

Abstract
Catechol-O-methyltransferase (COMT), an enzyme that metabolizes catecholamines, has recently been implicated in the modulation of pain. Our group demonstrated that human genetic variants of COMT are predictive for the development of Temporomandibular Joint Disorder (TMJD) and are associated with heightened experimental pain sensitivity (Diatchenko et al. 2005). Variants associated with heightened pain sensitivity produce lower COMT activity. Here we report the mechanisms underlying COMT-dependent pain sensitivity. To characterize the means whereby elevated catecholamine levels, resulting from reduced COMT activity, modulate heightened pain sensitivity, we administered a COMT inhibitor to rats and measured behavioral responsiveness to mechanical and thermal stimuli. We show that depressed COMT activity results in enhanced mechanical and thermal pain sensitivity. This phenomenon is completely blocked by the nonselective β-adrenergic antagonist propranolol or by the combined administration of selective β2- and β3-adrenergic antagonists, while administration of β1-adrenergic, α-adrenergic, or dopaminergic receptor antagonists fail to alter COMT-dependent pain sensitivity. These data provide the first direct evidence that low COMT activity leads to increased pain sensitivity via a β2/3-adrenergic mechanism. These findings are of considerable clinical importance, suggesting that pain conditions resulting from low COMT activity and/or elevated catecholamine levels can be treated with pharmacological agents that block both β2- and β3-adrenergic receptors.
Keywords: epinephrine, norepinephrine, catecholamines, allodynia, hyperalgesia, carrageenan
Catecholamines and enzymatic pathways that regulate the bioavailability of catecholamines influence persistent pain. Adrenergic systems contribute to the pathogenesis of rheumatoid arthritis in animals, as denervation of sympathetic noradrenergic fibers (Levine et al. 1986a) and depletion of peripheral epinephrine (Coderre et al. 1990) attenuate arthritic responses. Additionally, chronic administration of β-adrenergic receptor (βAR) agonists produces a painful arthritis-like syndrome (Vyden et al. 1971). These findings are consistent with clinical observations that sympathetic blockade or administration of the βAR antagonist propranolol reduces the severity of arthritis and joint responses to injury (Baerwald et al. 1997; Kaplan et al. 1980; Levine et al. 1986b).
A sustained elevation in catecholamines is also associated with common chronic musculoskeletal pain conditions. Fibromyalgia patients have higher basal norepinephrine levels and exaggerated norepinephrine responses to interleukin-6 (Torpy et al. 2000). Additionally, individuals with myofacial pain conditions have higher catecholamine levels and augmented sympathetic responses to stressors (Evaskus and Laskin 1972; Perry et al. 1989). As with arthritis, βAR antagonists provide significant pain relief for patients with fibromyalgia (Wood et al. 2005) and TMJD (Bhalang et al. 2004).
Abnormalities in catecholamine physiology are associated with diminished activity of catechol-O-methyltransferase (COMT), a ubiquitously expressed enzyme that metabolizes catecholamines (epinephrine, norepinephrine, and dopamine) L-dopa, catecholestrogens, ascorbic acid, and dihydroxyindolic intermediates of melanin (Mannisto and Kaakkola 1999). Facial pain patients exhibit lower COMT activity relative to controls (Marbach and Levitt 1976). Furthermore, functional polymorphisms in the COMT gene resulting in 3- to 15-fold reductions in COMT activity (Lotta et al. 1995; Nackley et al. 2005a) are associated with fibromyalgia (Gursoy et al. 2003), TMJD onset (Diatchenko et al. 2005), experimental pain sensitivity (Diatchenko et al. 2005; Zubieta et al. 2003), and morphine efficacy in cancer pain treatment (Rakvag et al. 2005). Collectively, these results suggest that elevated catecholamime levels promote persistent pain states.
To characterize the means whereby elevated catecholamine levels, resulting from reduced COMT activity, modulate pain sensitivity, we administered a COMT inhibitor to rats and measured behavioral responsiveness to mechanical and thermal stimuli. The degree of pain sensitivity produced by COMT inhibition was compared to that produced by intraplantar carrageenan, an inflammatory agent that causes sensitization of peripheral and central neurons and results in increased pain sensitivity to noxious (hyperalgesia) and normally innocuous stimuli (allodynia) (Cooper 1993; Hedo et al. 1999). The carrageenan model of inflammatory pain produces a well-characterized increase in pain sensitivity, allowing for assessment of potential analgesics (Kehl and Fairbanks 2003; Kocher et al. 1987; Stanfa and Dickenson 1995). Thus, we used this model as a standard for estimating the extent of COMT inhibitor effects on pain.
Here we show that depressed COMT activity results in profound increases in mechanical and thermal pain sensitivity, similar to that produced by intraplantar carrageenan. This enhanced sensitivity to noxious stimuli is blocked by the nonselective βAR antagonist propranolol or by combined administration of selective β2- and β3AR antagonists. Our findings provide evidence for a novel animal model that mimics the persistent pain observed in TMJD and fibromyalgia patients and elucidates the key importance of β-adrenergic pathways in pain states associated with reduced COMT activity.
Materials and Methods
Subjects
Two-hundred and twenty-eight adult male Sprague-Dawley rats (250–320g; Charles River Laboratories, Raleigh, NC) were used in these experiments. All procedures were approved by the University of North Carolina Animal Care and Use Committee and adhered to the guidelines of the Committee for Research and Ethical Issues of the IASP (Zimmermann 1983).
Drugs and Chemicals
OR486 and RO41-0960 were dissolved in dimethylsulfoxide (DMSO) and diluted in 0.9% saline pH 7.5 (3:2). Phentolamine, propranolol, SCH23390, and spiperone were dissolved in ethanol and diluted in 0.9% saline pH 3.5 (1:4). Betaxolol, ICI118,551, and SR59230A were dissolved in DMSO and diluted in 0.9% saline (1:4). Lambda carrageenan (3%) was dissolved in 0.9% saline and administered in a volume of 100 μl. OR486, RO41-0960, phentolamine, propranolol, and carrageenan were obtained from Sigma Aldrich (St. Louis, MO), while SCH23390, betaxolol, ICI118,551, SR59230A, salmeterol, and CL316243 were purchased from Tocris (Ellisville, MO).
General Experimental Methods
Rats were handled and habituated to the testing environment 2 – 3 days prior to establishing baseline responsiveness to mechanical and thermal stimuli. On test days, rats were placed in plexiglass cages positioned over an elevated perforated stainless steel platform and habituated to the environment for 15 – 20 min prior to testing. After stable baseline measures were obtained, pharmacological manipulations were performed and behavior reassessed.
In the first study, separate groups of rats received an intraperitoneal (i.p.) injection of the COMT inhibitor OR486 or RO41-0960 or vehicle. In subsequent studies designed to establish which receptors mediate COMT-dependent pain behavior, separate groups of rats received i.p. injections of antagonist or vehicle 10 min prior to i.p. administration of OR486 or vehicle. Behavioral responses to mechanical stimuli were reassessed every 30 min over a 2 hr time period and responses to thermal stimuli were reassessed at 2.25 hrs following administration of the COMT inhibitor. In all studies, the experimenter was blind to the experimental condition.
Generation of Inflammation-induced Hyperalgesia
Ten min following i.p. administration of the COMT inhibitor OR486 or vehicle, rats received a unilateral intraplantar (i.pl.) injection of carrageenan (3%, 100 μl) or saline. Behavioral responses to mechanical stimuli were reassessed every 30 min over a 2 hr time period and responses to thermal stimuli were reassessed at 2.25 hrs following carrageenan administration. Time points were selected based on previous work showing that the development of carrageenan-induced hindpaw edema is composed of an early phase beginning immediately and lasting 20–60 min post-injection and a late phase which is maximal 2–3 hrs post injection (Doherty and Robinson 1975).
Assessment of Tactile Allodynia and Mechanical Hyperalgesia
Tactile allodynia was assessed using the up-down method (Chaplan et al. 1994) to determine the threshold for paw withdrawal to punctate mechanical stimulation. A series of nine calibrated filaments (with bending forces of 0.40, 0.68, 1.1, 2.1, 3.4, 5.7, 8.4, 13.2, and 25.0 g; Stoelting) with approximately equal logarithmic spacing between stimuli (Mean ± SEM: 0.232 ± 0.04 units) were presented to the hind paw in successive order, whether ascending or descending. Filaments were positioned in contact with the hindpaw for a duration of 3 s or until a withdrawal response occurred. Testing was initiated with the middle hair of the series (3.4 g). In the absence of a paw withdrawal response, an incrementally stronger filament was presented and in the event of a paw withdrawal, an incrementally weaker filament was presented. After the initial response threshold was crossed, this procedure was repeated in order to obtain a total of six responses in the immediate vicinity of the threshold. The pattern of withdrawals (X) and absence of withdrawals (O) was noted together with the terminal filament used in the series of six responses. The 50% g threshold = (10[Xf + kδ])/10,000, where Xf = value (in log units) of the final von Frey hair used; k = tabular value of pattern of positive (X) and negative (O) responses, and δ = mean difference (in log units) between stimuli.
Immediately following determination of the response threshold, paw withdrawal frequency (%) to punctate mechanical stimulation was assessed. A von Frey monofilament with a calibrated bending force of 25 g was presented to the hind paw ten times for a duration of 1 s with an interstimulus interval of approximately 1 s. Mechanical hyperalgesia was defined as an increase in the percentage frequency ([# of paw withdrawals/10] x 100) of paw withdrawal evoked by stimulation with von Frey monofilaments.
Assessment of Thermal Hyperalgesia
Thermal hyperalgesia was evaluated using the radiant heat method (Hargreaves et al. 1988) in the same animals evaluated for responsiveness to von Frey monofilaments. Radiant heat was presented through the floor of the stainless steel platform to the midplantar region of the hind paw. Stimulation was terminated upon paw withdrawal or after 20 s if the rat failed to withdraw from the stimulus. Paw withdrawal latencies to the thermal stimulus were recorded in triplicate.
Statistical Analysis
All 50% paw withdrawal threshold data sets passed the D’Angostino-Pearson omnibus normality test, verifying that the data were sampled from a Gaussian population. Thus, mechanical behavioral data were analyzed by analysis of variance (ANOVA) for repeated measures or paired t-test where appropriate. Thermal behavioral data were analyzed by ANOVA or paired t-test. Post hoc comparisons were performed using the Bonferroni test. P < 0.05 was considered to be statistically significant.
Results
COMT Inhibition Increases Pain Sensitivity
To evaluate the effects of depressed levels of COMT on pain behavior, the COMT inhibitor OR486 or RO41-0960 was administered to separate groups of rats and their effects on tactile allodynia, mechanical hyperalgesia, and thermal hyperalgesia were determined. As OR486 and RO41-0960 have distinct chemical structures, their effects on pain sensitivity can be attributed directly to COMT inhibition. Behavioral responsiveness to mechanical and thermal stimuli did not differ between groups prior to pharmacological manipulations. I.p. administration of the COMT inhibitor OR486 (30 mg/kg) or RO41-0960 (30 mg/kg) produced tactile allodynia (F2,6 = 253.6, P < 0.0001; Fig. 1A), mechanical hyperalgesia (F2,6 = 120.1, P < 0.0001; Fig. 1B), and thermal hyperalgesia (F2,21 = 33.14, P < 0.0001; Fig. C) compared to administration of vehicle. COMT-dependent increases in pain sensitivity were observed 30 min following drug administration and lasted throughout the duration of the experimental procedure.
Fig. I.
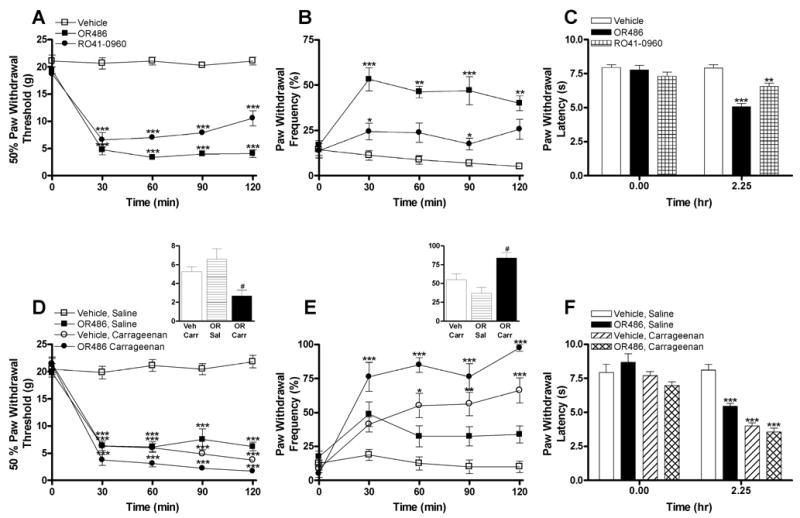
COMT inhibition produces tactile allodynia, mechanical hyperalgesia, and thermal hyperalgesia. Administration of the COMT inhibitors OR486 (30 mg/kg) or RO41-0960 (30 mg/kg) (A) decreased paw withdrawal threshold to mechanical stimuli (4.04 ± 0.32 g and 8.01 ± 0.56 g for animals receiving OR486 and RO41-0960, respectively, relative to controls with a paw withdrawal threshold of 20.76 ± 0.37 g), (B) increased paw withdrawal frequency to repeated presentation of a 25 g monofilament (46.56 ± 2.82 % and 22.81 ± 2.33 % for animals receiving OR486 and RO41-0960, respectively, relative to controls with a paw withdrawal frequency of 7.97 ± 1.10 %), and (C) decreased paw withdrawal latency to thermal stimuli relative to vehicle (5.09 ± 0.24 s and 6.56 ± 0.25 s for animals receiving OR486 and RO41-0960, respectively, compared to controls with a paw withdrawal latency of 7.92 ± 0.25 s). Animals receiving OR486 (30 mg/kg) + saline or vehicle + carrageenan (3%) produced a similar degree of (D) tactile allodynia and (E) mechanical hyperalgesia relative to those receiving vehicle + saline. Furthermore, administration of OR486 + carrageenan produced an additive effect on behavioral responsiveness to mechanical stimuli relative to those receiving either vehicle + carrageenan or OR486 + saline (panel D and E insets). (F) Animals receiving OR486 + saline, vehicle + carrageenan, or OR486 + carrageenan also produced a similar degree of thermal hyperalgesia relative to those receiving vehicle + saline. N = 8 per group. Data are Mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05 different from vehicle (A–C) or vehicle + saline (D–F). #P < 0.05 different from vehicle + carrageenan and OR486 + saline.
In order to evaluate the degree of pain sensitivity produced by COMT inhibition, the COMT inhibitor OR486 was administered to rats and its effects on pain behavior were compared to those produced in a classic inflammatory pain model. Separate groups of rats received i.p. injections of the COMT inhibitor OR486 (30 mg/kg) or vehicle 10 min prior to i.pl. carrageenan (3%) or saline. Compared to animals receiving vehicle + saline, those receiving OR486 + saline, vehicle + carrageenan, or OR486 + carrageenan exhibited tactile allodynia (F3,9 = 268.6, P < 0.0001; Fig. 1D), mechanical hyperalgesia (F3,9 = 44.78, P < 0.0001; Fig. 1E), and thermal hyperalgesia (F3,28 = 49.02, P < 0.0001; Fig. 1F). The degree of allodynia and hyperalgesia produced by OR486 was remarkable as it was comparable to that produced by intraplantar carrageenan. Behavioral responsiveness to mechanical and thermal stimuli did not significantly differ between animals receiving vehicle + carrageenan or OR486 + saline.
Additionally, administration of OR486 augmented carrageenan-induced pain sensitivity. Administration of OR486 + carrageenan produced significantly greater tactile allodynia and mechanical hyperalgesia relative to all comparison groups. The additive effects of OR486 + carrageenan versus vehicle + carrageenan and OR486 + saline on tactile allodynia (F2,6 = 23.78, P < 0.002) and mechanical hyperalgesia (F2,6 = 23.19, P < 0.002) are summarized in the Fig. 1D inset and Fig. 1E inset, respectively. Administration of OR486 + carrageenan also produced significantly greater thermal hyperalgesia relative to administration of OR486 + saline (F3,28 = 49.02, P < 0.0001; Fig. 1F).
The Nonselective βAR Antagonist Propranolol Completely Blocks COMT-dependent Increases in Pain Sensitivity
COMT acts peripherally and centrally to metabolize epinephrine, norepinephrine, and dopamine. Thus, pharmacological methods were used to establish which receptor class mediates the COMT-dependent increases in pain behavior. Separate groups of rats received i.p. injections of the αAR antagonist phentolamine (3 mg/kg), the βAR antagonist propranolol (3 mg/kg), the D1-like DAR antagonist SCH23390 (0.2 mg/kg), the D2-like DAR antagonist spiperone (0.2 mg/kg), or vehicle 10 min prior to i.p. administration of OR486. Doses of adrenergic (Hord et al. 2001; Kim et al. 1993; Lee et al. 1997; Safieh-Garabedian et al. 2002; Tonussi and Ferreira 1992) and dopaminergic (Hu and Jin 1999; Krowicki 1991) receptor antagonists were selected based on the ability of comparable doses to produce analgesia in other rat models. The development of OR486-induced increase in pain sensitivity was completely blocked by the β-adrenergic antagonist propranolol, which attenuated the development of tactile allodynia (F5,15 = 305.9, P < 0.0001; Fig. 2A), mechanical hyperalgesia (F5,15 = 93.96, P < 0.0001; Fig. 2B), and thermal hyperalgesia (F5,42 = 24.47, P < 0.0001; Fig. 2C) relative to animals receiving phentolamine, SCH23390, spiperone, or vehicle prior to OR486. The αAR, D1-like DAR, and D2-like DAR antagonists failed to block the effects of OR486. Antagonists administered in the absence of OR486 did not alter pain sensitivity (Fig. 2D–F). These results provide strong evidence for β-adrenergic signaling in COMT-dependent hyperalgesia, while evidence for α-adrenergic and dopaminergic signaling was not observed.
Fig. II.
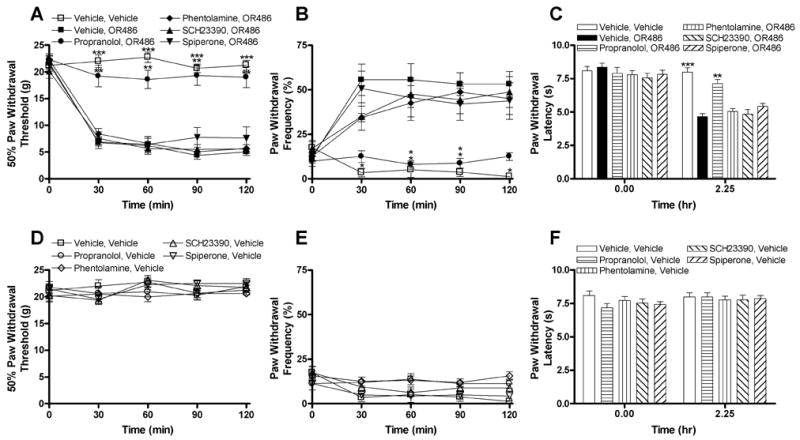
Administration of the nonselective βAR antagonist propranolol completely blocks OR486-induced pain sensitivity. Administration of propranolol (3 mg/kg) prior to OR486 (30 mg/kg) normalized (A) paw withdrawal threshold to mechanical stimuli, (B) paw withdrawal frequency to a noxious punctate stimulus, and (C) paw withdrawal latency to radiant heat relative to animals receiving the α-adrenergic antagonist phentolamine (3 mg/kg), D1-like dopamine antagonist SCH23390 (0.2 mg/kg), D2-like dopamine antagonist spiperone (0.2 mg/kg), or vehicle prior to OR486. Administration of phentolamine, propranolol, SCH23390, or spiperone 10 min prior to i.p. administration of vehicle failed to affect (D) paw withdrawal threshold to mechanical stimuli, (E) paw withdrawal frequency to a noxious punctate stimulus, or (F) paw withdrawal latency to radiant heat relative to vehicle. N = 8 per group. Data are Mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05 different from vehicle + OR486.
Selective β2- or β3AR Antagonists Partially Block COMT-dependent Increases in Pain Sensitivity
Propranolol is a nonselective βAR antagonist. Doses similar to that used in the present study block activity of β1- and β2ARs (O'Donnell et al. 1994) and reduce the activity of β3ARs (Tsujii and Bray 1998). Thus additional studies were conducted in order to identify the βAR subtype(s) that mediate the development of OR486-induced increases in pain sensitivity. Rats received i.p. injections of the β1 antagonist betaxolol (0.1, 1.0, or 10 mg/kg), the β2 antagonist ICI118,551 (0.05, 0.5, or 5 mg/kg), the β3 antagonist SR59230A (0.5, 5, or 50 mg/kg), or vehicle 10 min prior to i.p. administration of OR486. Administration of the β2AR antagonist ICI118,551 or the β3AR antagonist SR59230A, but not the β1AR antagonist betaxolol, reduced the heightened pain sensitivity produced by OR486 in a dose-dependent fashion (Fig. 3). Administration of the middle or high dose of ICI118,551 prior to OR486 partially blocked the development of tactile allodynia (F4,12 = 281.2, P < 0.0001; Fig. 3A) and mechanical hyperalgesia (F4,12 = 87.61, P < 0.0001; Fig. 3B), while the high dose of ICI118,551 was necessary to partially block thermal hyperalgesia (F4,25 = 9.87, P < 0.0001; Fig. 3C). Administration of the middle or high dose of SR59230A prior to OR486 partially blocked the development of tactile allodynia (F4,12 = 151.4, P < 0.0001; Fig. 3D), mechanical hyperalgesia (F4,12 = 104.6, P < 0.0001; Fig. 3E), and thermal hyperalgesia (F4,25 = 10.65, P < 0.0001; Fig. 3F). It is important to note that no differences in COMT-dependent pain sensitivity were observed between animals receiving the middle and high dose of ICI118,551 or between animals receiving the middle and high dose of SR59230A. Administration of betaxolol prior to OR486 failed to affect tactile allodynia (Fig. 3G), mechanical hyperalgesia (Fig. 3H), or thermal hyperalgesia (Fig. 3I). Selective βAR antagonists administered in the absence of OR486 did not alter pain sensitivity (Fig. 3J–L).
Fig. III.
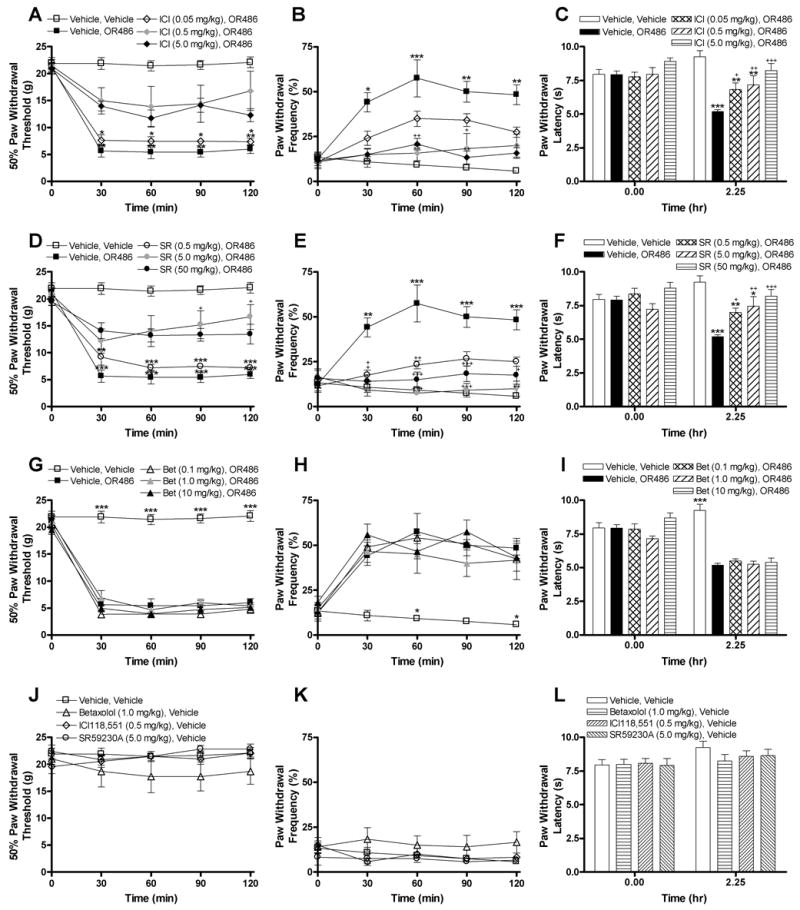
Administration of selective antagonists for β2- or β3ARs reduces OR486-induced pain sensitivity. Administration of the middle (0.5 mg/kg) or high (5.0 mg/kg) dose of the β2 antagonist ICI118,551 prior to OR486 (30 mg/kg) (A) increased paw withdrawal threshold and (B) decreased paw withdrawal frequency to a noxious punctate stimulus relative to animals receiving vehicle or the low (0.05 mg/kg) dose of ICI118,551. (C) Administration of the high dose of ICI118,551 also increased paw withdrawal latency to radiant heat relative to animals receiving vehicle prior to OR486. Administration of the middle (5 mg/kg) or high (50 mg/kg) dose of the β3 antagonist SR59230A prior to OR486 (30 mg/kg) (D) increased paw withdrawal threshold to mechanical stimuli, (E) decreased paw withdrawal frequency to a noxious punctate stimulus, and (F) increased paw withdrawal latency to thermal stimuli relative to animals receiving vehicle. (G–I) Administration of the β1AR antagonist betaxolol (0.1, 1.0, or 10 mg/kg) prior to OR486 failed to alter pain sensitivity. Administration of ICI118,551, SR59230A, or betaxolol 10 min prior to i.p. administration of vehicle failed to affect (J) paw withdrawal threshold to mechanical stimuli, (K) paw withdrawal frequency to a noxious punctate stimulus, or (L) paw withdrawal latency to radiant heat relative to vehicle. N = 8 per group. Data are expressed as Mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05 different from vehicle + vehicle. +++P < 0.001, ++P < 0.01, +P < 0.05 different from vehicle + OR486.
Coadministration of both β2- and β3AR Antagonists Blocks COMT-dependent Increases in Pain Sensitivity
Administration of β2- or β3AR antagonists reduced the development of increased pain sensitivity produced by COMT inhibition; however, only a partial blockade of OR486-induced increased pain sensitivity was achieved. These results suggest that while β2- and β3ARs independently contribute to COMT-dependent pain sensitivity, their combined activation is necessary to produce the maximum degree of COMT-dependent pain sensitivity. To test this hypothesis, ICI118,551 and SR59230A were administered together prior to administration of OR486. Coadministration of β2- and β3AR antagonists completely blocked the development of OR486-induced tactile allodynia (t3 = 23.69, P < 0.0003; Fig. 4A), mechanical hyperalgesia (t3 = 42.77, P < 0.0001; Fig. 4B), and thermal hyperalgesia (t7 = 10.60, P < 0.0001; Fig. 4C).
Fig. IV.
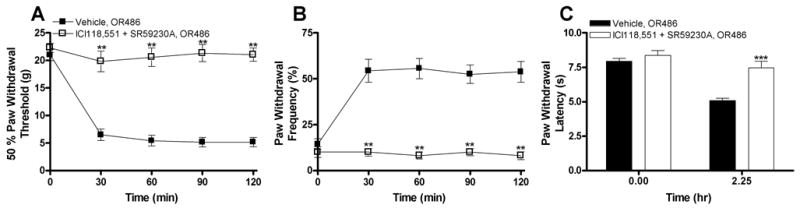
Coadministration of selective antagonists for β2- and β3ARs completely blocks OR486-induced pain sensitivity. Concurrent administration of ICI118,551 (0.5 mg/kg) and SR59230A (5.0 mg/kg) prior to OR486 (30 mg/kg) completely normalized (A) paw withdrawal threshold to mechanical stimuli, (B) paw withdrawal frequency to a noxious punctate stimulus, and (C) paw withdrawal latency to radiant heat. N = 8 per group. Data are expressed as Mean ± SEM. ***P < 0.001, **P < 0.01 different from vehicle + OR486.
Discussion
COMT Inhibition Increases Pain Behavior via β2 and β3 Adrenergic Receptors
COMT inhibition results in a remarkable enhancement of mechanical and thermal pain sensitivity, comparable to that produced by carrageenan, a chemical well known for its ability to sensitize nociceptors and increase pain behavior (Hedo et al. 1999). Furthermore, COMT inhibition augments carrageenan-induced pain sensitivity. COMT-dependent increases in pain sensitivity are completely blocked by the nonselective β-adrenergic antagonist propranolol or by the combined administration of selective β2- and β3-adrenergic antagonists. Administration of β2- or β3AR antagonists alone prior to OR486 produce only a partial blockade of enhanced pain sensitivity, thus suggesting that COMT-dependent pain sensitivity is mediated through coincident β2/3-adrenergic signaling processes.
Our findings elucidate the key importance of β2/3-adrenergic pathways in the development of pain states resulting from reduced COMT activity. β2ARs are predominantly located on vascular, uterine, and airway smooth muscle (Dixon et al. 1986) and mononuclear leukocytes in the periphery (Landmann 1992) as well as cerebellar and thalamic neurons (Nicholas et al. 1996; Rainbow et al. 1984) and glial cells (Salm and McCarthy 1992; Stone and Ariano 1989) in the central nervous system. Moderate levels of expression have also been observed in adipocytes (Kobilka et al. 1987) and spinal dorsal horn neurons (Nicholson et al. 2005). The contribution of β2ARs to enhanced pain sensitivity is in line with results from previous studies demonstrating that epinephrine activates β2ARs located on primary afferent nociceptors and produces a hyperalgesic state in rats (Aley et al. 2001; Khasar et al. 1999a; Khasar et al. 1999b; Khasar et al. 2003). Additionally, common variants of the human β2AR gene, coding for differences in receptor expression and internalization, are associated with the development of TMJD (Diatchenko et al. 2006). Furthermore, there is evidence that β2ARs play a functional role in the development of opioid-induced hyperalgesia, a syndrome characterized by increased sensitivity to noxious stimuli following acute and chronic opioid administration (Liang et al. 2006; Nackley et al. 2006). Our findings together with this work suggest that the alterations in COMT-dependent pain sensitivity and μ-opioid responses observed by Zubieta and colleagues (2003) may also be influenced by β2-adrenergic receptor stimulation.
The contribution of β3ARs is particularly novel as this is the first demonstration that they mediate the development of pain. Until now, β3ARs, which are mainly expressed in brown and white adipose tissue, have been primarily known for their ability to regulate norepinephrine-induced changes in energy metabolism and thermogenesis (Strosberg 1997). β3ARs are unique in that they do not undergo desensitization (Cao et al. 2000; Liggett et al. 1993); thus, their activation and the stimulation of downstream effectors likely continue for prolonged periods. Collectively, these findings suggest that suppressing β2/3-adrenergic systems will attenuate pain by reducing the activity of catecholamines that engage peripheral and/or central processes to promote mechanical allodynia and thermal hyperalgesia.
The present work employed an animal model of pain behavior designed to mimic human persistent pain conditions associated with diminished COMT activity. Although our findings elucidate the importance of β2/3ARs in the development of acute COMT-dependent pain sensitivity, sustained administration of a COMT inhibitor is necessary to characterize the role of β2/3ARs in the development of persistent pain conditions associated with prolonged reductions in COMT activity. Additional studies are also required to elucidate the role of peripheral versus central β2/3-adrenergic systems in the development of COMT-dependent pain sensitivity. Although epinephrine and norepinephrine have previously been shown to sensitize nociceptors located on small diameter primary afferents in the periphery (Hu and Zhu 1989; Khasar et al. 1999b; Shyu et al. 1989), elevated levels of central catecholamines are generally associated with descending inhibition of pain via actions at α2ARs or D2DARs in the spinal dorsal horn (Millan 2002). Antidepressants used extensively in the treatment of persistent pain conditions, are thought to inhibit pain transmission at the spinal level by increasing synaptic levels of norepinephrine and serotonin (Sanchez and Hyttel 1999) as well as by blocking tetrodotoxin-resistant sodium channels (Brau et al. 2001). Furthermore, catecholamine-induced elevations in blood pressure have been shown to produce analgesia through the stimulation of arterial baroreceptors (Maixner and Randich 1984; Randich and Maixner 1984; Zamir and Maixner 1986). Taken together, these findings are in line with the view that catecholamines can exert divergent influences on nociception as a function of localization and net influence on neuronal excitability. The bidirectional role of a single transmitter can be explained in part by diversity in receptor heterodimerization and second messenger systems. As catecholamines engage different receptor signaling networks in different animal pain models and clinical pain conditions, future studies are required to identify the anatomical sites and signaling mechanisms whereby β2- and β3ARs mediate COMT-dependent pain sensitivity.
Potential Signaling Mechanisms Whereby β2/3 Receptors Mediate Pain Sensitivity
Both β2- and β3ARs belong to the superfamily of G protein-coupled receptors and couple to Gs to stimulate adenylyl cyclase (Dixon et al. 1986; Strosberg 1997) or to Gi/s to stimulate the mitogen-activated protein kinase (MAPK) cascade (Daaka et al. 1997; Schmitt and Stork 2000; Soeder et al. 1999). Recent studies have also identified a novel Gi/s-independent mechanism whereby β2- and β3ARs activation can stimulate extracellular signal-regulated kinase (ERK; a type of MAPK) through a novel Gi/s-independent mechanism (Azzi et al. 2003; Cao et al. 2000). Activation of β2ARs located on nociceptors has been shown to increase pain sensitivity via Gs/adenylyl cyclase-driven protein kinase A- and protein kinase C-dependent pathways or via Gi/MAPK-dependent pathways (Aley et al. 2001; Khasar et al. 1999a; Khasar et al. 1999b). Upon stimulation, MAPKs regulate pain sensitivity through both transcriptional and non-transcriptional means (Obata and Noguchi 2004).
Within recent years it has become increasingly recognized that proinflammatory mediators, including cytokines (e.g., TNFα, IL-1β, and IL-6), prostaglandins, and reactive oxygen species, also play an important role in mediating pain sensitivity (Burysek and Houstek 1997; Kress 2004; Maimone et al. 1993; Marchand et al. 2005; Sommer and Kress 2004). Activation of β2ARs on cells in the periphery (Frost et al. 2004) or central nervous system (Maimone et al. 1993) promotes the synthesis and release of IL-6. Additionally, activation of β3ARs on adipocytes stimulates IL-6 transcription (Burysek and Houstek 1997). We have extended these findings in a recent study showing that systemic COMT inhibition increases circulating plasma levels of TNFα, IL-1β, and IL-6 in rat in a β2- and β3AR-dependent manner (Nackley et al. 2005b). Once these proinflammatory cytokines are synthesized and released, they can activate and sensitize nociceptors via direct receptor-mediated actions or through the recruitment of additional mediators (Kress 2004; Sommer and Kress 2004). Importantly there is emerging evidence that stimulation of MAPKs induces the transcription of TNFα, IL-1β, and IL-6 (Chi et al. 2004; Koj 1996). Moreover, a recent report demonstrates that β2ARs located on macrophages leads to IL-1β and IL-6 production through ERK1/2- and p38-dependent activation of ATF-1 and ATF-2 transcription factors (Tan et al. 2006). Thus, β2/3AR-mediated increases in protein kinase stimulation and proinflammatory cytokine production are not independent processes. Further studies are required to identify the cellular pathways through which β2- and β3AR stimulation increases pain sensitivity.
Conclusions and Implications
Taken together, these data provide evidence that COMT inhibition results in increased pain sensitivity comparable to that produced by carrageenan, via β2/3-adrenergic mechanisms. We previously identified polymorphisms in the COMT gene that are associated with low COMT enzymatic activity and TMJD onset (Diatchenko et al. 2005). Here, we report the mechanism whereby low levels of COMT lead to exacerbated pain states. Elevated levels of norepinephrine and epinephrine, resulting from depressed COMT activity, activate β2/3ARs to produce heightened pain sensitivity. These findings have important clinical implications, suggesting that β2- and β3AR antagonists may benefit patients suffering from pain conditions resulting from low COMT activity and/or elevated catecholamine levels.
Acknowledgments
We thank Ollie Monbureau for the development of the behavioral testing hardware and software. We thank B. Lambeth and J.M. Faison, Jr. for assistance with experiment preparation and sample collection. This work was funded by the NIH/NICHHD K12 HD052191 to A.N., NIH/NIDCR R01 DE016558 to L.D., and NIH/NINDS P01 NS045685 to W.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–9. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci U S A. 2003;100:11406–11. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerwald CG, Laufenberg M, Specht T, von Wichert P, Burmester GR, Krause A. Impaired sympathetic influence on the immune response in patients with rheumatoid arthritis due to lymphocyte subset-specific modulation of beta 2-adrenergic receptors. Br J Rheumatol. 1997;36:1262–9. doi: 10.1093/rheumatology/36.12.1262. [DOI] [PubMed] [Google Scholar]
- Bhalang K, Light K, Maixner W. The IADR/AADR/CADR 82nd General Session. Hawaii: 2004. Effect of propranolol on TMD and fibromyalgia pain: preliminary findings. [Google Scholar]
- Brau ME, Dreimann M, Olschewski A, Vogel W, Hempelmann G. Effect of drugs used for neuropathic pain management on tetrodotoxin-resistant Na(+) currents in rat sensory neurons. Anesthesiology. 2001;94:137–44. doi: 10.1097/00000542-200101000-00024. [DOI] [PubMed] [Google Scholar]
- Burysek L, Houstek J. beta-Adrenergic stimulation of interleukin-1alpha and interleukin-6 expression in mouse brown adipocytes. FEBS Lett. 1997;411:83–6. doi: 10.1016/s0014-5793(97)00671-6. [DOI] [PubMed] [Google Scholar]
- Cao W, Luttrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM, Lefkowitz RJ, Collins S. Direct binding of activated c-Src to the beta 3-adrenergic receptor is required for MAP kinase activation. J Biol Chem. 2000;275:38131–4. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chi DS, Fitzgerald SM, Pitts S, Cantor K, King E, Lee SA, Huang SK, Krishnaswamy G. MAPK-dependent regulation of IL-1- and beta-adrenoreceptor-induced inflammatory cytokine production from mast cells: implications for the stress response. BMC Immunol. 2004;5:22. doi: 10.1186/1471-2172-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Basbaum AI, Dallman MF, Helms C, Levine JD. Epinephrine exacerbates arthritis by an action at presynaptic B2-adrenoceptors. Neuroscience. 1990;34:521–3. doi: 10.1016/0306-4522(90)90160-6. [DOI] [PubMed] [Google Scholar]
- Cooper B. Contribution of edema to the sensitization of high-threshold mechanoreceptors of the goat palatal mucosa. J Neurophysiol. 1993;70:512–21. doi: 10.1152/jn.1993.70.2.512. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Anderson AD, Slade GD, Fillingim RB, Shabalina SA, Higgins T, Sama S, Belfer I, Goldman D, Max MB, Weir BS, Maixner W. Three major haplotypes of the β2 adrenergic receptor define psychological profile, blood pressure, and the risk for development of a common musculoskeletal pain disorder. American Journal of Medical Genetics. 2006 doi: 10.1002/ajmg.b.30324. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–43. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–9. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- Doherty NS, Robinson BV. The inflammatory response to carrageenan. J Pharm Pharmacol. 1975;27:701–3. doi: 10.1111/j.2042-7158.1975.tb09537.x. [DOI] [PubMed] [Google Scholar]
- Evaskus DS, Laskin DM. A biochemical measure of stress in patients with myofascial pain-dysfunction syndrome. J Dent Res. 1972;51:1464–6. doi: 10.1177/00220345720510053501. [DOI] [PubMed] [Google Scholar]
- Frost RA, Nystrom GJ, Lang CH. Epinephrine stimulates IL-6 expression in skeletal muscle and C2C12 myoblasts: role of c-Jun NH2-terminal kinase and histone deacetylase activity. Am J Physiol Endocrinol Metab. 2004;286:E809–17. doi: 10.1152/ajpendo.00560.2003. [DOI] [PubMed] [Google Scholar]
- Gursoy S, Erdal E, Herken H, Madenci E, Alasehirli B, Erdal N. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23:104–7. doi: 10.1007/s00296-002-0260-5. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hedo G, Laird JM, Lopez-Garcia JA. Time-course of spinal sensitization following carrageenan-induced inflammation in the young rat: a comparative electrophysiological and behavioural study in vitro and in vivo. Neuroscience. 1999;92:309–18. doi: 10.1016/s0306-4522(98)00734-9. [DOI] [PubMed] [Google Scholar]
- Hord AH, Denson DD, Stowe B, Haygood RM. alpha-1 and alpha-2 Adrenergic antagonists relieve thermal hyperalgesia in experimental mononeuropathy from chronic constriction injury. Anesth Analg. 2001;92:1558–62. doi: 10.1097/00000539-200106000-00042. [DOI] [PubMed] [Google Scholar]
- Hu JY, Jin GZ. Supraspinal D2 receptor involved in antinociception induced by l-tetrahydropalmatine. Zhongguo Yao Li Xue Bao. 1999;20:715–9. [PubMed] [Google Scholar]
- Hu SJ, Zhu J. Sympathetic facilitation of sustained discharges of polymodal nociceptors. Pain. 1989;38:85–90. doi: 10.1016/0304-3959(89)90077-8. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Robinson CA, Scavulli JF, Vaughan JH. Propranolol and the treatment of rheumatoid arthritis. Arthritis Rheum. 1980;23:253–5. doi: 10.1002/art.1780230220. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Fairbanks CA. Experimental animal models of muscle pain and analgesia. Exerc Sport Sci Rev. 2003;31:188–94. doi: 10.1097/00003677-200310000-00006. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999a;24:253–60. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999b;81:1104–12. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao FJ, Gear RW, Green PG, Levine JD. Vagal modulation of bradykinin-induced mechanical hyperalgesia in the female rat. J Pain. 2003;4:278–83. doi: 10.1016/s1526-5900(03)00631-x. [DOI] [PubMed] [Google Scholar]
- Kim SH, Na HS, Sheen K, Chung JM. Effects of sympathectomy on a rat model of peripheral neuropathy. Pain. 1993;55:85–92. doi: 10.1016/0304-3959(93)90187-T. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ. cDNA for the human beta 2-adrenergic receptor: a protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc Natl Acad Sci U S A. 1987;84:46–50. doi: 10.1073/pnas.84.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher L, Anton F, Reeh PW, Handwerker HO. The effect of carrageenan-induced inflammation on the sensitivity of unmyelinated skin nociceptors in the rat. Pain. 1987;29:363–73. doi: 10.1016/0304-3959(87)90051-0. [DOI] [PubMed] [Google Scholar]
- Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1996;1317:84–94. doi: 10.1016/s0925-4439(96)00048-8. [DOI] [PubMed] [Google Scholar]
- Kress MaSC. Neuroimmunology and Pain: Peripheral Effects of Proinflammatory Cytokines. In: KaH Brune HO, editor. Hyperalgesia: Molecular Mechanisms and Clinical Implications, Progress in Pain Research Management. Vol. 30. IASP Press; Seattle: 2004. pp. 57–65. [Google Scholar]
- Krowicki ZK. Dopamine receptor antagonists block the effect of Tyr-MIF-1 (Tyr-Pro-Leu-Gly-NH2) on the opiate form of footshock-induced analgesia. Neuropeptides. 1991;19:281–5. doi: 10.1016/0143-4179(91)90095-z. [DOI] [PubMed] [Google Scholar]
- Landmann R. Beta-adrenergic receptors in human leukocyte subpopulations. Eur J Clin Invest. 1992;22(Suppl 1):30–6. [PubMed] [Google Scholar]
- Lee DH, Chung K, Chung JM. Strain differences in adrenergic sensitivity of neuropathic pain behaviors in an experimental rat model. Neuroreport. 1997;8:3453–6. doi: 10.1097/00001756-199711100-00008. [DOI] [PubMed] [Google Scholar]
- Levine JD, Dardick SJ, Roizen MF, Helms C, Basbaum AI. Contribution of sensory afferents and sympathetic efferents to joint injury in experimental arthritis. J Neurosci. 1986a;6:3423–9. doi: 10.1523/JNEUROSCI.06-12-03423.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Fye K, Heller P, Basbaum AI, Whiting-O'Keefe Q. Clinical response to regional intravenous guanethidine in patients with rheumatoid arthritis. J Rheumatol. 1986b;13:1040–3. [PubMed] [Google Scholar]
- Liang D, Liao G, Wang J, Usuka J, Guo YY, Peltz G, Clark JD. A Genetic Analysis of Opioid-induced Hyperalgesia in Mice. Anesthesiology. 2006 doi: 10.1097/00000542-200605000-00023. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett SB, Freedman NJ, Schwinn DA, Lefkowitz RJ. Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proc Natl Acad Sci U S A. 1993;90:3665–9. doi: 10.1073/pnas.90.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–10. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- Maimone D, Cioni C, Rosa S, Macchia G, Aloisi F, Annunziata P. Norepinephrine and vasoactive intestinal peptide induce IL-6 secretion by astrocytes: synergism with IL-1 beta and TNF alpha. J Neuroimmunol. 1993;47:73–81. doi: 10.1016/0165-5728(93)90286-8. [DOI] [PubMed] [Google Scholar]
- Maixner W, Randich A. Role of the right vagal nerve trunk in antinociception. Brain Res. 1984;298:374–7. doi: 10.1016/0006-8993(84)91441-0. [DOI] [PubMed] [Google Scholar]
- Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- Marbach JJ, Levitt M. Erythrocyte catechol-O-methyltransferase activity in facial pain patients. J Dent Res. 1976;55:711. doi: 10.1177/00220345760550043801. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–32. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Maixner W, Diatchenko L. Perspectives on the genetic basis of opioid-induced hyperalgesia. Anesthesiology. 2006 doi: 10.1097/00000542-200605000-00004. In Press. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Shabalina SA, Chivileva IE, Satterfield KS, Korchynskyy O, Maixner W, Diatchenko L. American Society for Human Genetics. Salt Lake City; Utah: 2005a. Common human catechol-O-methyltransferase haplotypes modulate RNA stability, protein expression, and enzymatic activity. [Google Scholar]
- Nackley AG, Tan KS, Fecho K, Flood P, Maixner W, Diatchenko L. COMT modulates pain sensitivity and cytokine production through both β2 and β3 adrenergic receptors. Society for Neuroscience; Washington, DC: 2005b. [Google Scholar]
- Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol Sci. 1996;17:245–55. doi: 10.1016/0165-6147(96)10022-5. [DOI] [PubMed] [Google Scholar]
- Nicholson R, Dixon AK, Spanswick D, Lee K. Noradrenergic receptor mRNA expression in adult rat superficial dorsal horn and dorsal root ganglion neurons. Neurosci Lett. 2005;380:316–21. doi: 10.1016/j.neulet.2005.01.079. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–53. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- O'Donnell JM, Frith S, Wilkins J. Involvement of beta-1 and beta-2 adrenergic receptors in the antidepressant-like effects of centrally administered isoproterenol. J Pharmacol Exp Ther. 1994;271:246–54. [PubMed] [Google Scholar]
- Perry F, Heller PH, Kamiya J, Levine JD. Altered autonomic function in patients with arthritis or with chronic myofascial pain. Pain. 1989;39:77–84. doi: 10.1016/0304-3959(89)90177-2. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci U S A. 1984;81:1585–9. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakvag TT, Klepstad P, Baar C, Kvam TM, Dale O, Kaasa S, Krokan HE, Skorpen F. The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain. 2005;116:73–8. doi: 10.1016/j.pain.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Randich A, Maixner W. Interactions between cardiovascular and pain regulatory systems. Neurosci Biobehav Rev. 1984;8:343–67. doi: 10.1016/0149-7634(84)90057-5. [DOI] [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Haddad JJ, Massaad CA, Jabbur SJ, Saade NE. The role of the sympathetic efferents in endotoxin-induced localized inflammatory hyperalgesia and cytokine upregulation. Neuropharmacology. 2002;42:864–72. doi: 10.1016/s0028-3908(02)00028-x. [DOI] [PubMed] [Google Scholar]
- Salm AK, McCarthy KD. The evidence for astrocytes as a target for central noradrenergic activity: expression of adrenergic receptors. Brain Res Bull. 1992;29:265–75. doi: 10.1016/0361-9230(92)90056-4. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467–89. doi: 10.1023/a:1006986824213. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJ. beta 2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275:25342–50. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- Shyu BC, Olausson B, Andersson SA. Sympathetic and noradrenaline effects on C-fibre transmission: single-unit analysis. Acta Physiol Scand. 1989;137:85–91. doi: 10.1111/j.1748-1716.1989.tb08723.x. [DOI] [PubMed] [Google Scholar]
- Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, Collins S. The beta3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J Biol Chem. 1999;274:12017–22. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–7. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Stanfa L, Dickenson A. Spinal opioid systems in inflammation. Inflamm Res. 1995;44:231–41. doi: 10.1007/BF01782974. [DOI] [PubMed] [Google Scholar]
- Stone EA, Ariano MA. Are glial cells targets of the central noradrenergic system? A review of the evidence. Brain Res Brain Res Rev. 1989;14:297–309. doi: 10.1016/0165-0173(89)90015-5. [DOI] [PubMed] [Google Scholar]
- Strosberg AD. Structure and function of the beta 3-adrenergic receptor. Annu Rev Pharmacol Toxicol. 1997;37:421–50. doi: 10.1146/annurev.pharmtox.37.1.421. [DOI] [PubMed] [Google Scholar]
- Tan KS, Nackley-Neely AG, Satterfield KS, Maixner W, Diatchenko L, Flood P. β2-adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kB-independent mechanisms. Cellular Signalling. 2006 doi: 10.1016/j.cellsig.2006.06.007. (in press) [DOI] [PubMed] [Google Scholar]
- Tonussi CR, Ferreira SH. Rat knee-joint carrageenin incapacitation test: an objective screen for central and peripheral analgesics. Pain. 1992;48:421–7. doi: 10.1016/0304-3959(92)90095-S. [DOI] [PubMed] [Google Scholar]
- Torpy DJ, Papanicolaou DA, Lotsikas AJ, Wilder RL, Chrousos GP, Pillemer SR. Responses of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis to interleukin-6: a pilot study in fibromyalgia. Arthritis Rheum. 2000;43:872–80. doi: 10.1002/1529-0131(200004)43:4<872::AID-ANR19>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Tsujii S, Bray GA. A beta-3 adrenergic agonist (BRL-37,344) decreases food intake. Physiol Behav. 1998;63:723–8. doi: 10.1016/s0031-9384(97)00518-0. [DOI] [PubMed] [Google Scholar]
- Vyden JK, Groseth-Dittrich MF, Callis G, Laks MM, Weinberger H. Arthritis and Rheumatology Abstracts. 1971;14 [Google Scholar]
- Wood PB, Kablinger AS, Caldito GS. Open trial of pindolol in the treatment of fibromyalgia. Ann Pharmacother. 2005;39:1812–6. doi: 10.1345/aph.1G014. [DOI] [PubMed] [Google Scholar]
- Zamir N, Maixner W. The relationship between cardiovascular and pain regulatory systems. Ann N Y Acad Sci. 1986;467:371–84. doi: 10.1111/j.1749-6632.1986.tb14641.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–10. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–3. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]