

Abstract
An unusual peroxide base promoted isomerization was uncovered. Saturated endoperoxides derived from fulvenes give rise to 2-vinyl-2-cyclopentenones upon treatment with DBU in CH2Cl2 in a one-pot reaction. This methodology was applied to a convenient synthesis of dihydrojasmone. Moreover, functional groups placed on the side chain at C-6 participate in the base catalyzed isomerizations via conjugate attack at the enone moiety to give 2-cyclopentenones carrying oxygen heterocycles at C2.
1. Introduction
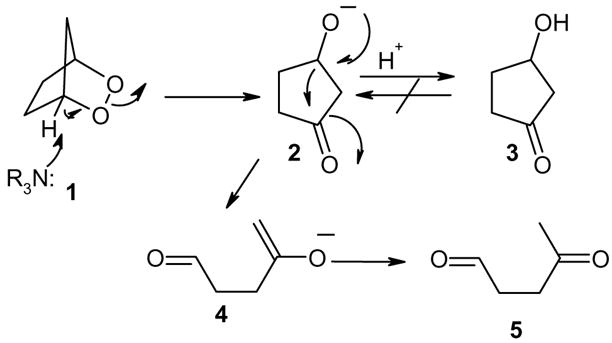
Endoperoxides, readily available in most cases from 1,3-dienes by way of a Diels-Alder reaction with singlet oxygen1 are sensitive to thermal, photochemical, and reductive conditions.2 In particular, base-catalyzed rearrangements, also known as Kornblum- De la Mare reactions are well-known.3 Treatment of bridged bicyclic endoperoxides typically result in the formation of hydroxy ketones. The mechanism of these reactions have been thoroughly studied. In particular, the base-catalyzed disproportionation of 2,3-dioxabicyclo[2.2.1]heptane (1) to 3-hydroyxcyclopentanone (3) and levulinaldehyde (5) by amine catalysis has been reported by Salomon and Zagorski in an elegant study.4 Isotope labeling studies show that the most likely mechanism for the disproportionation involves initial abstraction of the bridgehead proton in the rate-determining step (Scheme 1).
Scheme 1.
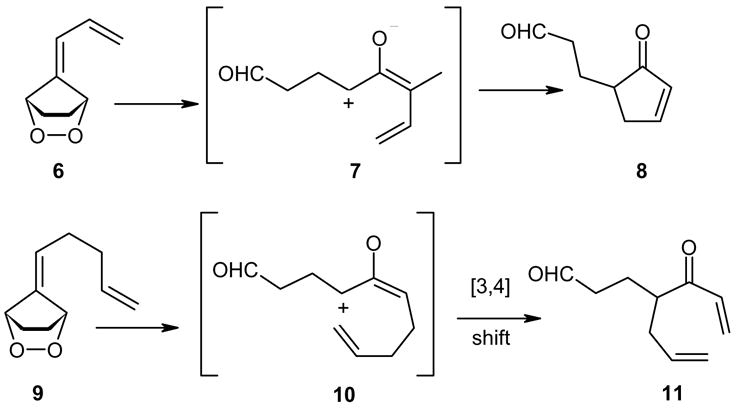
Previously, we studied the singlet oxygenations of fulvenes and low-temperature diazene reduction of the unstable endoperoxides5 and reported on some novel mechanisms encountered during the thermal isomerizations of saturated fulvene endoperoxide, e.g., 23, 32, 42 and 46.6, 7 We have explored the synthetic utility of the allene oxides/cyclopropanones8 to prepare functionalized cyclopentenones derived from saturated fulvene endoperoxides, e.g., 6→7→8 9 and also disclosed the first aliphatic examples of [3,4]-sigmatropic shifts on suitably substituted cyclopropanones derived from the corresponding endoperoxides by thermolysis (9→10→11) (Scheme 2) .10
Scheme 2.
2. Results and Discussion
We here report a new and synthetically useful facet of base-catalyzed endoperoxide isomerizations that we observed on saturated fulvene endoperoxides. The outcome of these reactions has proved particular to the nature of the base. Thus, treatment of fulvene endoperoxides with triethylamine at 0°-r.t. in CH2Cl2 results in the formation of the corresponding hydroxyketone as expected.5 However, the use of the stronger base, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), gave 2-vinyl-2-cyclopentenones, e.g., 14, in high yields (Scheme 3).
Scheme 3.
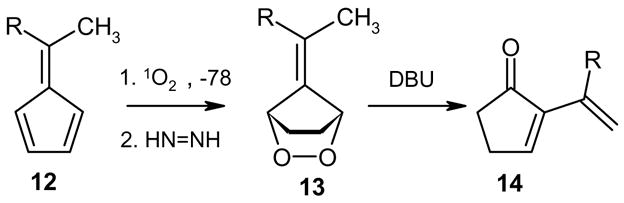
The mechanism presumably commences with initial proton abstraction at the bridgehead. However, the resulting hydroxyketone is prone to further proton abstraction at the allylic position. The timing of the proton elimination is subject to discussion and further studies, and two alternative mechanisms may be advanced for the dehydration step. One possible dehydration pathway would involve deprotonation of the γ-H, enolate formation, followed by hydroxide elimination (presumably via initial protonation by BH+), an E1Cb mechanism (Scheme 4). However, the fact that the reaction proceeds in CH2Cl2, an aprotic solvent that does not encourage ionic species, points to a concerted 1,4-elimination (E2). Regardless of the mechanism of the dehydration step, the resulting compounds from this tandem isomerization-dehydration process are 2-vinylcyclopentenones, a substance class for which there are very few synthetic methods.11-15 The ease with which the 2-vinylcyclopentenones can be constructed from the readily available fulvenes in a one-pot reaction makes this approach particularly attractive. Table 1 depicts some selected systems we have prepared so far.
Scheme 4.

Table 1.
DBU-catalyzed isomerizations/dehydrations of fulvene endoperoxides
| Entry | Fulvene | Endoperoxide | Cyclopentenone | Yield |
|---|---|---|---|---|
| 1 |
|
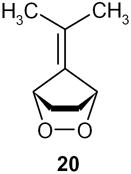
|
|
76% |
| 2 |
|
|
|
83% |
| 3 |
|
|
|
81% |
| 4 |
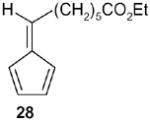
|
|
|
69% |
| 5 |
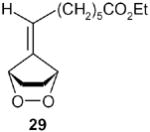
|
|
|
82% |
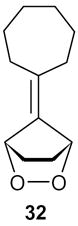
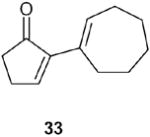
| 6 |
|
|
|
66% |
| 7 |
|
|
|
68% |
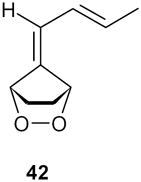
| 8 |
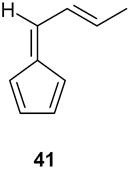
|
|
|
66% |
| 9 |
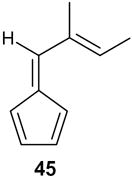
|
|
|
59% |
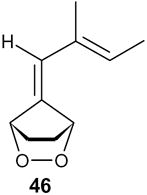
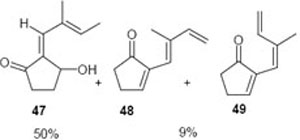
In one case, where the starting fulvene carried a 6-vinyl group, the reaction with DBU led to a mixture of the expected hydrox ketone 47 (50%) and a mixture of the two isomers 48 and 49 (9%). It appears that the protons at the remote methyl group are just acidic enough to permit abstraction, resulting in the formation of a highly conjugated system (Scheme 5).
Scheme 5.
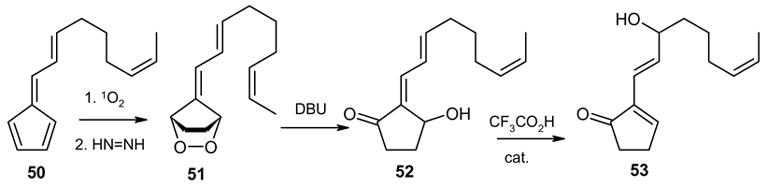
In another example, we subjected the hydroxy ketone 52, obtained from 51 by treatment with DBU to reaction with catalytic trifluoroacetic acid, in hopes of affecting dehydration. The product from this reaction was the alcohol 53, obviously stemming from a process involving dehydration, double bond shift and rehydration.
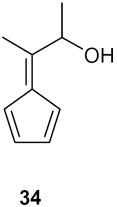
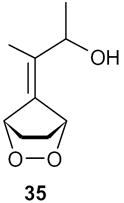
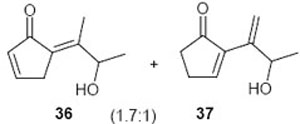
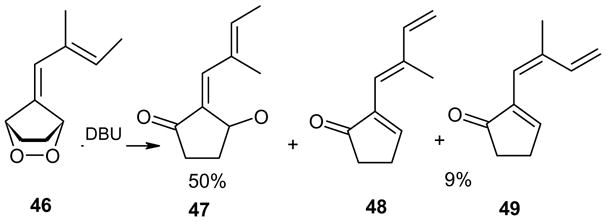
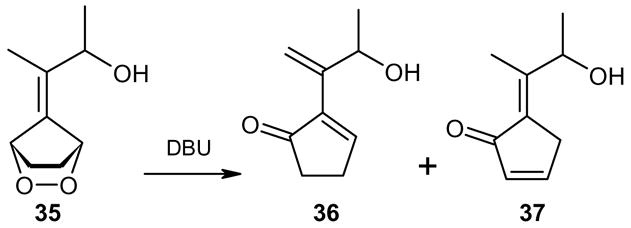
The presence of functionality on side chain of the exocyclic double bond was tested in the case of 37 (entry 6). The treatment of the endoperoxide 35 with DBU delivered a 3:2 mixture of the dehydration products 36 and 37 (Scheme 7). Obviously the hydroxyl group in the side chain did not suffer elimination. The two products were identified in the mixture by means of their 1H and 13C NMR spectra.
Scheme 7.
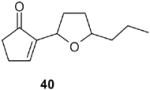
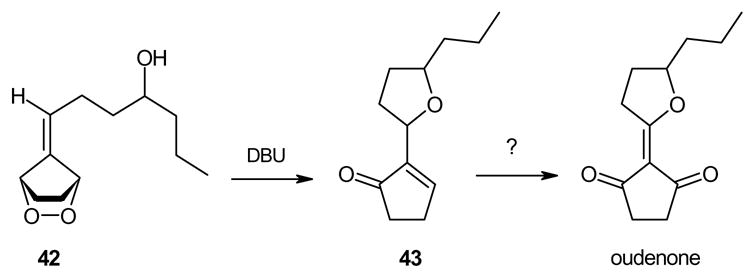
When the hydroxyl group was placed at C-3 on the side chain of the exocyclic double bond in the starting fulvene (entry 7), treatment of the saturated endoperoxides 42 with DBU furnished the tetrahydrofuran derivative 40 exclusively. Compound 40 appears to be a suitable precursor of the hypotensive drug oudenone,16 and this approach is currently being explored (Scheme 8).
Scheme 8.
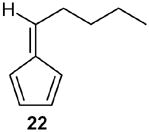
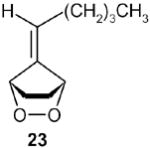
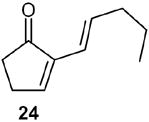
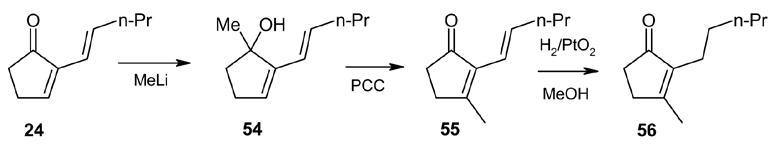
Finally, the fulvene endoperoxide-DBU isomerization-dehydration methodology was implemented in a short synthesis of the naturally occurring fragrant compound dihydrojasmone.17 Starting with fulvene 22 (entry 2), readily available from valeraldehyde in 77% yield was photooxygenated in CH2Cl2 at −78 °C, followed by diazene reduction, generated in situ from azodicarboxylate and acetic acid at low temperatures, endoperoxide 23 was obtained which without isolation was treated with DBU to give the cyclopentenone 24 in 83% yield. The conversion of 24 to dihydrojasmone was achieved by the well-documented protocol 18, 19 involving methyllithium addition to the carbonyl group and subsequent treatment with PCC in CH2Cl2 to furnish 56 in 50% overall yield from 24 (Scheme 9).
Scheme 9.
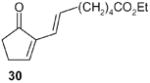
Compound 30 (entry 4) can also be obtained in this manner in a very short sequence from the readily available fulvene. The former represents an important precursor of certain prostaglandins and related compounds. These aspects are currently being explored.
It is noteworthy, that the respective unsaturated fulvene endoperoxides do not suffer dehydration after base-catalyzed isomerization with DBU, presumably owing to the unstable nature of the products (cyclopentadienones). Endoperoxide 57, derived from 6,6-dimethylfulvene for instance, gave a mixture of the hydroxyketones 58 and 59 in a ratio of 5:1 (Scheme 10).20
Scheme 10.
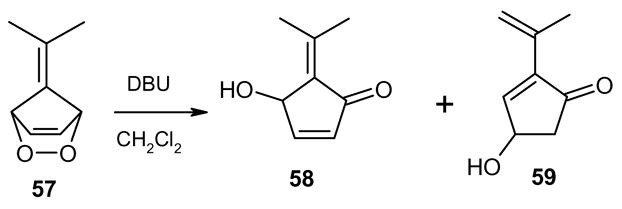
Compound 58 is the expected primary isomerization product, however, 59 is a secondary product that most likely forms by further isomerization of 58 by a mechanism outlined in Scheme 11.
Scheme 11.
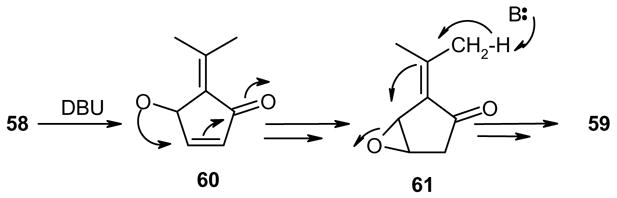
It is interesting to note, that the base-catalyzed decomposition of unsaturated fulvene endoperoxides appears to be entirely analogous to those of the saturated counterparts, except for the dehydration step in the latter. Intermediates 16 (Scheme 2) and 61 (Scheme 11) differ only in the nature of the leaving groups (alcohol, versus epoxide). The epoxide suffers ring opening to the allyllic alcohol, whereas the hydroxyl group is eliminated in 16.
3. Conclusion
In summary, we have uncovered a new useful facet of base-catalyzed endoperoxide rearrangements that result in a Kornblum-De la Mare isomerization followed by dehydration to give 2-vinyl-2-cyclopentenones which are highly desirable synthetic intermediates. In cases where there are oxygen nucleophiles on the side chain, conjugate attack at the β-carbon of the exocyclic double bond occurs to give 2-cyclopentenones that carry a heterocycle at C2. Implementation of this methodology in the synthesis of a variety of targets is underway.
4. Experimental
1H and 13C NMR spectra were recorded on a General Electric QE-Plus 300 MHz and Bruker Avance DRX 300 MHz spectrometers, using CDCl3 as solvent and TMS as internal standard, unless specified otherwise. IR spectra were obtained on a Nicolet Avance 370 DTGS FT-IR spectrometer. Column chromatographic separations were carried out with Davison 6–200 Mesh silica gel. For preparative TLC, Merck silica gel (grade 60 PF254) was used. All reactions were conducted under an atmosphere of dry nitrogen or argon. Fulvenes were stored under argon in the freezer at −30 °C. Non-deuterated solvents were dried and distilled prior to use. All fulvenes in this study were prepared according to published procedures.20, 21 Signal multiplicities in the NMR spectra are reported as follows: s-singlet, d-doublet, t-triplet, dd-doublet of doublets, dt-doublet of triplets, q-quartet, quin-quintuplet, sext-sextet, m-multiplet, br-broad.
Spectroscopic data for the synthesis of those fulvenes not found in literature are described below.
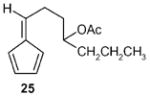
Fulvene 25 was prepared from 38 (vide infra) by stirring with acetic anhydride in the presence of pyridine for 48 hours followed by aqueous work-up in 86% yield. 1H NMR (CDCl3, TMS): δ 6.55 (m, 1H, 6.46 (m, 2H), 6.37 (t, J= 8.0 Hz, 1H), 6.12 (dt, 1H), 4.95 (m, 1H), 2.55 (m, 2H), 2.02 (s, 3H), 1.8 (m, 2H), 1.55 (m, 2H), 1.33 (m, 2H), 0.9 (t, J= 7.2 Hz, 3H); 13C NMR (CDCl3, TMS): δ 170.8, 146.2, 141.5, 133.4, 131.1, 125.6, 118.9, 73.5, 36.3, 33.9, 27.1, 21.2, 18.7, 14.0 ppm; Anal. Calcd for C14H20O2: C 76.33; H 9.15. Found: C 76.28; H 9.13.
Fulvene 28 (78%). 1H NMR (CDCl3, TMS): δ 6.6-6.4 (m, 4H), 6.2 (m, 1H), 4.1 (q, J= 7.5 Hz, 2 H), 2.54 (q, J= 7.5 Hz, 2H), 2.30 (t, J= 7.2 Hz, 2H), 1.65 (m, 2H), 1.50 (m, 2H), 1.4 (m, 2H), 1.25 (t, J= 7.5 Hz, 3H); 13C NMR (CDCl3, TMS): δ 173.7, 146.1, 142.8, 133.1, 130.8, 125.6, 119.1, 60.2, 34.2, 30.9, 29.1, 28.8, 24.8, 14.3 ppm; Anal. Calcd for C14H20O2: C 76.33; H 9.15. Found: C 76.31; H 9.15.
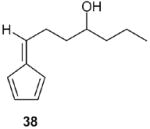
Fulvene 38 (49% yield): 1H NMR (CDCl3, TMS): δ 6.53 (m, 2H), 6.45 (m, 1H), 6.40 (t, J= 7.5 Hz, 1H), 6.2 (m, 1H), 3.63 (m, 1H), 2.65 (q, J= 7.5 Hz, 2H), 1.67 (m, 2H), 1. 44 (m, 2H), 1.30 (m, 4H), 0.9 (t, J= 7.5 Hz, 3H); 13C NMR (CDCl3, TMS): δ 146. 2, 142.6, 1332, 130.8, 125.6, 119.1, 71.3, 37.6, 36.9, 31.9, 27.5, 25.3, 22.7, 14.1 ppm. Anal. Calcd for C12H18O: C 80.85; H 10.18. Found: C 80.82; H 10.23.
Fulvene 50 (39% yield): 1H NMR ((CDCl3, TMS): δ 675 (m, 2H), 6.5-6.2 (m, 5 H), 5.4 (m, 2H), 2.3 (m, 2H), 2.2 (m, 2H), 2.05 (m, 2H), 1.0 (t, J= 7.5 Hz, 3H) ppm. Anal. Calcd for C14H18: C 90.26; H 9.74. Found: C 90.25; H 9.73.
Typical procedure for the preparation of the saturated endoperoxides
A solution of 6,6-dimethylfulvene (1.06 g, 10 mmol) and 5 mg of TPP (meso-tetraphenylporphyrin) in 200 mL CH2Cl2 was cooled to −78 in a flat Dewar containing dry ice-acteone. The mixture was stirred while being irradiated with a 250 W high-pressure sodium lamp for 3 hours under an oxygen atmosphere (a ballon filled with oxygen served this purpose). Then the lamp was turned off, 8.7 g (45 mmol) of potassium azodicarboxylate was added to the mixture in small portions, a pressure-equalizing dropping funnel fitted with a drying tube containing drierite was attached to the flask. A solution of 5.1 g (85 mmol) of acetic acid in 20 mL CH2Cl2 was added dropwise to the stirred slurry at −78 °C, and the mixture was slowly warmed up to −35 °C and stirred at this temperature for 1h. The mixture was then slowly warmed up to 0 °C for an additional 30 minutes. Then the salt was filtered by suction, the filtercake washed with cold CH2Cl2, while keeping the filter flask in an ice bath. The filtrate was washed with saturated aqueous NaHCO3, dried over MgSO4, filtered into a 250 mL round-bottomed flask. An aliquote was concentrated at reduced pressure (caution: saturated fulvene endoperoxides -when neat- are potentially explosive!), and dissolved in CDCl3 for NMR spectroscopy. The product was practically pure according to its 1H NMR spectrum.5 For the DBU-catalyzed isomerizations, the endoperoxides need not be isolated.
Typical procedure for the DBU-catalyzed isomerizations
Three drops of DBU were added to a solution of endoperoxide 57, obtained as described above, in 250 mL CH2Cl2 at 0 °C, and the solution was stirred at room temperature for 12–15 hours. Then the mixture was concentrated at reduced pressure, and the residue chromatographed on SiO2, eluting with hexane/ether (3:1).
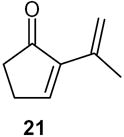
2-Isopropenyl-2-cyclopentenone (21): 1H NMR (CDCl3, TMS): δ 7.5 ((t, J= 3.0 Hz, 1H), 6. 07 (s, 1H), 5.16 (s, 1H), 2.60 (m, 2H), 2.47 (m, 2H), 1.94 (s, 3H); 13C NMR (CDCl3, TMS): δ 208.5, 158.5, 143.7, 135.1, 117.3, 36.7, 26.3, 22.0 ppm. FT-IR (film): 2973, 2927, 2855, 1710, 1453, 1387, 1262, 1177, 1038, 913, 802 cm−1. Anal. Calcd for C8H10O: C 78.65; H 8.25. Found: C 78.61; H 8.24.
(E)-(2)-Pent-1-enyl-2-cyclopentenone (24): 1H NMR (CDCl3, TMS): δ 7.4 (t, J= 3.0 Hz, 1H), 6.6 (dt, J= 16.0, 7.0 Hz, 1H), 6.1 (d, J= 16.0 Hz, 1H), 2.6 (m, 2H), 2.45 (m, 2H), 2.1 (q, J= 7.0 Hz, 2H), 1.50 (sextet, J= 7.0 Hz, 2H), 0.94 (t, J= 7.0 Hz, 3H); 13C NMR (CDCl3, TMS): δ 208.3, 156.5, 141.2, 135.7, 119.8, 35.57, 35.52, 26.2, 22.2, 13.7 ppm. Anal. Calcd for C10H14O: C 79.96; H 9.39. Found: C 79.95; H 9.38.
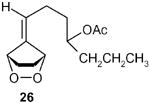
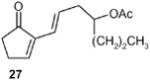
(E)-1-(5-Oxocylopent-1-enyl)hept-1-en-4yl acetate (27): 1H NMR (CDCl3, TMS): δ 7.3 (br s, 1H), 6.45 (dt, J= 15.6, 7.2 Hz, 1H), 6.0 (d, J= 15.6 Hz, 1H), 4.85 (quin, J= 6.0 Hz, 1H), 2.5 (m, 2H), 2.35 (m, 2H), 2.26 (m, 2H), 1.94 (s, 3H), 1.45 (m, 2H), 1.24 (m, 2H), 0.82 (t, J= 7.2, 3H); 13C NMR (CDCl3, TMS): δ 207.8, 170.4, 157.4, 157.2, 140.6, 129.9, 122.3, 72.9, 38.0, 35.3, 26.0, 20.1, 18.3, 13.7, 13.5 ppm. Anal. Calcd for C14H23O3: C 70.20; H 9.69. Found: C 70. 17; H 9.66.
(E)-Ethyl 7-(5-oxocyclopent-1-enyl)hept-6-enoate (30): 1H NMR (CDCl3, TMS): δ 7.3 (t, J= 3Hz, 1H), 6.5 (dt, J= 16.0, 7.2 Hz, 1H), 4.0 (q, J= 7.2 Hz, 2H), 2.5 (m, 2H), 2.35 (m, 2H), 2.2 (m, 2H), 2.05 (m, 2H), 1.60 (m, 2H), 1.35 (m, 2H), 1.7 (t, J= 7.2 Hz, 3H);. 13 C NMR (CDCl3, TMS): δ 208.4, 173.6, 157.0, 141.0, 135.1, 120.1, 60.2, 35.6, 34.1, 33.1, 28.4, 26.3, 24.5, 14.2 ppm. Anal. Calcd for C14H20O3: C 70.20; H 9.69.Found: C 70.19; H 9.66.
2-Cycloheptenylcyclopent-2-one (33): 1H NMR (CDCl3, TMS): δ 7.4 (t, J= 3.0 Hz, 1H), 6.7 (t, J= 6.6 Hz, 1H), 2.56 (m, 2H), 2.45 (m, 2H), 2.37 (m, 2H), 2.25 (m, 2H), 1.8 (m, 2H), 1.55 (m, 4H); 13C NMR (CDCl3, TMS): δ 208.2, 155.5, 145.0, 135.8, 133, 36.0, 32.2, 31.1, 28.4, 26.4, 26.3, 25.3 ppm. Anal. Calcd for C12 H16O: C 81.77; H 9.15. Found: C 81.77; H 9.14.
(E)-5-(3-Hydroxybutan-2-ylidene)cyclopent-2-enone (36): 1H NMR (CDCl3, TMS): δ 7.54 (m, 1H), 6.30 (m, 1H), 5.2 (br s, 1H, OH), 5.05 (q, J= 7.0 Hz, 1H), 3.2 (s, 2H), 1.32 (d, J= 7.0 Hz, 3H) ppm.
2-(3-Hydroxybut-1-en-2-yl)cyclopent-2-enone (37): ): 1H NMR (CDCl3, TMS): δ 7.7 (t, J= 3.0 Hz, 1H), 5.65 (s, 1H), 5.4 (s, 1H), 4.5 (q, J= 6.6, 1H), 3.9 (br s, 1H, OH), 2.7 (m, 2H), 2.5 (m, 2H), 1.26 (d, J= 6.6 Hz, 3H); 13C NMR (CDCl3, TMS, 36+37): δ 209.1, 197.4, 160.7, 156.0, 156.4, 143.5, 142.5, 137.0, 128.5, 114.7, 68.9, 68.2, 35.2, 35.0, 26.3, 22.0, 21.0, 18.2 ppm.
2-(5-Propyltetrahydrofuran-2-yl)cyclopent-2-enone (40, both epimers): 1H NMR (CDCl3, TMS): δ 7. 5 (t, J= 2.7 Hz, 1H), 4.7 and 4.61 (m, 1H), 4.1 and 3.8 (m, 1H), 2.6 (m, 2H), 2.42 (m, 2H), 2.3-1.9 (m, 2H), 1.2–1.8 (m, 6 H), 0.9 (m, 3H); 13C NMR (CDCl3, TMS): δ 208.7, 208.5, 157.8, 157.5, 148.4, 148.2, 79.7, 79.4, 74.1, 73.8, 38.25, 38.16, 35.6, 32.0, 31.9, 31.4, 26.63, 26.6, 19.5, 14.3 ppm. Anal. Calcd for C12H18O2: C 74.19; H 9.34. Found: C 74.14; H 933.
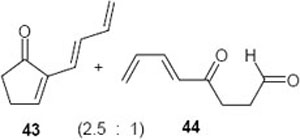
(E)-(2-Buta-1,3-dienyl)cyclopent-2-enone (43): 1H NMR (CDCl3, TMS): δ 7.5 (t, J= 3..0 Hz, 1H), 7.16 (dd, J= 17.0, 10.5 Hz, 1H), 6.2–6.5 (m, 2H), 5.35 (d, J= 17.0 Hz, 1H), 5.17 (d, J= 10.5 Hz, 1H), 2.6 (m, 2H), 2.46 (m, 2H); 13C NMR (CDCl3, TMS): δ 208.6, 159.0, 141.6, 138.0, 134.3, 123.0, 120.0, 36.3, 27.2 ppm. HRMS m/z calcd for C9H10O: 134.0732. Found: 134.0733.
(5E, 7E)-4-Oxonona-5,7-dienal (44): 1H NMR (CDCl3, TMS): δ 9.8 (s, 1H), 7.35 (m, 1H), 6.4 (t, J= 11.1 Hz, 1H), 6.1 (m, 1H), 5.9 (d, J= 11.1 Hz, 1H), 2.75 (m, AA’BB’ pattern, 4H), 1.8 (dd, J= 6.6, 1.0 Hz, 3H); 13C NMR (CDCl3, TMS): δ 201.3, 198.7, 144.2, 141.5, 130.9, 127.8, 38.3, 33.2, 19.5 ppm. HRMS m/z calcd for C 8H10O2: 138.0681. Found: 138.0677.
(E)-3-Hydroxy-2-((E)-2-methylbut-2-enylidene)cyclopentanone (47): 1H NMR (CDCl3, TMS): δ 7.0 (s, 1H), 6.2 (q, J= 7.2 Hz, 1H), 5.2 (d, J= 4.8 Hz, 1H), 2.6 (m, 2H), 3.3 (br s, 1H, OH), 2.23 (m, 2H), 2.0 (s, 3H), 1.8 (d, J=7.2, 3H) ppm. Anal. Calcd for C10H14O2: C 72.26; H 8.49. Found: C 72.35; H 8.50.
(E) and (Z)-2-(2-Methylbuta-1,3-dienyl)cyclopent-2-enone and (48 and 49): 1H NMR (CDCl3, TMS): δ 7.6 (t, J= 3.0 Hz, 1H), 7.5 (t, J= 3.0 Hz, 1H), 6.8 (dd, J= 17.1, 10.8 Hz, 1H), 6.5 (dd, J= 17.4, 10.8 Hz, 1H), 6.2 (s, 1H), 6.13 (s, 1H), 5.4 (dd, J= 17.4, 10.8, 1H), 5.2 (dd, J= 17.1, 10.8 Hz, 1H), 2.7 (m, 2H), 2.4 (m, 2H), 1.97 (s, 3H), 1.95 (s, 3H) ppm.
(E)-3-Hydroxy-2-((2E, 6Z)-octa-2,6-dienylidene)cyclopentanone (52): 1H NMR (CDCl3, TMS): δ 7.0 (d, J= 11.4 Hz, 1H), 6.5 (m, 1H), 6.3 (m, 1H), 5.3 (m, 2H), 5.1 (m, 1H), 2.6 (m, 2H), 1.9–2.4 (m, 8H), 0.9 (t, J= 7.5 Hz, 3H) ppm; 13C NMR (CDCl3, TMS): δ 206.8, 149.4, 137.3, 137.2, 133.5, 128.0, 126.8, 70.2, 36.4, 34.3, 31.0, 26.9, 21.2, 15.0 ppm. Anal. Calcd for C14H20O2: C 76.33; H 9.15. Found: C 76.36; H 9.12.
2-((1E, 6Z)-3-Hydroxyocta-1,6-dienyl)cyclopent-2-enone (53): 1H NMR (CDCl3, TMS): δ 7.5 (t, J= 3.0 Hz, 1H), 6.8 (dd, J= 16.0, 8.0 HZ, 1H), 6.4 (d, J= 16 Hz, 1H), 5.4 (m, 2H), 5.3 (m, 1H), 2.64 (m, 2H), 2.5 (m, 2H), 1.7–2.2 (m, 6H), 0.9 (t, J= 7.5 Hz, 3H). Anal. Calcd for C14H20O2: C 76.33; H 9.15. Found: C 76.29; H 9.14.
(E)-1-Methyl-2-(pent-1-enyl)cyclopent-2-enol (54). To a stirred solution of 1.00 g (6.66 mmol) of 24 in 50 mL of dry ether at −78 °C was added dropwise through a syringe 5.23 mL of an ethereal solution of methyllithium (1.4 M). The reaction mixture was allowed to warm up to room temperature and stirred for 1 hour. A saturated aqueous solution of NH4Cl solution (10 mL) was added dropwise, the layers separated and the aqueous layers extracted twice with 10-mL portions of ether. The combined organic layers were washed twice with 10 mL of brine each, and the organic layer dried over anhydrous MgSO4. The solvent was removed at reduced pressure and the residue chromatographed on SiO2 (3:1 hexanes/EtOAc), providing 0.8 g (72% yield) of 54. 1H NMR (CDCl3, TMS): δ 6.15 (dt, J= 16.2, 6.6 Hz, 1H), 5.95 (d, J= 16.2 Hz, 1H), 5.6 (narrow m, 1H), 2.4-1.9 (m, 6H), 1.45 (sext, J= 7.2 Hz, 2H), 1.40 (s, 3H), 0.91 (t, J= 7.2 Hz, 3H); 13C NMR (CDCl3, TMS) δ 146.6, 132.1, 127.5, 123.2, 83.2, 42.5, 35.5, 28.5, 25.8, 22.5, 13.7 ppm.
(E)-3-Methyl-(2-pent-1-enyl)cyclopent-2-enone (55). A solution of 1.0 g (6 mmol) of the tertiary alcohol 54 in 5 mL of CH2Cl2 was added dropwise to a stirred suspension of 2.58 g (12.0 mmol) of pyridinium chlorochromate (PCC) in 20 mL CH2Cl2 at 0 °C. The mixture was stirred at room temperature for 1 hour, and was diluted with 25 mL of ether. The ethereal solution was decanted from the solid, which was washed with three 20 mL portions of ether. The combined ethereal phases were washed successively with two 25 mL portions of 5% NaOH, 5% HCl, and two 10 mL portions of saturated aqueous NaHCO3, and dried over MgSO4. The solvent was removed at reduced pressure and the residue purified by column chromatography on SiO2, eluting with 3:1 hexanes/EtOAc, to give 0.74 (69%) of 55 as a pale yellow oil. 1H NMR (CDCl3, TMS): δ 6.7 (dt, J= 16.0, 7.2 Hz, 1H), 6.06 (d, J= 16 Hz, 1H), 2.55 (m, 2H), 2.35 (m, 2H), 2.12 (s, 3H), 2.0 (m, 2H), 1.5 (m, 2H), 0.9 (t, J= 7.2 Hz, 3H). These data are almost identical to those reported for 55.13
4-Hydroxy-2-(propen-1-en-2-yl)cyclopent-2-one (59). 1H NMR (CDCl3, TMS): 7.3 (d, 1H), 6.1 (s, 1H), 5.2 (s, 1H), 4.9 (m, 1H), 3.4 (br, OH), 2.9 (dd, B part of an ABX system, 2J= 18.6 Hz, 1H), 2.38 (d, 2J= 18.6 Hz, 1H), 1.93 (s, 3H); 13C NMR (CDCl3, TMS): δ 204.9, 156.2, 144.3, 134.4, 119.9, 68.0, 48.9, 22.5 ppm.
3-Methyl-2-pentylcyclopent-2-enone (dihydrojasmone, 56). Ketone 55 (0.5g, 3.0 mmol) was dissolved in 10 mL of methanol, 2% PtO2 was added, and the mixture was hydrogenated at atmospheric pressure at room temperature. Hydrogenation was interrupted after the uptake of one equivalent of H2, and the catalyst filtered off, the residue concentrated at reduced pressure to give 56 in quantitative yield. Its physical and spectroscopic data were identical in all respects with the reported values in literature.
Scheme 6.
Acknowledgments
This work was supported by funds from the National Science Foundation (CHE-9729001), the National Institutes of Health, MBRS-SCORE Program-NIGMS (Grant No. GM52588) and in part from a grant (P20 MD) from the Research Infrastructure in Minority Institutions Program, NCMHD, NIH.
Footnotes
This paper is dedicated to my teacher, friend and associate, Professor Armin de Meijere, on the occasion of his choice
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wasserman HH, Murray RW, editors. Singlet Oxygen. Academic Press; New York: 1979. [Google Scholar]
- 2.Balci M. Chem Rev. 1981;81:91–108. [Google Scholar]
- 3.Kornblum N, De La Mare HE. JAmChemSoc. 1951;73:880. [Google Scholar]
- 4.Zagorski MG, Salomon RG. J AmChemSoc. 1980;102:2501. [Google Scholar]
- 5.Adam W, Erden I. Angew Chem. 1978;90:223. [Google Scholar]; Angew Chem Int Ed Engl. 1978;17:210. [Google Scholar]
- 6.Erden I, Amputch M. Tetrahedron Lett. 1987;28:3779. [Google Scholar]
- 7.Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:1255. [Google Scholar]
- 8.Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:2291. [Google Scholar]
- 9.Erden I, Xu F, Drummond J, Alstad R. J Org Chem. 1993;58:3611. [Google Scholar]
- 10.Erden I, Xu F, Cao W. Angew Chem. 1997;109:1557. [Google Scholar]; Angew Chem Int Ed Engl. 1997;36:1516. [Google Scholar]
- 11.Ishihara H, Inomata K, Mukaiyama T. Chem Lett. 1975:531. [Google Scholar]
- 12.Funel JA, Prunet J. J Org Chem. 2004;69:4555. doi: 10.1021/jo049557d. [DOI] [PubMed] [Google Scholar]
- 13.Shimada J, Hashimoto K, Kim BH, Nakamura E, Kuwajima I. J Am ChemSoc. 1984;106:1759. [Google Scholar]; Nakamura E, Shimada J, Kuwajima I. J Chem Soc, Chem Comm. 1983;9:498. [Google Scholar]
- 14.Stetter H, Leinen HT. ChemBer. 1983;116:254. [Google Scholar]
- 15.Johnson CR, Adams JP, Braun MP, Senayake CBW. Tetrahedron Lett. 1992;33:919. [Google Scholar]
- 16.For a recent synthesis of oudenone, see Flynn BL, Silveira Claudio C, de Meijere A. Synlett. 1995:812.
- 17.For a review on Jasmonoids, see Ho T-L. Syn Commun. 1974;4:265.
- 18.Dauben WG, Michno DM. J Org Chem. 1977;42:682. [Google Scholar]
- 19.Seo J, Fain H, Blanc JB, Montgomery J. J Org Chem. 1999;64:6060. [Google Scholar]
- 20.Though 2(3H)-oxepinones are the sole products from thermal isomerizations of unsaturated fulvene endoperoxides in aprotic, non-nucleophilic solvents, , 57 has been reported to give rise to considerable amounts of 58 in methanol; see refs. 23 and 24.
- 21.Stone KJ, Little RD. J Org Chem. 1984;49:1849. [Google Scholar]
- 22.Erden I, Xu F, Sadoun A, Smith W, Sheff G, Ossun M. J Org Chem. 1995;60:813. [Google Scholar]
- 23.Harada N, Suzuki S, Uda H, Ueno H. J Am Chem Soc. 1971;94:1777. [Google Scholar]; Harada N, Uda H, Ueno H, Utsumi S. Chem Lett. 1973:1173. [Google Scholar]
- 24.a) Skorianetz W, Schulte-Elte KH, Ohloff G. Angew Chem. 1972;84:3111. [Google Scholar]; Angew Chem Int Ed Engl. 1972;11:330. [Google Scholar]; b) Skoriantez W, Schulte-Elte KH, Ohloff G. Helv Chim Acta. 1971;54:1913. [Google Scholar]