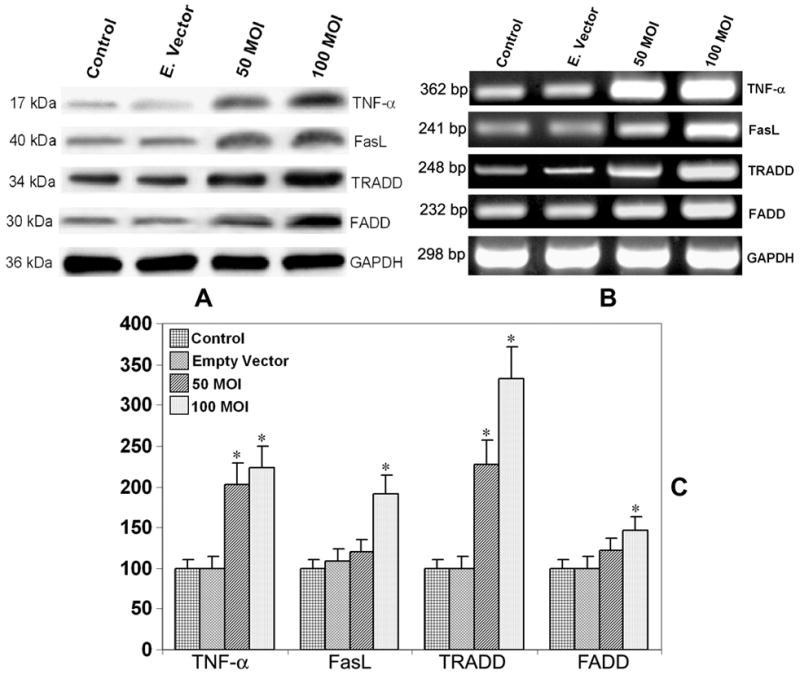
(A) Western blot analysis for the expression of TNF-α, FasL, FADD and TRADD in rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector was transfected at a concentration of 100 MOI and used as a vector control. The membranes were reprobed for GAPDH content to demonstrate that similar amounts of protein were loaded in each lane. (B) Semiquantitative RT-PCR analysis for the expression of TNF-α, FasL, TRADD and FADD mRNA in rAAV-TFPI-2-transfected U251 cells using gene specific primers. U251 cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI. An empty vector transfected at a concentration of 100 MOI was used as a vector control. Total cellular RNA was isolated using RNeasy kit and approximately 100 μg were amplified on a PCR machine. Glyceraldehyde-3-phosphate dehydrogenase mRNA was used as a housekeeping gene. (C) Real-Time RT-PCR analysis using SYBR green and gene specific primers for the quantitative evaluation of TNF-α, FasL, TRADD and FADD mRNA expression. U251 cells were transfected with rAAV-TFPI-2 at concentrations of 50 and 100 MOI. AAV-CMV transfected at a concentration 100 MOI was used as an empty vector control. The data are mean ± S.D. of 6 samples (*p<0.001).
Increased mRNA expression of death factors and death domains
In order to study the expression of TNF-α, FasL and related death domains at the transcriptional level, we analyzed the mRNA level of TNF-α, FasL, TRADD and FADD using semiquantitative RT-PCR after TFPI-2 restoration. The results in Figure 6B indicate significant increases in the mRNA levels of TNF-α, FasL, TRADD and FADD after transfection of U251 cells with rAAV-TFPI-2. In contrast, we detected no significant difference in the mRNA levels of any molecules studied cells transfected with the empty vector and parental cells. The increase in TNF-α mRNA was the most remarkable in comparison with the three other molecules measured. These results clearly indicate that TFPI-2 restoration plays a prominent role in the increased expression of TNF-α, FasL and related death domains in U251 cells. Again, GAPDH was used to ensure equal loading. Quantitative real-time RT-PCR analysis for the expression of TNF-α, FasL, TRADD and FADD mRNA demonstrated a similar pattern as of semi-quantitative RT-PCR (Fig. 6C). The expression of all the genes screened was significantly elevated at 100 MOI with a maximum increase up to 330% in the case of TRADD. At 50 MOI the increase was significant only with TNF-α and TRADD.
Increased activity of caspases and executioners
In the present study, we observed increased levels of Bax and released cytochrome c (cytosolic fraction) after TFPI-2 restoration (Fig. 7A). B-cell lymphoma 2 (Bcl-2), a major anti-apoptotic molecule that inhibits mitochondrial cytochrome c release, was decreased significantly in TFPI-2-restored U251 cells (Fig. 7A). We also observed significant increases in the expression of the cleaved fraction of lamin A, PARP and the activated deoxyribonuclease (DFF40), the three major apoptotic executioner molecules (Fig. 7B). Western blot analysis of major procaspases and cleaved caspase molecules involved in the apoptotic pathways are shown in Figure 7C. The activities of all cleaved forms of caspases 10, 9, 8, 7, 6 and 3 were increased after transfection of U251 cells with rAAV-TFPI-2. The increased levels of Bax, released cytochrome c, activated caspases, cleaved lamin, cleaved PARP and DFF40 along with other results clearly indicate enhanced apoptosis after TFPI-2 restoration in U251 glioblastoma cells. Stripping and reprobing of the nitrocellulose membrane for GAPDH indicated equal loading of proteins in all the samples analyzed.
Figure 7.
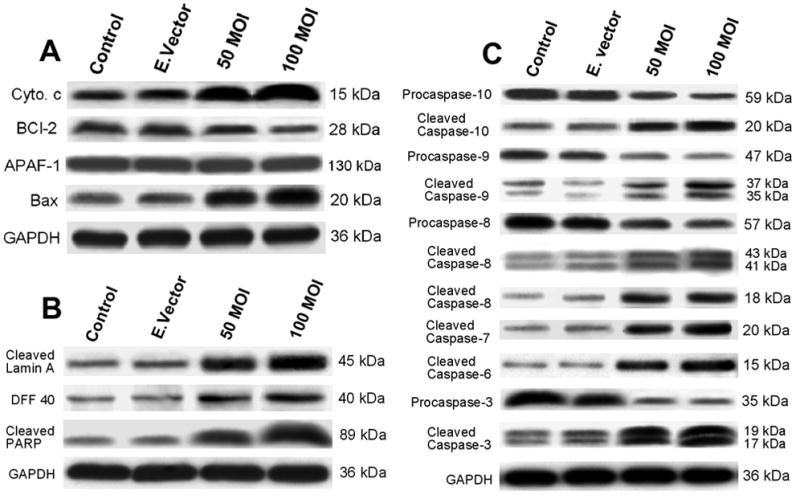
(A) Western blot analysis for cytochrome c, Bcl-2, APAF and Bax in rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector transfected at a concentration of 100 MOI was used as a vector control. Mitochondria were isolated from the total cell lysates and the cytosol fraction was used to measure released cytochrome c. The nitrocellulose membranes were reprobed and analyzed for GAPDH content to demonstrate that similar amounts of protein were loaded in each lane. (B) Western blot analysis for cleaved lamin A, DFF 40 and cleaved PARP in rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector transfected at a concentration of 100 MOI was used as a vector control. The nitrocellulose membranes were reprobed and analyzed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content to demonstrate that similar amounts of protein were loaded in each lane. (C) Western blot analysis for caspases 10, 9, 8, 6 and 3 in the cell lysates of rAAV-TFPI-2-transfected U251 cells. Cells were transfected with 50 and 100 MOI of rAAV-TFPI-2. An empty vector transfected at a concentration of 100 MOI was used as a control. The nitrocellulose membranes were reprobed and analyzed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content to demonstrate that similar amounts of protein were loaded in each lane.
DISCUSSION
Apoptosis is the process of programmed cell death that occurs under numerous physiological and pathological conditions. It plays an important role in regulating cell growth, development, immune response and the clearing of redundant or abnormal cells in organisms. The induction and execution of apoptosis require the coordination of a series of molecules including signaling molecules, receptors, death domains, enzymes and gene regulating proteins. Resistance to apoptosis is a hallmark of cancer and failure to execute apoptosis due to mutations of several genes provides cancer cells with an ability to survive and proliferate. Activation of apoptosis in cancer cells offers a novel and potentially useful approach to improve patient responses to conventional chemotherapy.
The imbalance between matrix-degrading proteases and their inhibitors such as TFPI-2 plays a crucial role in tumor invasion and metastasis. The aberrant hypermethylation of TFPI-2 may be a common mechanism that contributes to the aggressive phenotype of brain tumor cells. It has been reported that the apoptosis of LSCC cells after restoration of TFPI-2 gene may be connected with matrix metalloproteinases (MMPs) down-regulation (34). In addition to their role in ECM degradation, the proteinases also play an important role in regulating a wide variety of cellular functions such as release of growth factors (35), which in turn promote the proliferation of the developing tumors. Thus, restoration TFPI-2 can inhibit tumor progression in a variety of ways through regulating the activity of proteinases such as MMPs and plasmin. Furthermore, we have observed a total absence of MMP-9 activity in cultured HS-683 cells where TFPI-2 is highly expressed. These observations clearly indicate the significant role of TFPI-2 in the arrest of tumor cell invasion and metastasis.
In the present study, we successfully restored TFPI-2 protein in U251 cells, in which the protein is totally absent due to lack of gene expression, through the use of highly efficient, replication-deficient adenoviral vectors expressing TFPI-2 cDNA. Various experiments, including the TUNEL assay for apoptosis and FACS analysis for DNA fragmentation, showed increased apoptosis after TFPI-2 restoration. Activity assay, double immunofluorescence and western blot analyses for various caspases showed increased expression of the cleaved active subunits of the caspases involved in the caspase cascade leading to apoptosis. Furthermore, western blots and semiquantitative RT-PCR analyses demonstrated increased expression of the death factors and death domains after restoration of TFPI-2 in U251 cells. The study further depicted increased activity of apoptosis executioner molecules such as cleaved PARP, cleaved lamin A and DFF40. Our study is the first study with results that clearly demonstrate the mechanism of enhanced apoptosis in U251 cells after restoration of TFPI-2 protein levels.
TNF-α is an important cytokine that plays a critical role in inflammatory responses and apoptosis (36). Other studies have reported that TNF-α is directly toxic to vascular endothelial cells that play a major role in angiogenesis during tumor invasion and metastasis (37). TNF-α induces apoptosis in cultured cerebral endothelial cells through the cleavage of caspase 3 (37). The function of TNF-α is mediated through two distinct cell surface receptors (TNF Receptor I and TNF Receptor II). Upon ligand activation to the receptor, TNF-R1 associates with death domain containing adaptor protein TRADD. TRADD, in turn, associates with FADD, which contains an amino terminal death effector domain (DED) and binds to initiator caspase 8 (20). In the present study, we observed increased expression of both TNF-α and TRADD at both the mRNA and protein levels.
We have previously demonstrated that restoration of TFPI-2 through adeno-associated viral vectors could prevent angiogenesis and tumorigenesis both in vitro and in vivo in SNB19 glioblastoma cells (13). Further, we observed the downregulation of MMP-9 and VEGF as well as decreased angiogenesis and reduced cell invasion both in vitro and in vivo after restoration of TFPI-2 in U251 cells (unpublished data). These data suggest exertion of death stimulus after restoration of TFPI-2 through Bax, which in turn increases mitochondrial membrane permeability and leads to the release of cytochrome c from mitochondria. Bcl-2 exerts a survival function in response to apoptotic stimuli through inhibition of mitochondrial cytochrome c release (38). Here, we noted increased protein levels of Bax and released cytochrome c. These data suggest enhanced apoptotic stimuli after restoration of TFPI-2 in U251 glioblastoma cells.
Caspases are ICE-family proteases that initiate apoptosis in mammalian cells. Caspases are divided into initiator and effector (executioner) caspases depending on their site of action. Initiator caspases cleave inactive proforms of effector caspases, thereby activating them. Effector caspases, in turn, cleave other apoptotic executioner molecules such as lamin A, DFF45 and PARP, resulting in cellular disassembly and DNA fragmentation. In the present study, we observed increased protein levels of cleaved (active) caspases 10, 9, 8, 7, 6 and 3, which include both effector and executioner caspases after stable transfection of TFPI-2 in U251 cells, where TFPI-2 protein is usually absent. Increased levels of executioner caspases, which are directly responsible for the cleavage of apoptotic executioner molecules, clearly demonstrate enhanced apoptosis in U251 cells after TFPI-2 restoration.
Lamins are intermediate filament proteins that form a matrix on the inner surface of the nuclear envelope. Lamin A is cleaved by caspase 6 and serves as a marker for caspase 6 activation. During apoptosis, the 70 kDa lamin A is specifically cleaved to a large 45 kDa fragment and a small 28 kDa fragment. Human DDF45 and its mouse homologue ICAD serve as chaperones for caspase-activated deoxyribonuclease during its synthesis (39). Caspase 3 is the primary enzyme responsible for processing DFF45 and release of its C-terminal fragment (40). Upon cleavage of DFF45/ICAD by activated caspase 3, DFF40/CAD is released and eventually causes DNA fragmentation, which is the hallmark of apoptotic cell death. Reports are not available about the role of TFPI-2 either on lamin-A or DFF40/CAD. In the present study, we observed increased protein levels of both lamin A and DFF40 indicating enhanced nuclear disassembly and induction of apoptosis after TFPI-2 restoration.
Poly (ADP-ribose) polymerase (PARP) is a highly conserved nuclear enzyme present in higher eukaryotes. PARP is a DNA-binding protein that recognizes DNA strand breaks and is implicated in the DNA repair that occurs during apoptotic response. PARP cleavage has been shown to occur early in the apoptotic response as a result of caspase 3 activity (41). PARP cleavage correlates well with chromatin condensation, has been shown to be associated with condensed chromatin in apoptotic cells, and precedes the ability to detect actual DNA fragmentation (42). Here, we observed a significant increase of cleaved PARP accompanied by chromatin condensation and DNA fragmentation, the hallmarks of apoptosis.
In conclusion, the results of the present study demonstrate that TFPI-2 restoration in a highly invasive glioblastoma cell line induces both intrinsic and extrinsic caspase-mediated pathway leading to apoptosis. Our study suggests that recombinant AAV-mediated gene expression offers a novel and potential tool for cancer gene therapy.
Acknowledgments
We thank Shellee Abraham for secretarial assistance and Diana Meister and Sushma Jasti for reviewing the manuscript.
Footnotes
This research was supported by National Cancer Institute Grant CA 75557, CA 92393, CA 95058, CA 116708 and N.I.N.D.S. NS47699 and NS5729, and Caterpillar, Inc., OSF Saint Francis, Inc., Peoria, IL (to J.S.R.).
Reference List
- 1.Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–98. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 2.Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24:1266–72. doi: 10.1200/JCO.2005.04.6599. [DOI] [PubMed] [Google Scholar]
- 3.Combs SE, Widmer V, Thilmann C, Hof H, Debus J, Schulz-Ertner D. Stereotactic radiosurgery (SRS): treatment option for recurrent glioblastoma multiforme (GBM) Cancer. 2005;104:2168–73. doi: 10.1002/cncr.21429. [DOI] [PubMed] [Google Scholar]
- 4.Chand HS, Schmidt AE, Bajaj SP, Kisiel W. Structure-function analysis of the reactive site in the first Kunitz-type domain of human tissue factor pathway inhibitor-2. J Biol Chem. 2004;279:17500–7. doi: 10.1074/jbc.M400802200. [DOI] [PubMed] [Google Scholar]
- 5.Rao CN, Peavey CL, Liu YY, Lapiere JC, Woodley DT. Partial characterization of matrix-associated serine protease inhibitors from human skin cells. J Invest Dermatol. 1995;104:379–83. doi: 10.1111/1523-1747.ep12665851. [DOI] [PubMed] [Google Scholar]
- 6.Herman MP, Sukhova GK, Kisiel W, et al. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J Clin Invest. 2001;107:1117–26. doi: 10.1172/JCI10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugiyama T, Ishii S, Yamamoto J, et al. cDNA macroarray analysis of gene expression in synoviocytes stimulated with TNFalpha. FEBS Lett. 2002;517:121–8. doi: 10.1016/s0014-5793(02)02588-7. [DOI] [PubMed] [Google Scholar]
- 8.Lino M, Foster DC, Kisiel W. Quantification and characterization of human endothelial cell-derived tissue factor pathway inhibitor-2. Arterioscler Thromb Vasc Biol. 1998;18:40–6. doi: 10.1161/01.atv.18.1.40. [DOI] [PubMed] [Google Scholar]
- 9.Rao CN, Reddy P, Liu Y, et al. Extracellular matrix-associated serine protease inhibitors (Mr 33,000, 31,000, and 27,000) are single-gene products with differential glycosylation: cDNA cloning of the 33-kDa inhibitor reveals its identity to tissue factor pathway inhibitor-2. Arch Biochem Biophys. 1996;335:82–92. doi: 10.1006/abbi.1996.0484. [DOI] [PubMed] [Google Scholar]
- 10.Petersen LC, Sprecher CA, Foster DC, Blumberg H, Hamamoto T, Kisiel W. Inhibitory properties of a novel human Kunitz-type protease inhibitor homologous to tissue factor pathway inhibitor. Biochemistry. 1996;35:266–72. doi: 10.1021/bi951501d. [DOI] [PubMed] [Google Scholar]
- 11.Chand HS, Du X, Ma D, et al. The effect of human tissue factor pathway inhibitor-2 on the growth and metastasis of fibrosarcoma tumors in athymic mice. Blood. 2004;103:1069–77. doi: 10.1182/blood-2003-06-1930. [DOI] [PubMed] [Google Scholar]
- 12.Konduri SD, Rao CN, Chandrasekar N, et al. A novel function of tissue factor pathway inhibitor-2 (TFPI-2) in human glioma invasion. Oncogene. 2001;20:6938–45. doi: 10.1038/sj.onc.1204847. [DOI] [PubMed] [Google Scholar]
- 13.Yanamandra N, Kondraganti S, Gondi CS, et al. Recombinant adeno-associated virus (rAAV) expressing TFPI-2 inhibits invasion, angiogenesis and tumor growth in a human glioblastoma cell line. Int J Cancer. 2005;115:998–1005. doi: 10.1002/ijc.20965. [DOI] [PubMed] [Google Scholar]
- 14.Hajra KM, Liu JR. Apoptosome dysfunction in human cancer. Apoptosis. 2004;9:691–704. doi: 10.1023/B:APPT.0000045786.98031.1d. [DOI] [PubMed] [Google Scholar]
- 15.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–45. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 16.Wajant H. CD95L/FasL and TRAIL in tumour surveillance and cancer therapy. Cancer Treat Res. 2006;130:141–65. doi: 10.1007/0-387-26283-0_7. [DOI] [PubMed] [Google Scholar]
- 17.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 18.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–56. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 19.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 20.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 21.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 23.Susin SA, Zamzami N, Castedo M, et al. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–41. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 25.Chandrasekar B, Vemula K, Surabhi RM, et al. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004;279:20221–33. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 26.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, et al. The Ced-3/interleukin 1beta converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2alpha are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996;271:27099–106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- 28.Catchpoole DR, Lock RB. The potential tumour suppressor role for caspase-9 (CASP9) in the childhood malignancy, neuroblastoma. Eur J Cancer. 2001;37:2217–21. doi: 10.1016/s0959-8049(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 29.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 30.Joseph B, Marchetti P, Formstecher P, Kroemer G, Lewensohn R, Zhivotovsky B. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene. 2002;21:65–77. doi: 10.1038/sj.onc.1205018. [DOI] [PubMed] [Google Scholar]
- 31.Levkovitz Y, Gil-Ad I, Zeldich E, Dayag M, Weizman A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J Mol Neurosci. 2005;27:29–42. doi: 10.1385/JMN:27:1:029. [DOI] [PubMed] [Google Scholar]
- 32.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 33.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–65. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y, Xie M, Liu M, Jin D, Li P. Growth suppression of human laryngeal squamous cell carcinoma by adenovirus-mediated tissue factor pathway inhibitor gene 2. Laryngoscope. 2006;116:596–601. doi: 10.1097/01.mlg.0000205589.84020.d2. [DOI] [PubMed] [Google Scholar]
- 35.McCawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13:534–40. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 36.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 37.Kimura H, Gules I, Meguro T, Zhang JH. Cytotoxicity of cytokines in cerebral microvascular endothelial cell. Brain Res. 2003;990:148–56. doi: 10.1016/s0006-8993(03)03450-4. [DOI] [PubMed] [Google Scholar]
- 38.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7:102–11. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 39.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 40.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 41.Rosen A, Casciola-Rosen L. Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem. 1997;64:50–4. doi: 10.1002/(sici)1097-4644(199701)64:1<50::aid-jcb8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 42.Rosenthal DS, Ding R, Simbulan-Rosenthal CM, Vaillancourt JP, Nicholson DW, Smulson M. Intact cell evidence for the early synthesis, and subsequent late apopain-mediated suppression, of poly(ADP-ribose) during apoptosis. Exp Cell Res. 1997;232:313–21. doi: 10.1006/excr.1997.3536. [DOI] [PubMed] [Google Scholar]