

Abstract
Women are at high risk of dying from unrecognized cardiovascular disease. Many differences in cardiovascular disease between men and women appear to be mediated by vascular smooth muscle cells (SMC). Since estrogen reduces the proliferation of SMC, we hypothesized that activation of estrogen receptor alpha (ERα) by agonists or by growth factors altered SMC function. To determine the effect of growth factors, estrogen, and ERα expression on SMC differentiation, human aortic SMC were cultured in serum-free conditions for 10 days. SMC from men had lower spontaneous expression of ERα and higher levels of the differentiation markers calponin and smooth muscle α-actin than SMC from women. When SMC containing low expression of ERα were transduced with a lentivirus containing ERα, activation of the receptor by ligands or growth factors reduced differentiation markers. Conversely, inhibiting ERα expression by small interfering (si) RNA in cells expressing high levels of ERα enhanced the expression of differentiation markers. ERα expression and activation reduced the phosphorylation of Smad2, a signaling molecule important in differentiation of SMC, and initiated cell death through cleavage of caspase-3. We conclude that ERα activation switched SMC to a dedifferentiated phenotype and may contribute to plaque instability.
Keywords: apoptosis, cardiovascular disease, gene expression, nuclear receptors, smooth muscle differentiation
Introduction
Since 1984, more women than men died from cardiovascular diseases, although the prevalence of diagnosed disease is lower among women 1. Women have higher rates of stable angina, high blood pressure, congestive heart failure and stroke, but have less angiographic evidence of atherosclerotic plaques and have fewer myocardial infarctions than men 1. The inhibition of collagen production, smooth muscle proliferation and endothelial dysfunction by estrogen may delay the formation of plaques in women until after menopause 2. Hormone replacement therapy started more than ten years after menopause increases a woman’s risk for myocardial infarction 3, 4, although therapy initiated near menopause may be more effective in preventing coronary heart disease 5. Additionally, because of gender differences in symptoms during acute coronary events and in response to interventional strategies, it is difficult to correctly diagnose and treat women 6, 7.
A few studies comparing vascular wall properties and disease presentation of men and women with symptoms of coronary artery disease provide insight into the complicated effects of female hormones and their receptors in vascular cells. Although women with acute coronary syndromes are often free of angiographically-visible stenoses, testing of coronary flow reserve demonstrates endothelial and smooth muscle dysfunction 8, 9. Younger women who die from coronary artery thrombosis are more likely than men or postmenopausal women to have plaque erosion, rather than rupture of a lipid-rich plaque 10, 11. Plaque erosions are characterized by loss of endothelial cells covering a non-occlusive, smooth muscle cell (SMC)- and hyaluronan-rich plaque with few inflammatory cells or type I collagen 12. It is speculated that migration of dedifferentiated SMC and expression of hyaluronan weakens endothelial cell adhesion and predisposes the coronary arteries for thrombotic events 12.
After menopause, women experience a dramatic rise in aortic stiffness which may cause hypertension 13. In those who develop coronary artery disease, the plaques become more numerous with larger lipid cores and thinner fibrous caps marked by calcification 11. It is uncertain how matrix deposition and plaque stability are affected by the lack of estrogen or by growth factors which activate the estrogen receptor (ER) 2.
Because SMC are responsible for many of the differences in coronary disease noted between men and women such as microvessel dysfunction, plaque erosion, and matrix deposition; we sought to understand the role of ERα in smooth muscle differentiation in estrogen- or growth factor-rich environments to mimic gender or menopausal effects. In this study, we obtained aortic SMC from male and female donors and determined the effects of ERα expression, estrogen, and growth factors on differentiation, survival and adherence of these cells.
Methods
Explantation and SM α-actin detection
Following informed consent, sections of aorta were obtained from heart transplant donors and recipients at The Ohio State University Medical Center as approved by the institutional review board. Aortic slices were stripped of endothelium and adventitia, rinsed, and cut into small bits. The average age (±SEM) for females and males examined in this study was 32.6±6.39 and 47.4±8.62, respectively (n=5 each). No statistical difference in age was observed between the donors and recipients or between genders. The SMC were expanded in growth media with amphotericin and gentamicin (Clonetics/Cambrex, Walkersville, MD and Cascade Biologics, Portland, OR) then tested for smooth muscle (SM) α-actin expression using FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Cell populations containing at least 85% positive staining for SM α-actin were used for subsequent studies.
Real-Time PCR for ERα
ERα mRNA was analyzed by Real-Time PCR in SMC from five male donors and five female donors, starved for five days to allow ERα upregulation. TaqMan primers and probe designed by Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi14) were synthesized by Applied Biosystems (Foster City, CA). The following primers were used to detect ERα: forward 5′- agctcctcctcatcctctcc-3′, reverse 5′-tctccagcagcaggtcatag-3′, and probe 6FAM-5′-tcaggcacatgagtaacaaaggca-3′ TAMRA. RNA was isolated using NucleoSpin RNA II (BD Clontech, Mountain View, CA), and cDNA generated using random hexamers (Invitrogen, Carlsbad, CA). A 111 bp product from ERα was amplified over 40 cycles with 18S RNA as internal control using ABI PRISM 7700 Sequence Detection System (Applied Biosystems).
Cloning of ERα into EGFP-pLenti6/V5 plasmid and transduction
The pLenti6/V5-D-TOPO vector (Invitrogen) was engineered to contain an enhanced green fluorescent protein (EGFP) surrounded by additional restriction sites and designated pLenti-EGFP (generously provided by Mark Wewers, Ohio State University). cDNA for ERα was amplified by PCR from a pBK-CMV/ERα plasmid kindly provided by Robert Brueggemeier (Ohio State University), introducing EcoRI and EcoRV restriction sites. EGFP was removed from pLenti-EGFP by digestion with EcoRI and EcoRV and replaced with ERα to generate pLenti-ERα. pLenti vector lacking EGFP was used as a control. Purified pLenti-ERα or empty vector control (3 μg) were transfected with 2 μg pMD.G and 10 μg pCMVΔR8.2 helper plasmids (kindly provided by Dr. K. Boris-Lawrie, Ohio State University) into HEK293FT cells according to the directions for the ViraPower Lentiviral Expression System. Virus secreted into the media was concentrated (Vivaspin 100,000 MWCO, Vivascience, Germany) and titered in SMC cultures, with blasticidin (2 μg/mL) for selection. SMC were then transduced with the virus for each experiment at approximately 5 MOI and incubated overnight in growth media containing 6 μg/mL polybrene.
Transfection of siRNA plasmids
SMC (1×106) were transfected with 10 μg control or ERα siRNA plasmid (Panomics, Redwood City, CA) using nucleofection (Amaxa, Gaithersburg, MD). Transfection efficiency was monitored using 2 μg of pmaxGFP plasmid (Amaxa).
SMC Differentiation and activation
Differentiation experiments were performed on SMC in the following groups, seeded in an 8-well plate as noted: native cells expressing endogenous ERα (7 ×104 cells per well), cells with low expression of ERα to be transduced with ERα Lentivirus (8 ×104), and cells with high ERα levels transfected with ERα siRNA (1.8 ×105). After recovery, the cells were starved overnight in phenol red- and serum-free basal media (EBM-PRF, Clonetics/Cambrex) and exposed for 10 days to vehicle control (VEH, either HCL 4 μmol/L or ethanol 1:400,000 dilution), 17β-estradiol (ESTR 10 nmol/L, Sigma, St. Louis, MO), the ERα agonist propyl pyrazole triol (PPT 10 nmol/L, Tocris Cookson, Ellisville, MO), epidermal growth factor (EGF, 10 ng/mL, R&D Systems (Minneapolis, MN), platelet derived growth factor-BB (PDGF-BB, 10 ng/mL, R&D Systems), or transforming growth factor-β1 (TGFβ1, 5 ng/mL, R&D Systems) in EBM-PRF. Agonists or vehicle controls were added each day and then cells were lysed in cell lysis buffer (Cell Signaling Technology, Danvers, MA). Samples of the culture media at the end of the experiment were quantitated for active TGFβ1 by ELISA (Quantikine, R&D Systems). Activation studies were performed on SMC stably transduced with pLenti control or ERα (7×104 cells per well or 2×105 cells per 25cm2 flask), incubated for the times indicated using agonists as listed above, then lysed with cell lysis buffer or CelLytic NuCLEAR Extraction Kit (Sigma). Equal protein amounts (20–50 μg) were subjected to Western blot analysis and detected with West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL) and the Fluor S-Max system (Bio-Rad, Hercules, CA). Smooth muscle α-actin (SM α-actin), β-actin, and calponin antibodies were obtained from Sigma. Antibodies to phospho-ERα and cleaved caspase-3 were from Cell Signaling Technology. Cyclin D1 (clone DCS-6), Erk2 and ERα (HC20) antibodies were obtained from Santa Cruz (Santa Cruz, CA).
ERα transcriptional activation
Stably transduced SMC were transfected with an estrogen response element (ERE) reporter construct producing secreted alkaline phosphatase (SEAP) (Clontech/BD Biosciences) using Effectene (Qiagen, Valencia, CA). The SEAP signal was obtained over three days and normalized as a percent of the maximum signal achieved.
Immunofluorescence for ERα
Virally transduced SMC were fixed in 70% ETOH, permeabilized and blocked with 0.05% triton/1% goat serum. Cells were incubated overnight with ERα antibody (Ab-16, Lab Vision-Neomarkers, Fremont, CA) in 1% goat serum. ERα was detected with Alexa Fluor 568 anti-rabbit secondary antibody (Molecular Probes, Invitrogen) and a DP-11 digital camera connected to an IX-50 inverted microscope with 10X objective (Olympus, Melville, NY).
Cell Density
Phase contrast images were taken using identical settings at day 10 of the differentiation experiments using the DP-11 digital camera and IX-50 inverted microscope with 4X objective (Olympus). Quantity One colony counting software (Bio-Rad) was used to detect live cells (gray) but exclude apoptotic cells (white). Numbers were normalized to VEH control samples for each cell population or control transfection/transduction cells.
Statistics
Real-Time PCR results for ERα expression were analyzed using longitudinal regression over ten experiments to test the difference in delta cycle times, which are normally distributed. Western blot densitometry ratios for contractile proteins in starved or PDGF-stimulated SMC from six people were compared using a mixed model regression to account for correlation within cell lines. Densitometry values from the remaining immunoblots were normalized to loading controls, and by the VEH control sample for the control group, compared by two-factor analysis of variance with interaction (ANOVA, Stata, version 9, StataCorp, College Station, Texas) and pairwise comparisons were adjusted using the Holm’s method 15.
Results
Low level expression of ERα in human aortic smooth muscle
Estrogen receptors are present in healthy aortic SMC and regulate growth 2. Because genes that affect cell growth often change cell differentiation, and ERα enhances proliferation in transformed cells, we hypothesized that the expression and activation of SMC ERα modulated cell differentiation. The ERα mRNA level, stated as a fold-induction above the SMC population containing the lowest level of ERα, was about 4.3 times higher on average for female donors than for male donors (p<0.001, Figure 1A). By comparison, serving as a positive control, the ERα level for the breast cancer line MCF7 was about 1000-times higher than SMC containing the lowest ERα levels, while as a negative control, the colon cancer cell line HT29 had little to no ERα detected by PCR. For subsequent studies, we used SMC from either the female donor with the greatest amount of ERα or the male donor with the lowest ERαexpression. We confirmed proportional ERα protein expression in these two cell populations (Figure 1B).
Figure 1. Aortic SMC from female donors have more ERα than cells from male donors.
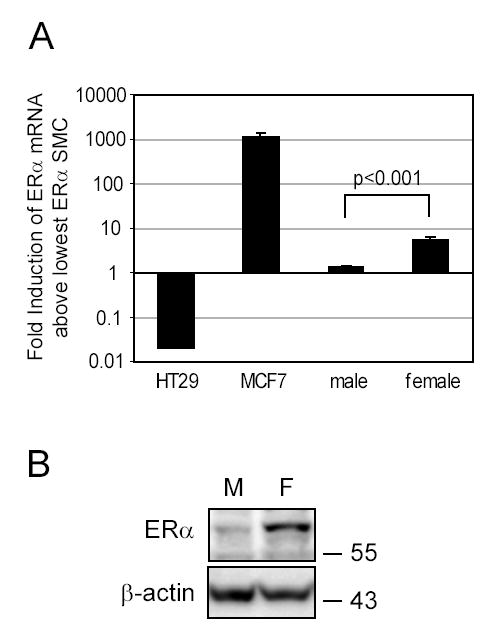
A. Aortic SMC from five male and five female tissue donors were lysed after 5 days of starvation to analyze ERα mRNA levels by Real-Time PCR. ERα mRNA levels for SMC, the colon cancer cell line HT29 (negative control) and the ERα-positive breast cancer cell line MCF7 (positive control) were expressed as fold-induction above the cell population from the male SMC which consistently had the lowest levels of ERα expression. B. SMC expressing the lowest and highest levels of mRNA for ERα (M: male, F: female) were starved in basal media for 10 days and Western blotted for ERα expression. β-actin was detected as a loading control.
SMC containing ERα failed to differentiate
Because growth factors activate ERα and TGFβ1 causes differentiation of SMC 16, 17, SMC expressing the highest ERα levels (HI) and the lowest ERα levels (LO) were treated with these growth factors as well as ERα ligands (PPT and 17β-estradiol). As shown, cells with high ERα levels (HI) had little expression of the differentiation markers SM α-actin or calponin except in the presence of TGFβ1 (Figure 2A). In contrast, cells with lower levels of ERα (LO) retained both SM α-actin and calponin in all conditions except when incubated with EGF or PDGF (Figure 2A). We observed that low ERα cells had significantly greater amounts of calponin (p<0.0001 overall), and greater amounts of SM α-actin (p=0.0001 overall) compared to high ERα expressing cells (Figure 2B). Individual comparisons are as shown in Figure 2B.
Figure 2. Differentiation of aortic SMC varies inversely to ERα expression.
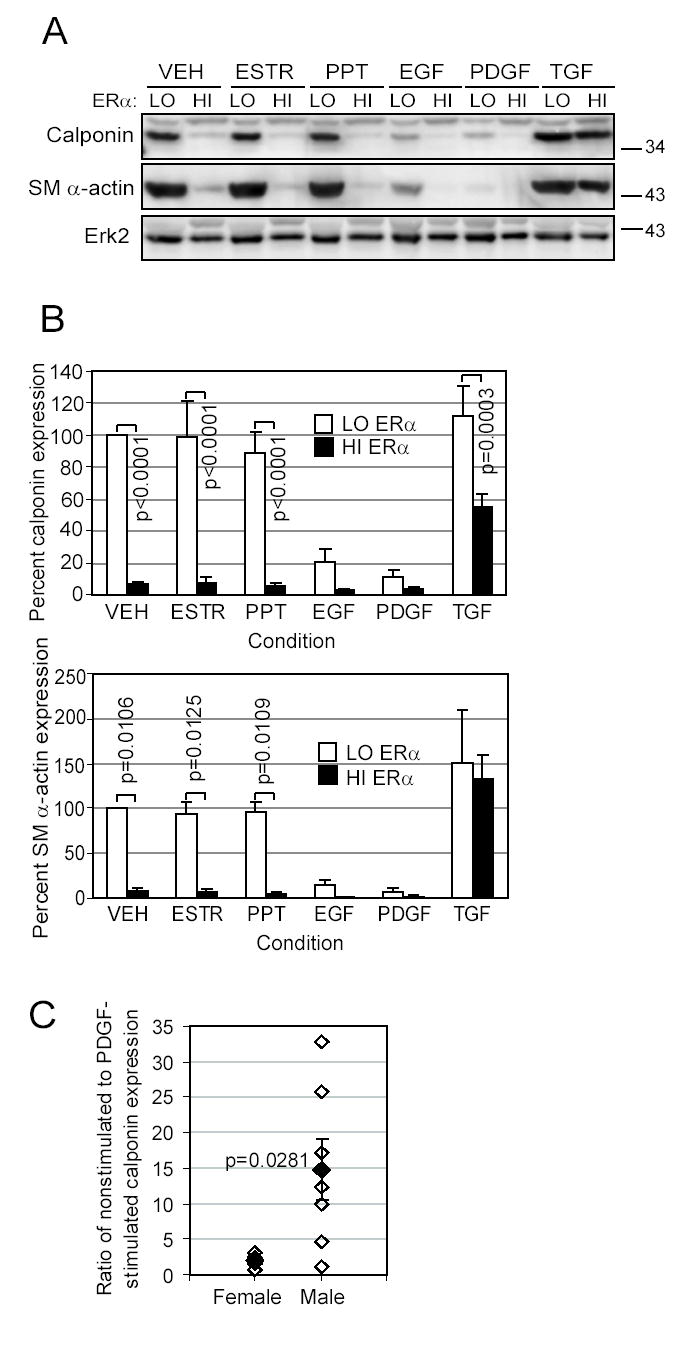
SMC with the highest (HI) or the lowest (LO) ERα expression were exposed for 10 days in EBM-PRF to VEH control, ESTR (10nmol/L), PPT (10nmol/L), EGF (10 ng/mL), PDGF-BB (10 ng/mL), or TGFβ1 (5 ng/mL). A. Western blotting was performed on protein lysates to determine expression of contractile proteins calponin and SM α-actin, and Erk2 as a loading control. A representative blot is shown. B. Protein expression was quantitated by densitometry, normalized by the Erk2 band, and stated as a percent of the low-ERα VEH control condition (□low-ERα SMC, ■high-ERα SMC, mean±SEM of 4 replicates). C. SMC from three male donors and three female donors were grown for 10 days in EBM-PRF with either PDGF-BB or VEH control. Western blotting was performed to detect calponin, and differentiation was calculated as a ratio of the intensity for the VEH-treated to the PDGF-treated samples (multiple replicates: ⋄n=6 female, n=7 male, ♦mean±SEM).
Since SMC from the low ERα (male) donor had more differentiation markers than the high ERα (female) donor, we further characterized basal differentiation of SMC from other male or female donors (n=3 each). As shown in Figure 2C, VEH control-stimulated SMC from male donors had high levels of calponin, but lost much of this marker upon PDGF stimulation, similar to cells in Figure 2A. In contrast, female donor SMC expressed only low levels of calponin in either condition. Consequently, the average calponin ratio was significantly higher for SMC from men than for SMC from women (p=0.0281).
Transduction of ERα inhibited SMC differentiation
Since ERα expression correlated with SMC dedifferentiation, we examined whether induced expression of ERα in low ERα-containing cells directly inhibited SMC differentiation. Transduction efficiency was determined by ERα immunofluorescent staining (Figure 3A).Transduction of ERα lowered calponin (p<0.0001) and SM α-actin (p<0.0001) expression overall. ERα-expression reduced SM α-actin in response to ESTR, PPT and TGFβ1 treatment, and reduced calponin in response to VEH, ESTR or PPT (Fig 3B and C, p values as shown). In contrast to these cell markers, cyclin D increased upon transduction of ERα (p=0.0001 overall).
Figure 3. SMC dedifferentiate following transduction of ERα.
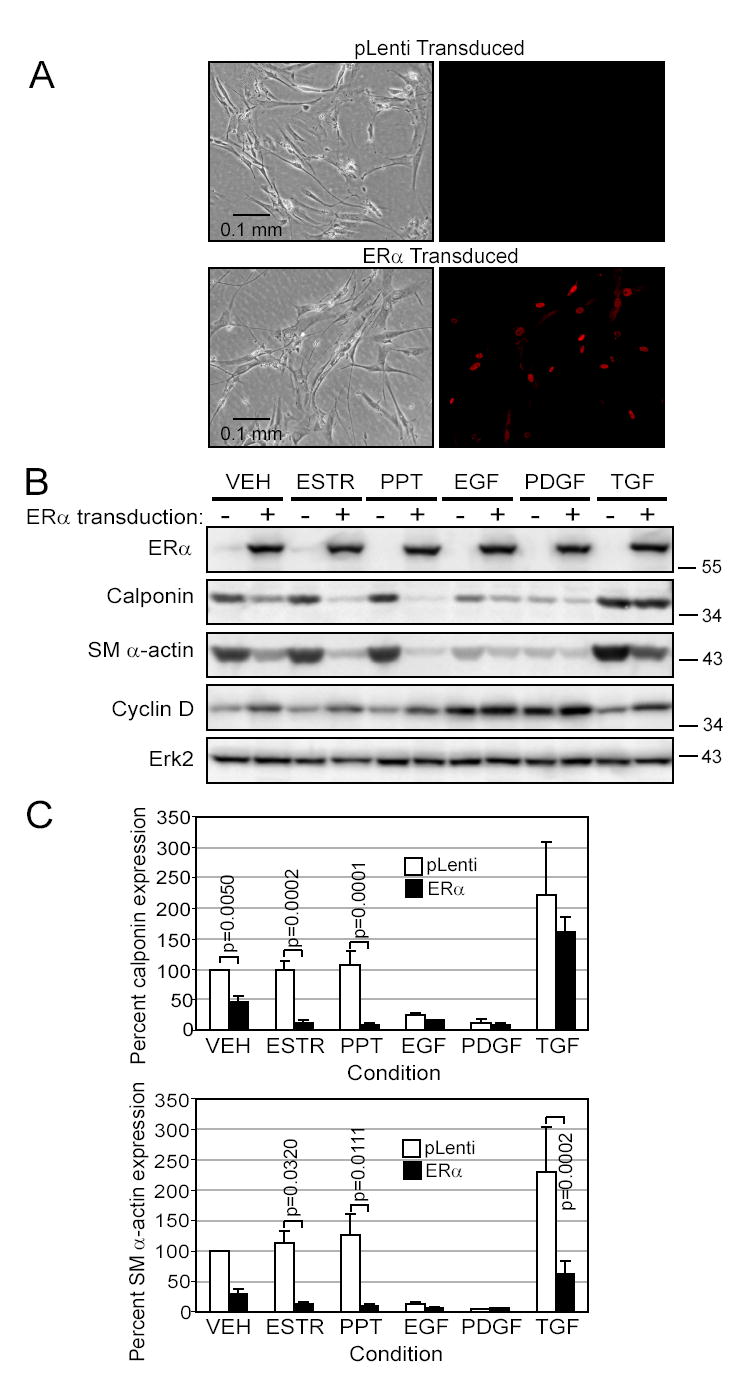
SMC transduced with either an empty vector (pLenti) or ERα cDNA were exposed for 10 days in EBM-PRF with various stimuli as indicated in methods and Figure 2 A. Immunofluorescent staining of ERα illustrates the efficiency of transduction. B. Western blots demonstrate expression of ERα, loss of calponin and SM α-actin contractile proteins and upregulation of cyclin D1 in the ERα-transduced cells. C. Calponin and SM α-actin densitometry were expressed as a percent of the pLenti VEH control (□pLenti or ■ERα, mean±SEM of 4 (calponin) or 5 (SM α-actin)).
Interruption of ERα by siRNA augmented TGFβ1-induced differentiation
Since high native levels of ERα correlated with low levels of SMC differentiation markers, we next reduced ERα expression through siRNA to enhance their differentiation program. High transfection efficiency was obtained using pmaxGFP plasmid DNA in co-transfections (Figure 4A). A reduction in ERα protein was confirmed in the siRNA-transfected cells compared to the empty vector control (Figure 4A).
Figure 4. Reduction of ERα expression by siRNA induces differentiation.
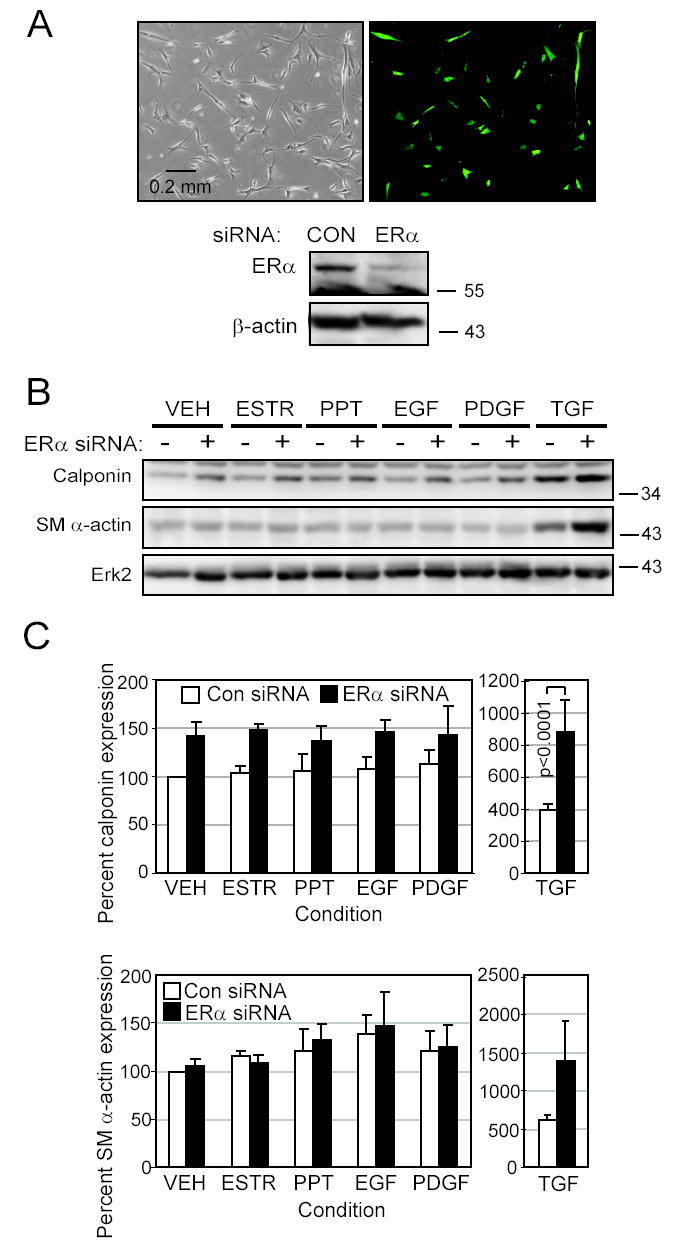
SMC expressing high levels of ERα were transfected with either empty vector or plasmid producing ERα siRNA. A. Transfection of pmaxGFP into high-ERα SMC illustrated the high transfection efficiency. Reduction in ERα protein levels by siRNA in cells starved for 10 days was demonstrated by Western blotting. β-actin was used as a loading control. B. Transfected cells were exposed for 10 days in EBM-PRF with stimuli as indicated in methods. Western blotting was performed to detect the differentiation markers calponin and SM α-actin. C. Calponin and SMα-actin densitometry as a percent of the empty vector VEH control condition was calculated (mean±SEM of 4 replicates, transfected with either □empty vector or ■ERα siRNA).
We found that reduced ERα expression led to higher levels of differentiation markers. ERα siRNA upregulated calponin expression overall (p=0.0038), with significant pairwise difference occurring in TGFβ1-treated cells (Figure 4B and C). Although ERα siRNA slightly raised SM α-actin levels in cells incubated with TGFβ1, the increase was not significant (p=0.1542).
Ligand activation of ERα inhibited Smad2 phosphorylation
We next investigated ligand-dependent ERα activation by ESTR and PPT, and the ligand-independent activation by EGF and PDGF. A low level of ERα phosphorylation was observed in the ERα-transduced cells in the VEH-treated condition, whereas ESTR and PPT preferentially phosphorylated ERαS118, and EGF and PDGF phosphorylated ERαS167 (Figure 5A). TGFβ1 caused no activation above VEH control of either serine residue.
Figure 5. Ligand activation of ERα causes nuclear translocation and inhibition of Smad2 phosphorylation.
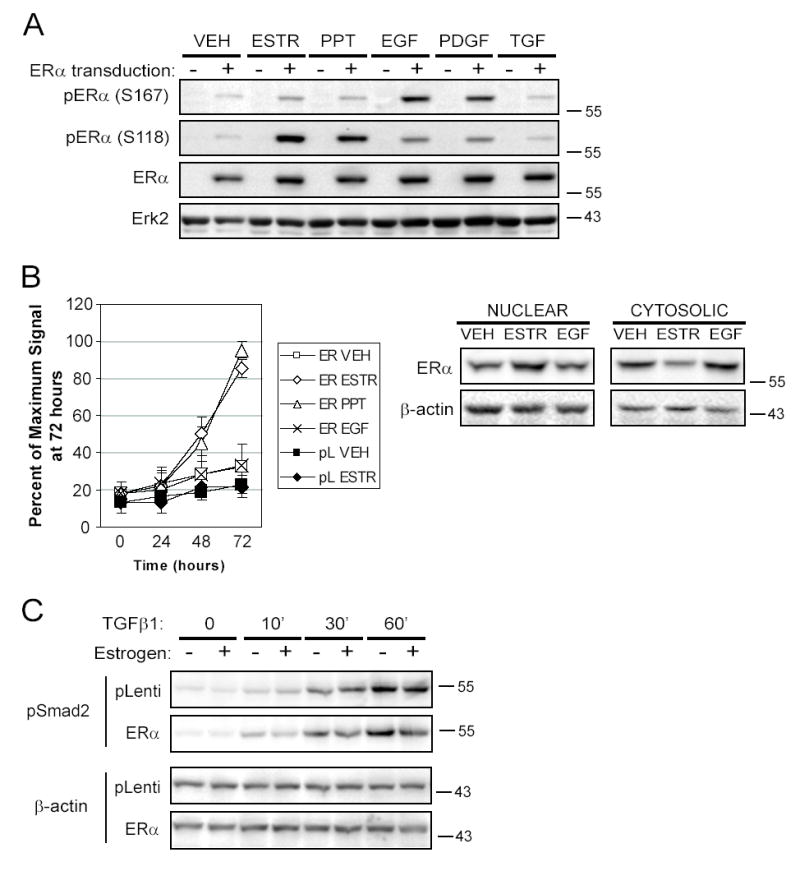
SMC transduced with pLenti or ERα virus were stably selected with blasticidin and activated with the various stimuli as indicated previously. A. Western blots from cells activated for 20 minutes were sequentially immunoblotted with antibodies to phospho-ERα (S167), phospho-ERα (S118), ERα, and Erk2, with stripping of the membrane between antibodies. Shown is a representative figure from two experiments. B. Stably transduced cells were transfected with an estrogen response element reporter construct producing secreted alkaline phosphatase (SEAP) and were treated as described for up to three days. Aliquots of the media were analyzed for SEAP each day and luminescent signal was normalized as a percent of the maximum signal achieved on day 3 (mean±SEM of 4 experiments). Western blotting of nuclear and cytosolic lysates from SMC transduced with ERα and activated for 20 minutes as indicated demonstrated nuclear translocation of ERα when activated by ESTR. C. SMC stably transduced with pLenti or ERα were pre-incubated with ESTR (+) or ETOH (−) for 30 minutes, then activated with TGFβ1 for the time indicated. Phosphorylated Smad2 and β-actin as a loading control were detected by Western blotting (representative of two experiments).
In contrast to the activation by phosphorylation seen with EGF and PDGF, only ESTR and PPT activated transcription of an ERE reporter construct (Figure 5B, p=0.0013 for ESTR and p=0.0008 for PPT compared to VEH). Activation for up to ten days with EGF, PDGF-BB or TGFβ1 caused no detectable signal above vehicle control samples (data not shown). As a partial explanation for the transcriptional inactivity of ERα phosphorylated by growth factors, we found that stimulating the cells with ESTR, but not EGF, PDGF-BB or TGFβ1, for 20 to 60 minutes caused nuclear translocation of ERα (Figure 5B and data not shown).
Since our differentiation analysis suggested that TGFβ1 elevated SMC differentiation markers in the presence of ERα but full expression of these markers required lower levels of ERα, we examined whether ERα inhibited TGFβ signaling by interfering with Smad activation, as previously described18. Since ESTR or PPT potently reduced SMC differentiation, we determined whether ESTR inhibited TGFβ1 signaling through Smads. TGFβ1 induced the phosphorylation of Smad2 in pLenti or ERα-transduced SMC for up to 60 minutes (Figure 5C). However, Smad2 phosphorylation was reduced if the ERα-transduced cells were preincubated with ESTR 30 minutes before activation. The relevance of TGFβ1 to the differentiation of SMC was examined by measuring whether the cells spontaneously produced TGFβ1 and whether this production correlated to cellular differentiation. Active TGFβ1 was detected in the supernatant of pLenti-transduced SMC at 49.0±27.9 pg/ml in the VEH control condition and 67.2±45.4 and 110.7±42.1 after ESTR or PPT incubation, respectively (no significant differences, n=2, mean±SEM). Transduction of ERα in the cells did not alter TGFβ1 production suggesting ERα expression altered the response to TGFβ1 (45.6±23.1, 49.8±33.6 and 112.0±39.7 for VEH, ESTR, and PPT exposed cells, respectively, n=2).
Ligand Activation of Estrogen Receptor Initiated Apoptosis
Since estrogen inhibits the growth of SMC and causes apoptosis 2, 19, we examined the initiation of apoptosis in the presence of ERα agonists. Indeed, SMC transduced to express ERα had evidence of caspase-3 activity when stimulated with ESTR or PPT (Figure 6A, p=0.0231 and p=0.0646, respectively, n=2).
Figure 6. ERα ligands cause apoptosis of aortic SMC.
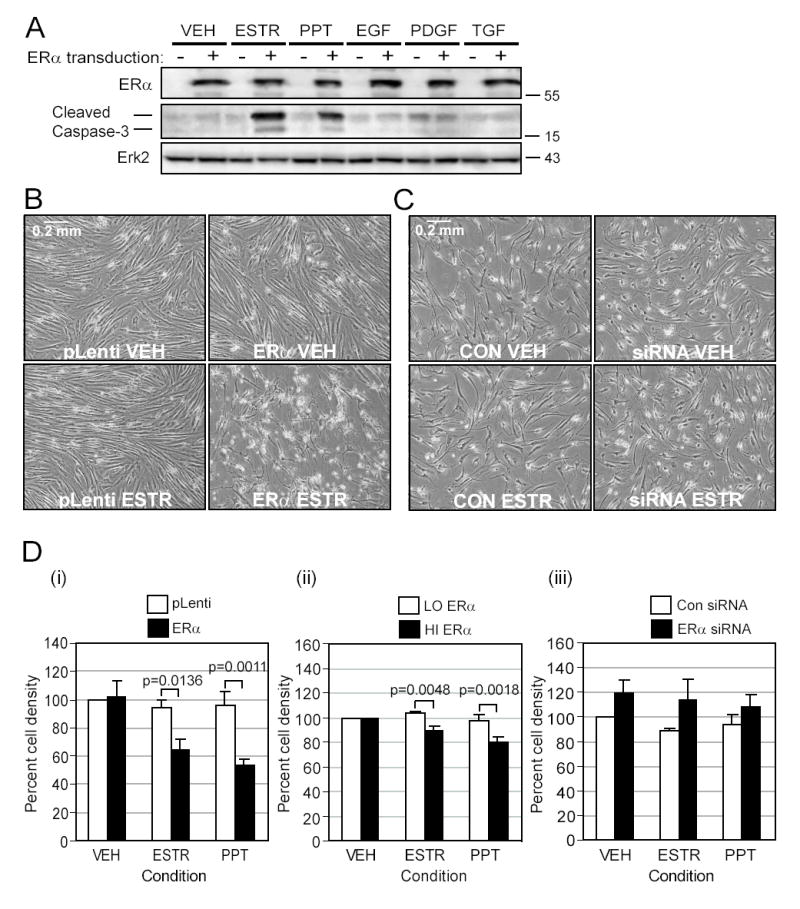
A. SMC virally transduced with either pLenti or pLenti-ERα were exposed for 10 days in EBM-PRF with various stimuli as indicated in methods. The cleaved or active form of caspase-3 was detected at 17 and 19kD by SDS-PAGE Western blots. B. Representative phase contrast photos were taken of SMC containing low ERα levels transduced with either pLenti or ERα and treated with VEH or ESTR for 10 days. C. Representative photos are shown of cells with high endogenous levels of ERα transfected with either control or ERα siRNA plasmid and treated with VEH or ESTR for 10 days. D. Phase contrast photos were quantitated for pixel density of live (gray) cells in the following groups: (i) pLenti- (□) or ERα-transduced cells (■) activated by ERα agonists (n=5); (ii) SMC natively expressing low ERα levels (□) or high ERα levels (■)(n=4); or (iii) SMC expressing high ERα levels transfected with empty plasmid (□) or siRNA plasmid (■) (n=4).
The effects of ERα expression on cell growth were apparent by cell detachment when ERα-transduced SMC were treated with ESTR (Figure 6B, lower right panel) or PPT (picture not shown) indicating that cells were undergoing apoptosis. Consequently, fewer ERα-transduced cells were counted after ESTR or PPT treatment compared to pLenti-transduced cells (p=0.0006 overall, Figure 6D (i)). Consistent with this observation, cells natively expressing high levels of ERα had significantly lower cell densities than the low-ERα cells when treated with ESTR or PPT (p=0.0003 overall, Figure 6D (ii)). Finally, a small increase in cell density was found overall (p=0.0274) when the high-ERα SMC were transfected with ERα siRNA, although no individual paired comparisons were significant (Figure 6C and D (iii)).
Discussion
The present study extends the role of ERα in vascular SMC beyond its ability to inhibit growth. To understand differences in SMC status between men and women, we characterized aortic SMC differentiation and ERα expression in these two groups. We detected significantly higher levels of ERα in SMC from our female donors compared to SMC from male donors. The inverse was true for differentiation markers, however, as cells from men expressed greater levels of SM α-actin and calponin protein under starved conditions, providing a connection between ERα expression and differentiation.
We analyzed the effect of two ERα ligands and three growth factors on cell populations containing the lowest and highest levels of ERα. SMC differentiation markers remained high for cells natively expressing low amounts of ERα whether incubated with VEH, ERα agonists, or TGFβ1. However, EGF and PDGF decreased SM α-actin and calponin levels in these cells, similar to published accounts 20. In contrast, cells expressing high native ERα had a low level of SM α-actin and calponin under most conditions except when treated with TGFβ1. Similar findings were observed in cells virally transduced with ERα, which resulted in their dedifferentiation. Only TGFβ1 could partially overcome the inhibitory effect of ERα. These data indicated that ERα may play a role in causing the low contractile protein levels detected in SMC from women. The ability of ERα to inhibit differentiation was unexpected, since ERα is known to inhibit growth and would be expected to induce differentiation. In contrast, ERα caused an increase in cyclin D1 expression, indicating that growth inhibition did not align with quiescence. To confirm this biological role for ERα, we found that reduction in ERα resulted in greater contractile protein expression, especially in the presence of TGFβ1.
Several possible pathways could be involved in the reduced SMC differentiation caused by ERα. Inhibition of cell cycle regulators and activation of proliferation genes such as cyclin D are known to occur in ERα-positive breast cancer cells exposed to ESTR 21. Similar changes in SMC could induce a phenotypic switch from differentiated to proliferating or migratory SMC. Alternatively, ERα may inhibit transcription by shunting coactivator proteins such as p300/CBP away from other transcription factors, some of which are necessary for smooth muscle gene expression 22–24. ERα activates transcription at estrogen response elements on DNA, but is known to suppress the TGFβ1/Smad pathway by binding to and repressing Smad 2 and 3, positive regulators of contractile protein transcription in SMC 18, 25. In agreement, Smad2 phosphorylation was inhibited by estrogen in ERα-transduced SMC in the current study. Since the SMC released detectable levels of active TGFβ1, the ability of ERα to inhibit Smad-regulated differentiation is a likely mechanism of action.
Cytoplasmic signaling pathways activated by ERα including phosphatidylinositol 3-kinase and Akt, growth factor receptor autophosphorylation, mitogen activated protein kinases (MAPK), and src kinases can contribute to SMC dedifferentiation 19, 26–28. A positive feedback loop also exists in which S118 of ERα is phosphorylated by ESTR and MAPK, whereas S167 of ERα is phosphorylated through the Akt pathway16, 29, 30. Depending on the stimulus, we saw preferential phosphorylation of ERα epitopes in SMC indicating that upstream and downstream signaling events likely differed in these cells. Only ERα ligands caused nuclear translocation and transcriptional activity at an ERE.
Many studies show that ESTR induces apoptosis through ERα in SMC 2, 19. We found that ESTR and PPT significantly reduced cell density of native and transduced cells expressing high levels of ERα, while inhibition of ERα by siRNA increased cell density.
Our results may explain some differences in coronary events in women and men. ERα activation in an affected coronary artery may cause the dedifferentiation and migration of SMC into the intima, causing microvessel dysfunction. Our observation of apoptosis of SMC after estrogen exposure could partly explain why post-menopausal hormone replacement therapy causes higher rates of myocardial infarction through thinning of collagen and rupture of plaques.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants (HL63800-05, HL67176-04, HL70294-03 and HL066108-04) to Clay B. Marsh and NIH Individual National Research Service Award, 5F32 HL09550, and American Heart Association Ohio Affiliate Postdoctoral Fellowship Award, 9920597V, to Christine Roos (Montague). The authors would like to acknowledge the advice of Arthur R. Strauch and Tim D. Eubank, both of Ohio State University.
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Conflict of Interest disclosure No conflict of interest exists among the authors and any company or advisory council relevant to this work.
References
- 1.American Heart Association . Heart Disease and Stroke Statistics - 2005 Update. Dallas, Texas: 2005. [Google Scholar]
- 2.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 3.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 4.Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, Vittinghoff E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605–613. doi: 10.1001/jama.280.7.605. [DOI] [PubMed] [Google Scholar]
- 5.Grodstein F, Manson JE, Stampfer MJ. Hormone Therapy and Coronary Heart Disease: The Role of Time since Menopause and Age at Hormone Initiation. J Womens Health (Larchmt ) 2006;15:35–44. doi: 10.1089/jwh.2006.15.35. [DOI] [PubMed] [Google Scholar]
- 6.Patel H, Rosengren A, Ekman I. Symptoms in acute coronary syndromes: does sex make a difference? Am Heart J. 2004;148:27–33. doi: 10.1016/j.ahj.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Clayton TC, Pocock SJ, Henderson RA, Poole-Wilson PA, Shaw TR, Knight R, Fox KA. Do men benefit more than women from an interventional strategy in patients with unstable angina or non-ST-elevation myocardial infarction? The impact of gender in the RITA 3 trial. Eur Heart J. 2004;25:1641–1650. doi: 10.1016/j.ehj.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 8.Bugiardini R, Bairey Merz CN. Angina with “normal” coronary arteries: a changing philosophy. JAMA. 2005;293:477–484. doi: 10.1001/jama.293.4.477. [DOI] [PubMed] [Google Scholar]
- 9.Hochman JS, Tamis JE, Thompson TD, Weaver WD, White HD, Van de WF, Aylward P, Topol EJ, Califf RM. Sex, clinical presentation, and outcome in patients with acute coronary syndromes. Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes IIb Investigators. N Engl J Med. 1999;341:226–232. doi: 10.1056/NEJM199907223410402. [DOI] [PubMed] [Google Scholar]
- 10.Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–1363. doi: 10.1161/01.cir.93.7.1354. [DOI] [PubMed] [Google Scholar]
- 11.Burke AP, Farb A, Malcom G, Virmani R. Effect of menopause on plaque morphologic characteristics in coronary atherosclerosis. Am Heart J. 2001;141:S58–S62. doi: 10.1067/mhj.2001.109946. [DOI] [PubMed] [Google Scholar]
- 12.Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN, Virmani R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler Thromb Vasc Biol. 2002;22:1642–1648. doi: 10.1161/01.atv.0000034021.92658.4c. [DOI] [PubMed] [Google Scholar]
- 13.Waddell TK, Dart AM, Gatzka CD, Cameron JD, Kingwell BA. Women exhibit a greater age-related increase in proximal aortic stiffness than men. J Hypertens. 2001;19:2205–2212. doi: 10.1097/00004872-200112000-00014. [DOI] [PubMed] [Google Scholar]
- 14.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology; 2000; Totowa, NJ.: Humana Press; [DOI] [PubMed] [Google Scholar]
- 15.Sankoh AJ, Huque MF, Dubey SD. Some comments on frequently used multiple endpoint adjustment methods in clinical trials. Stat Med. 1997;16:2529–2542. doi: 10.1002/(sici)1097-0258(19971130)16:22<2529::aid-sim692>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 16.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 17.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272:10948–10956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–42914. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- 19.Mori-Abe A, Tsutsumi S, Takahashi K, Toya M, Yoshida M, Du B, Kawagoe J, Nakahara K, Takahashi T, Ohmichi M, Kurachi H. Estrogen and raloxifene induce apoptosis by activating p38 mitogen-activated protein kinase cascade in synthetic vascular smooth muscle cells. J Endocrinol. 2003;178:417–426. doi: 10.1677/joe.0.1780417. [DOI] [PubMed] [Google Scholar]
- 20.Dandre F, Owens GK. Platelet-derived growth factor-BB and Ets-1 transcription factor negatively regulate transcription of multiple smooth muscle cell differentiation marker genes. Am J Physiol Heart Circ Physiol. 2004;286:H2042–H2051. doi: 10.1152/ajpheart.00625.2003. [DOI] [PubMed] [Google Scholar]
- 21.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 22.Speir E, Yu ZX, Takeda K, Ferrans VJ, Cannon RO., III Competition for p300 regulates transcription by estrogen receptors and nuclear factor-kappaB in human coronary smooth muscle cells. Circ Res. 2000;87:1006–1011. doi: 10.1161/01.res.87.11.1006. [DOI] [PubMed] [Google Scholar]
- 23.Cao D, Wang Z, Zhang CL, Oh J, Xing W, Li S, Richardson JA, Wang DZ, Olson EN. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol Cell Biol. 2005;25:364–376. doi: 10.1128/MCB.25.1.364-376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishihara A, Hanai JI, Okamoto N, Yanagisawa J, Kato S, Miyazono K, Kawabata M. Role of p300, a transcriptional coactivator, in signalling of TGF-beta. Genes Cells. 1998;3:613–623. doi: 10.1046/j.1365-2443.1998.00217.x. [DOI] [PubMed] [Google Scholar]
- 25.Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 26.Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003;278:2118–2123. doi: 10.1074/jbc.M210828200. [DOI] [PubMed] [Google Scholar]
- 27.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 28.Kawai-Kowase K, Sato H, Oyama Y, Kanai H, Sato M, Doi H, Kurabayashi M. Basic fibroblast growth factor antagonizes transforming growth factor-beta1-induced smooth muscle gene expression through extracellular signal-regulated kinase 1/2 signaling pathway activation. Arterioscler Thromb Vasc Biol. 2004;24:1384–1390. doi: 10.1161/01.ATV.0000136548.17816.07. [DOI] [PubMed] [Google Scholar]
- 29.Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, Coombes RC, Ali S. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002;21:4921–4931. doi: 10.1038/sj.onc.1205420. [DOI] [PubMed] [Google Scholar]
- 30.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]