

Abstract
We have prepared human recombinant antibody molecules against the glycoprotein antigen of the rabies virus (GPRV) based on the single chain variable fragment (scFv) format. Anti-GPRV scFvs were selected from a human synthetic scFv phage display library with a repertoire of approximately 109 specificities. After three rounds of selection against the PV11 strain of the virus, 40% of the clones tested recognized the rabies antigen. Of the 20 positive clones that were sequenced, five distinct sequences were identified. These distinct scFvs were cloned into a mammalian expression vector carrying the human IgG1 Fc region. The specificity of the resulting scFv-Fc molecules for GPRV was established by ELISA, dot blot and western blot analyses and membrane immunofluorescence. Two of the scFv-Fc fusion proteins neutralized the PV11 strain in a standard in vivo neutralization assay where the virus was incubated with the scFv-Fc molecules before intracranial inoculation in mice. These anti-GPRV scFv-Fc molecules have the potential to be used as an alternative to the presently available HRIG, for use in post-exposure preventive treatment.
Keywords: rabies, glycoprotein, human scFv, phage display, scFv-Fc, neutralizing
Introduction
Rabies, once established, is universally fatal. The most common mode of infection in man is through dog bite. It remains one of the major causes of mortality in India with about 30 000 deaths per year and another 2·5–3 million people requiring post-exposure preventive measures [1–3]. The most effective preventive measure is the timely administration of rabies immunoglobulin as well as vaccine. However, the recommended human rabies immunoglobulin (HRIG) is very expensive, not easily available in India and carries the risk of infections associated with human material. Equine anti-rabies immunoglobulin (ERIG) is produced in the country but the quantity falls far short of the requirement. Of the 1500 l of anti-rabies serum required in India for the estimated 0·3 million severe bite cases alone, about 120 l is produced in the country [Dr D.K. Sood, CRI Kasauli, India, personal communication; 4]. Equine antibodies also have the disadvantage of being foreign and therefore may cause severe hypersensitivity reactions.
Phage display technology [5–7] has facilitated the production of human antibodies or their fragments in vitro. Recombinant human antibody fragments have been proven promising in vitro as well as in phase I and II clinical trials in certain cancers [8–11]. However, the potential of antibody based recombinant molecules in the prevention and therapy of infectious diseases remains unexplored despite these diseases being the major cause of morbidity and mortality in developing countries.
We have exploited a library of synthetic single chain variable fragments (scFv) of human antibody molecules for the selection of scFvs against the glycoprotein antigen of the rabies virus (GPRV). The present paper describes the selection of these fragments and characterization of scFvs fused with the constant region of human IgG1. These constructs have the potential to be used in prevention and/or therapy of rabies.
Materials and methods
Virus and Antigen, PV11, a fixed strain of rabies virus obtained from the Central Research Institute, Kasauli, India, was grown according to the published protocol [12] in the Vero cell line (NFATCC, Pune, India). The supernantant was collected every three days and fresh medium added until the cells degenerated. Before collection, the cells were checked for virus infection by immunofluorescence. A single cell suspension of infected and uninfected Vero cells was distributed in the wells of Teflon coated slides and fixed in cold acetone at −20°C for 1 h. Uninfected Vero cells and cells after infection were checked for the presence of PV11 by an immunofluorescence assay. Commercially available human anti-rabies immunoglobulin, HRIG (Berirab® P, Marburg, Germany, marketed by Hoechst India Ltd) was used to detect the virus, followed by rabbit anti-human IgG conjugated with FITC (Dakopatts, Glostrup, Denmark). The slides were mounted in 50% glycerol (in PBS) and viewed under an epi-fluorescence microscope (Carl Zeiss, Jena, Germany) with the appropriate filters (Excitation filter 450–490, chromatic beam splitter 510, barrier filter 515–565).
Virus was concentrated using a 300-kD cutoff membrane (Sartorius, Gottingen, Germany) followed by ultracentrifugation at 50 000×g for 2 h at 4°C. The viral pellet was washed with PBS and ultracentrifuged again. The glycoprotein antigen of the rabies virus (GPRV) was isolated using Triton X-100 [13]. The protein concentrations of the virus preparation and GPRV were determined by the Lowry method [14] and the Bio-Rad detergent compatible protein estimation kit (Bio-Rad Laboratories, Hercules, USA), respectively.
E. coli strains
TG1 and HB2151 (The two strains are fully described in Hoogenboom et al. 1991) [15].
Selection of anti-GPRV scFv
A human synthetic scFv phage display library with approximately 109 antigen binding specificities (Griffin et al. unpublished observation, MRC Centre, Cambridge, UK) was used to select the anti-GPRV scFvs The purified PV11 virus was used as the selecting antigen. Four rounds of selection were carried out as described [16]. Immunotubes (Maxisorp, Roskilde, Nunc, Denmark) were coated at a concentration of 100 µg/ml, 50 µg/ml, 50 µg/ml and 25µg/ml of PV11 in 0·1 m NaHCO3 for the 1st, 2nd, 3rd and 4th selections, respectively. Titration for phage infectivity (transducing units, t.u.) was carried out after each round of selection and transduction [16].
Screening for anti-GPRV phage displayed scFvs
Supernatants from TG1 clones from the 3rd and 4th rounds of selection were screened by ELISA for PV11 binding phage. Briefly, 200 µl of 2X TY (16 g tryptone, 10 g yeast extract, 5 g NaCl (DIFCO Laboratories, Detroit, MI, USA) in 1 l double distilled water) containing 100 µg/ml ampicillin was added to each well of two 96 well round bottom microtitre plates. Colonies were picked from plates used for estimation of transducing units and inoculated into separate wells of the microtitre plates. Two wells in each plate were left uninoculated as controls in subsequent assays. Each colony was also replica plated onto TYE-Agar (10 g tryptone, 5 g yeast extract, 8 g NaCl, 15 g agar (DIFCO Laboratories, USA) in 1 l double distilled water) containing 100µg/ml ampicillin and incubated overnight at 37°C. The microtitre plates were incubated for 1 h, shaking at 37°C. M13KO7 helper phage was added at 109 plaque forming units (p.f.u.) per well and incubated standing at 37°C for 45min, followed by shaking for 45min at 37°C. Supernatants were discarded after centrifugation for 10min at 4000rpm. The bacterial pellets were resuspended in 200 µl 2xTY containing 100 µg/ml ampicillin and 25 µg/ml kanamycin and incubated shaking at 30°C overnight. The cells were spun down and the supernatants tested for binding to PV11 by ELISA.
Plates (High binding, Costar, Cambridge, MA, USA) were coated with PV11 at a concentration of 10 µg/ml in 0·1 m NaHCO3 overnight at 4°C followed by blocking with phosphate buffered saline (PBS) pH 7·2 containing 2% skimmed milk powder (MPBS) for 2 h at 37°C. Phage clone supernatants containing 2% skimmed milk (9:1 supernatant:20% skimmed milk in PBS) were detected by rabbit anti-fd IgG (Sigma-Aldrich, Steinheim, Germany) followed by goat anti-rabbit IgG-HRP conjugate (Bangalore Genei, Bangalore, India). After addition of the chromogenic substrate (O-phenylenediamine (OPD, 4 mg) and H2O2 (15µl of 30% H2O2) in 10 ml citrate phosphate substrate buffer (pH 5)), the reaction was stopped with 2·5N H2SO4 and the absorbance read at 492 nm.
Sequencing scFv DNA
Twenty clones with high absorbance readings were sequenced using the Perkin Elmer Big Dye Terminator Ready Reaction sequencing kit (PE Biosystems, Warrington, UK) with the ABI™ 373 DNA sequencer. The primers used to sequence the scFv inserts in the pHEN2 vector are described below. The sequences were analysed using the GCG Sequence Analysis Software. The variable region sequences obtained were analysed using the human immunoglobulin database available at http://www.mrc-cpe.cam.ac.uk/imt-doc/public/INTRO.htm
Primers
Primer used to sequence VH: FOR_LinkSeq 5′ GCCACCTCCGC CTGAACC
Primer used to sequence VL: pHEN-SEQ: 5′ CTATGCGGCC CCATTCA
Primers used to clone anti-rabies scFvs into the vector SIgpIgPlus:
Igfusfor 5′ TAGAGATATCACCTAGGACGGTCAG
Igfusbk 5′ GGATAAGCTTGAGGTGCAGCTGGTG
Soluble expression of scFv
The 20 phage clones that were sequenced were also selected for induction of soluble scFv production. Briefly, plasmid mini-preparations were made from each clone (QIAgen spin Mini-prep kit, USA) and electroporated into the nonsuppressor E. coli strain HB2151. One colony with an apparently intact scFv insert (checked by PCR, results not shown) from each set was selected. 50 ml exponential cultures (OD600 = 0·9) of each clone grown in 2xTY containing 100 µg/ml ampicillin, were induced with 1 mm isopropylthio-galactoside (IPTG, Promega, USA) overnight at 30°C and centrifuged at 6000rpm for 20min. Protein precipitates obtained after saturating the supernatants with 50% ammonium sulphate (overnight, 4°C) were centrifuged at 6000rpm at 4°C, resuspended in 2·5 ml PBS and dialysed against PBS overnight at 4°C. Periplasmic preparations were made by incubating the bacterial pellets on ice in PBS containing 1 mm EDTA for 1 h, followed by centrifugation at 6000rpm for 20 min at 4°C.
Screening by ELISA for soluble anti-PV11 scFvs
ELISA plates were coated with 50 µg/ml PV11. After blocking, soluble scFvs in concentrated supernatants and periplasmic preps were detected with the mouse monoclonal antibody 9E10 (recognizes the C-terminal c-myc tag of the scFv) followed by incubation with anti-mouse IgG-HRP (Dakopatts)
Western blot assay of periplasmic preparations
10 µl periplasmic samples were electrophoresed through a 15% resolving SDS-polyacrylamide gel and transferred onto PVDF membrane (Millipore, Bedford, MA, USA) [17,18]. The blots were blocked with 3% MPBS and incubated with 9E10 followed by anti-mouse IgG-HRP conjugate and developed using the BioRad chemiluminescence kit (Bio-Rad Laboratories).
Anti-PV11 – scFv-IgG1 Fc fusion proteins (scFv-Fc)
Primers were designed based on the scFv sequences, with restriction sites for the enzymes HindIII and EcoRV (see above) in the back and forward primers, respectively (GibcoBRL, Life Technologies, USA). Four distinct scFv sequences, amplified by PCR, were cloned into the mammalian expression vector Signal pIgPlus (Ingenius, USA). The plasmids were purified and transfected into E. coli strain TG1. Plasmid maxi-preps prepared from single clones (QIAgen, Hilden, Germany) were electroporated into COS-7 cells in order to express scFv fusion proteins containing the human IgG1 Fc portion at their C-termini. The cells were grown in DMEM supplemented with foetal calf serum (GibcoBRL) in the presence of the selective antibiotic G418 (Geneticin, GibcoBRL). Two clones (III-20 and IV-67) were randomly chosen for further characterization. Culture supernatants were concentrated 100 fold by precipitation with ammonium sulphate (60% saturation), dialysed against PBS and further concentrated through 3 kD cut-off membranes (Centriplus 3, Amicon, Beverley, MA, USA). Supernatant containing Fc alone (from vector alone transfectants) was similarly processed and used as a negative control.
Quantitative assay of recombinant protein
ELISA plates were coated with 10 µg/ml mouse anti-human IgG in 0·1 m NaHCO3 overnight at 4°C and blocked with 2% MPBS at 37°C for 2 h. Standards (human IgG1 (Sigma) serially diluted twofold) and concentrated supernatants were added to their respective wells and incubated at 37°C for 1 h, followed by rabbit anti-human IgG-HRP (Dakopatts). A standard curve was prepared (concentration of standards versus absorbance) and the concentrations of the samples were extrapolated from the curve.
Specificity of the fusion proteins scFv-Fc
All the assays included the commercially available human rabies immunoglobulin (HRIG, Hoechst, Germany) as positive control for the fusion proteins.
ELISA
50 µg/ml PV11 was coated overnight at 4°C. After blocking, concentrated recombinant proteins were added to their respective wells and detected with anti-human IgG-HRP conjugate.
Dot blot assay
5 µl concentrated supernatant and 0·5 µg BSA (5 µl) were dotted onto nitrocellulose membrane and the blot was developed using the BioRad chemiluminescence kit (BioRad Laboratories, USA) in accordance with the kit protocol. Anti-human IgG-HRP (Dakopatts, Denmark) was used to detect the Fc portion of the recombinant molecules.
Western blot analysis
GPRV and BSA were electrophoresed through a 10% SDS-PAG and transferred onto PVDF membrane. GPRV and BSA were probed with the fusion proteins III-20 Fc, IV-67 Fc, the negative control Fc and the positive control HRIG.
Membrane immunofluorescence
The two scFv-Fc preparations, III-20-Fc and IV-67-Fc, the Fc control and the positive control HRIG were added to PV11 infected (72 h post-infection, > 95% cells infected with the virus) and uninfected live Vero cells in suspension and incubated for 45min on ice. Cells were washed with PBS and similarly incubated with rabbit anti-human IgG-FITC (Sigma). The cells were washed, resuspended in 2% paraformaldehyde in PBS, mounted on glass slides and observed under an epi-fluorescence microscope (Carl Zeiss, Germany)
Neutralization assay
A standard in vivo neutralization assay was carried out in the mouse model as described by Fitzgerarald [19]. HRIG was used as the reference serum. Serial fivefold dilutions of HRIG were made ranging from 1/500 to 1/125000 in PBS. To this, 50 LD50 units of PV11 (titrated prior to the neutralization assay by Fitzgerald's method; LD50 calculated by the method of Reed and Muench [20]) at an equal volume were added resulting in the dilution of both serum and virus by a factor of 2. The final dilutions of HRIG ranged from 1/1000 to 1/625000 with the virus at 25LD50 units. Neat preparations of scFv-Fcs (III-20-Fc and IV-67-Fc) were also mixed with an equal volume of the virus at 50 LD50 units. Three dilutions of virus, 25 LD50, 2·5 LD50 and 0·25 LD50, were included as virus controls. Fusion proteins without virus were also included to rule out toxicity of the recombinant preparations. All the vials were incubated for 90 min at 37°C and then chilled on ice immediately. Each animal in each group consisting of 10 outbred Swiss mice (3–4-week-old, 14–16 g each) was inoculated intracerebrally with 25 µl of the respective preparations after anaesthetizing with ether. Deaths till day five were disregarded. The number of mice surviving in each group on day 14 was recorded. The 50% end point for neutralization for HRIG was calculated by the method of Reed and Muench [20].
Results
More than 95% Vero cells became infected with the PV11 strain of rabies virus 72 h after exposure to virus (Fig. 1). No fluorescence was detected in the control cells (not shown).
Fig. 1.

Immunofluorescence staining (FITC) of Vero cells 72 h after infection with the PV11 strain of rabies virus. Magnifications shown are as on the film. Single cell suspension of Vero cells (uninfected and infected with PV11 virus) were fixed in acetone for 1 h at −20°C. Virus was detected using the commercially available human rabies immunoglobulin (HRIG) followed by FITC labelled anti-human IgG. The cells were observed under an epifluorescence microscope
The protein concentrations of PV11 and GPRV were found to be 0·5 mg/ml and 1·3 mg/ml, respectively. These preparations were used throughout the study.
Anti-GPRV scFv selection and screening
The total number of bacterial colonies is proportional to the number of phage particles eluted from the PV11 coated immunotube, and is expressed as transducing units. It was found to be 1·92 × 107, 5·70 × 105, 1·05 × 108 and >10 8 after the first, second, third and fourth rounds of selection, respectively.
Supernatants of phage clones from the third and fourth selections were screened by ELISA for PV11 binding phage. An arbitrary cut-off of 0·5 OD492 above background (0·125) was considered positive (absorbance range of positive supernatants: 0·6–2·876). 37 of 92 supernatants (40·21%) from the third and 44 of 94 (45·83%) from the fourth selection bound to PV11.
Sequencing scFv insert DNA in the plasmid pHEN2
At least five distinct scFv DNA sequences were obtained from the 20 that were sequenced. Figure 2 shows the predicted scFv amino acid sequences. The VH and VL families to which the variable sequences belong are shown in Table 1.
Fig. 2.

Derived amino acid sequences of the five clones highlighted in Table 6. Clone IV-62 was not further characterized due to the presence of the amber codon (*). The upper and lower panels show sequences of the heavy and light chain variable regions, respectively.
Table 1.
VH and VL families of the anti-GPRV scFv sequences. The germline V gene locus which shows maximum homology with the V gene selected and its percentage homology with the segment is shown in brackets. The sequences were analysed using the human Immunoglobulin V-BASE database (http://www.mrc-cpe.cam.ac.uk/imt-doc/public/INTRO.html)
| Clone | VH (germline locus, % homology) | VL (germline locus, % homology) |
|---|---|---|
| IV-67 | VH3 (3–48, 98·98) | VL1 (1g, 97·61) |
| III-20 | VH3 (Orphon-HC16, 98·63) | VL3 (3l, 99·33) |
| III-30 | VH3 (3–20, 98·64) | VL1 (1g, 100) |
| III-35 | VH3 (Orphon-HC16, 98·63) | VL3 (3l, 98·97) |
Soluble expression of scFv
All periplasmic samples checked by Western Blotting were found to contain proteins of the correct size, approximately 26 kD (Fig. 3). However, binding to PV11 was not detectable by ELISA in either the concentrated supernatants or the periplasmic preparations (results not shown).
Fig. 3.
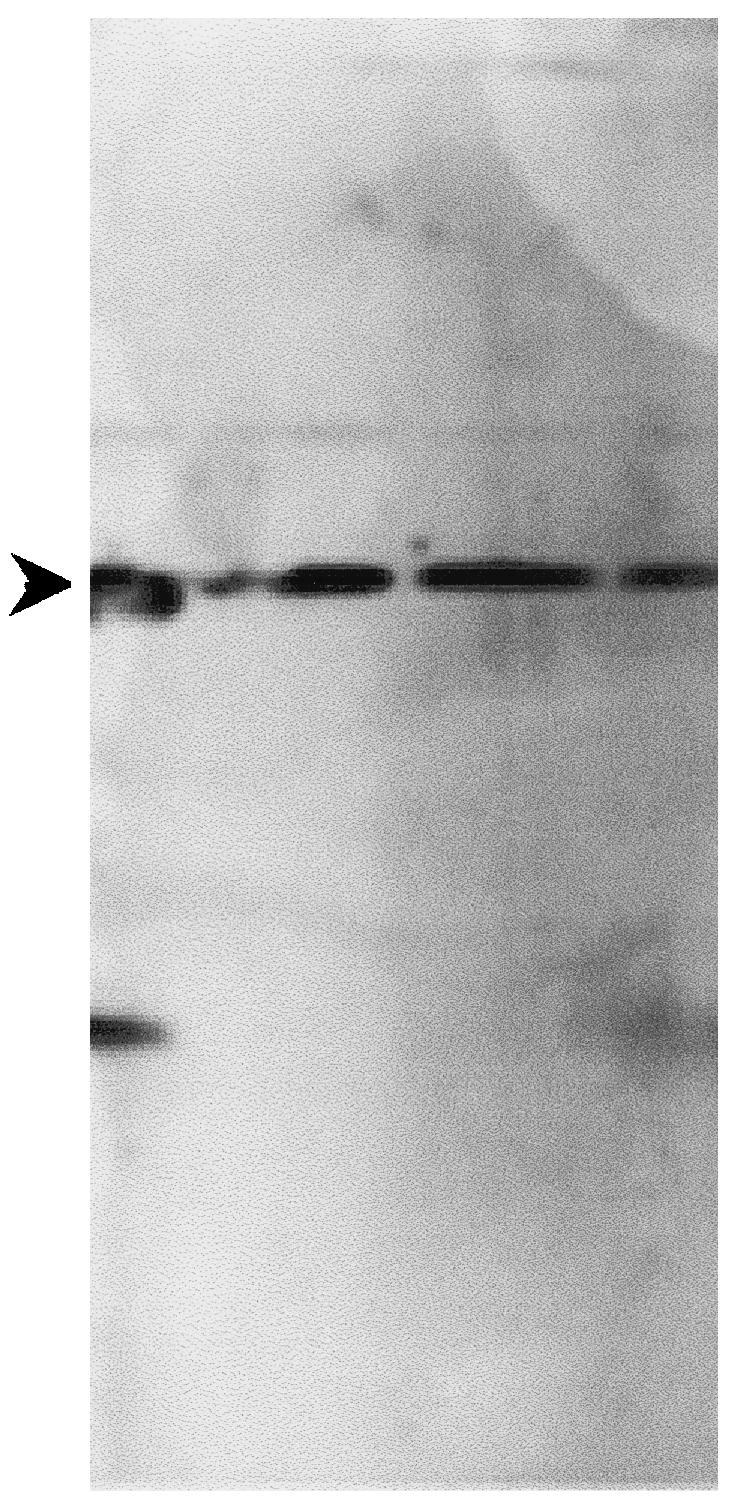
Western blot analysis of periplasmic preparations of HB2151 containing scFv inserts. One colony from each set of electroporation which had an intact scFv insert as determined from Fig. 4 was grown and induced with IPTG for scFv secretion. The periplasmic preparation was electrophoresed through a 15% SDS-polyacrylamide gel and transferred onto PVDF membrane. The c-myc tag at the 3′ end of scFvs was detected using the monoclonal antibody 9E10. All the colonies produced scFv as evidenced by bands at the correct position (approximately 26 kD), indicated by the arrow.
Anti-PV11 scFvñIgG1 Fc (scFv-Fc) fusion proteins
Clones III-20 and IV-67 were randomly picked for characterization. The presence of fusion proteins in concentrated COS-7 culture supernatants was confirmed by a band at approximately 55 kD, in a western blot assay with anti-human IgG-HRP (Fig. 4a). The corresponding band in a silver stained gel is shown in Fig. 4b.
Fig. 4.
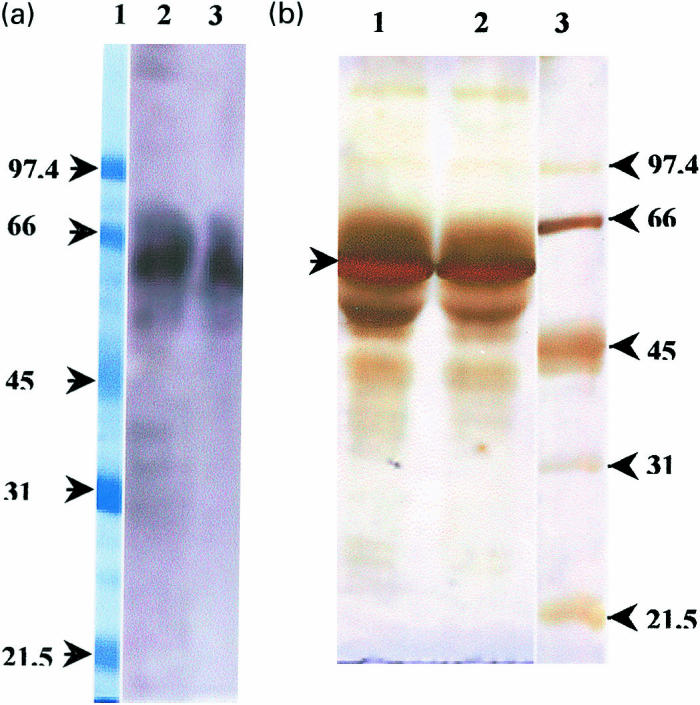
a, Western blot analysis showing the position of the scFv-Fc fusion protein. Lane 1, Molecular weight markers (97·4 kD-21·5 kD); Lane 2, III-20-Fc; Lane 3, IV-67-Fc. The fusion proteins were electrophoresed through a 10% SDS-PAG and transferred onto PVDF membrane. The Fc containing proteins were detected with HRP conjugated anti-human IgG and developed using chemiluminescence. b, Silver stained SDS-PAG showing the position of the scFv fusion protein as determined after the western blot analysis in Fig. 4(a). Lane 1, III-20-Fc; Lane 2, IV-67-Fc; Lane 3, Molecular weight markers. The samples were electrophoresed through a 10% SDS-PAG. The gel was stained with silver nitrate to detect proteins.
Table 2 shows the concentrations of the scFv-Fc recombinant proteins as determined by sandwich ELISA. This assay also confirmed that the Fc containing molecules were expressed and secreted.
Table 2.
Concentration of the fusion proteins III-20-Fc, IV-67-Fc and Fc control.
| Clone | Concentration |
|---|---|
| III-20-Fc | 600 ng/ml |
| IV-67-Fc | 3500 ng/ml |
| Fc | 1200 ng/ml |
The concentrations were determined by sandwich ELISA. Mouse anti-human IgG1at 10 µg/ml was used as the capture antibody coated in the solid phase. Purified human IgG1 was used to prepare the standard curve from which the concentrations of the recombinant proteins were determined.
Specificity of scFv-Fc molecules
The expressed proteins bound to PV11 in ELISA with absorbance492 values of 0·251, 0·337 and 0·127 for III-20-Fc, IV-67-Fc and Fc, respectively. These values, although low, are significantly positive. In the Dot Blot Analysis, the fusion proteins III-20-Fc and IV-67-Fc bound specifically to GPRV and PV11 as shown in Fig. 5. There was no binding to BSA, which was included as a negative control. On probing for GPRV using III-20-Fc, IV-67-Fc and the positive control HRIG, a single band at approximately 66–67 kD, corresponding to the molecular weight of GPRV, was detected in a Western Blot Analysis (Fig. 6) The fusion proteins did not bind to BSA in Lanes 5 and 6. III-20-Fc, IV-67-Fc and HRIG bound specifically to the membranes of PV11 infected Vero cells in an immunofluorescence assay (Fig. 7a–c, respectively). There was no fluorescence with the Fc control (not shown).
Fig. 5.

Dot blot analysis showing the specific binding of fusion proteins to GPRV and PV11·1µg GPRV, PV11 and BSA were dotted onto PVDF membrane. These were detected with III-20-Fc (lane 1), IV-67-Fc (lane 2), Fc (lane 3, negative control) and HRIG (lane 4, positive control) followed by HRP conjugated anti-human IgG. The blots were developed using chemiluminescence.
Fig. 6.

Western Blot Analysis to show the specific binding of the fusion proteins III-20-Fc, IV-67-Fc and HRIG to GPRV. Lane 1, III-20-Fc; Lane 2, IV-67-Fc; Lane 3, Fc control; Lane IV, HRIG; Lanes 5 & 6, BSA probed with III-20-Fc & IV-67-Fc, respectively. GPRV was electrophoresed through a 10% SDS-PAG and transferred onto PVDF membrane. The GPRV was probed using the fusion proteins III-20-Fc, IV-67-Fc. HRIG was used as the positive control. No bands were detected when probed with the Fc control or when BSA was used as the negative control for GPRV.
Fig. 7.

Membrane immunofluorescence of PV11 infected Vero cells with (a) III-20-Fc, (b) IV-67-Fc and (c) HRIG. Magnifications indicated are on the film. Live single cell suspensions of PV11 infected Vero cells were treated with either the fusion proteins, or HRIG as primary antibodies followed by FITC conjugated anti-human IgG. All incubations were carried out on ice and the stained cells were fixed with 2% paraformaldehyde after the final wash.
Neutralization assay
The virus controls demonstrated the expected symptoms in mice leading to their death. 25LD50 resulted in 100% deaths, which reduced with each 10fold dilution. The HRIG available commercially at 150 IU/ml neutralized the virus as expected with the 50% neutralization titre at 1/24507th dilution corresponding to 0·006 IU/ml (Table 3).
Table 3.
Data obtained in an in vivo neutralization assay to assess the neutralizing function of the fusion proteins
| Group | No. of mice alive after Day 5 | No. of mice alive after Day 14 | Percentage survival |
|---|---|---|---|
| 25 LD50, PV11 incubated with dilutions of HRIG as shown | |||
| 1/1000 | 10 | 10 | 100 |
| 1/5000 | 10 | 9 | 90 |
| 1/25000 | 9 | 1 | 11·1 |
| 1/125000 | 8 | 2 | 25 |
| 1/625000 | 10 | 1 | 10 |
| 25 LD50, PV11 incubated with fusion proteins | |||
| IV-67-Fc | 9 | 3 | 33·3 |
| III-20-Fc | 8 | 4 | 50 |
| Virus Control (positive control and virus check) | |||
| 25 LD50 | 10 | 0 | 0 |
| 2·5 LD50 | 10 | 5 | 50 |
| 0·25 LD50 | 10 | 7 | 70 |
| Negative control (fusion proteins with diluent) | |||
| IV-67-Fc | 10 | 10 | 100 |
| III-20-Fc | 9 | 8 | 88·8 |
Both scFv-Fc molecules neutralized the PV11 virus. 50% mice survived when the fusion protein III-20-Fc was incubated with the virus before inoculation while 33·3% survived with IV-67-Fc. The fusion proteins were not toxic for the mice when administered on their own.
Discussion
We have selected human scFvs against the glycoprotein antigen of the rabies virus from a synthetic scFv phage display library. The virus uses GPRV to enter host cells via specific receptors and it is therefore a critical molecule for viral infection and spread [21].
This is the only antigen of the rabies virus capable of inducing virus neutralizing antibodies (VNA) which confer immunity against a lethal challenge infection with the virus [21]. It is also the only virally encoded antigen expressed on the surface of infected cells making it an ideal target for the design of anti-rabies therapeutic molecules. PV11, a fixed strain of rabies virus, used for the entire study, belongs to the same genotype as the wild type rabies virus and shares the same antigenic properties [22].
At least five distinct anti-GPRV scFv sequences were selected. They have similar framework regions but differ more in the sequences of the CDRs (Fig. 2). This may indicate that the clones recognize the same epitope but with varying affinities or that the differences in the CDR sequences change the binding pocket such that some of them bind to different epitopes. Interestingly, one of the clones (IV-62, see Fig. 2) had a stop codon within the first codon of the CDR2 of VH and hence could only be expressed in the TG1 strain of E. coli which is a SupE amber suppressor strain. Therefore, a sequence that would normally be rejected in the human immune system and in nonsuppressor E. coli was picked up serendipitously using a suppressor strain. PV11 proved to be a good selecting agent as evidenced by the number and variety of clones selected. It is a large particle carrying approximately 2000 surface GPRV molecules per virion [23–25], which was undoubtedly the reason why the clones subsequently characterized were found to recognize GPRV.
All the heavy chain variable regions of the selected scFvs belong to the VH3 family. In a previous study where human anti-rabies monoclonal antibodies generated by EB virus transformation of B lymphocytes from immunized subjects were analysed, seven out of the nine monoclonal antibodies were found to utilize gene segments belonging to the VH3 family [26]. It therefore appears that the genes of the VH3 family are preferentially utilized against GPRV. There was no similar preferential use of a particular light chain variable gene family as shown in Table 1, also observed by Ikematsu et al. [27]. The light chain variable region may contribute only marginally to the overall binding architecture and kinetics. In an earlier experiment in our laboratory, certain GPRV binding phage clones were selected from the 2-lox human Fab library [28] which contained the VH DNA, but were found to have deleted the VL region (unpublished).
In the TG1 strain of E. coli, the scFvs are expressed on the phage surface fused to the minor coat protein, protein III, separated by an amber codon, facilitating the cycles of selection and infection. In order to achieve optimum expression of the scFv as a soluble product, phagemids isolated from positive clones were transfected into the amber nonsuppressor HB2151 strain of E. coli. The absence of binding to PV11 in ELISA despite the expression and secretion of the intact protein (Fig. 3) could mean that the proteins were not folding correctly or that their concentration was too low for detection in our assay. It may also indicate low affinity of the scFvs In the absence of further data, it is difficult to choose between the above possibilities.
In the whole antibody molecule, the constant region may provide structural support to the binding site and contribute to its conformation. The same may be true for the phage protein III, since phage particles bound well to PV11. We had intended from the outset to link the Fc portion of human IgG1 to the C terminus of the anti-GPRV scFvs in order to provide the construct with functional properties. In view of the failure of scFvs to bind to PV11 as soluble protein it was hoped that linkage to Fc would also confer the required structural stability. The scFvs from clones III-20 and IV-67 were expressed fused to human IgG1 Fc. These were found to bind to PV11. This may be a consequence of conformation stabilization, increased sensitivity, or increased affinity due to dimerization (avidity) or a combination of all three factors. The tag attached to the scFv provides only a single epitope, while the presence of Fc on the fusion protein provides multiple epitopes thus increasing the sensitivity of the assay. In any case, fusion of the scFvs with the Fc portion of the human IgG1 conferred antigen-binding activity. It is possible that this modification could be applied successfully to other scFvs that fail to bind to their antigen.
The specificity of the scFv-Fc molecules for GPRV was established by western blots and dot blots. Membrane immunofluorescence on infected Vero cells further verified their binding to GPRV, as the only viral antigen expressed on the external surface of rabies infected cells is GPRV [21,22,29]. We were limited by the low yield of fusion proteins and affinity measurements are yet to be carried out. However, given the large repertoire of the library, we expect to have a few reasonably high affinity binders among the many PV11 binders that were selected [30,31].
In view of the specificity and neutralizing ability of the scFv-Fcs, we have a preparation which, if available in sufficient quantities and further shown to be of high affinity and to protect against the wild rabies virus, could be advocated in place of the current HRIG/ERIG for prevention of rabies in man. It might also be used in therapy after infection as antibodies, in particular those with the IgG class, have been shown to be protective in the mouse model [32–34]. We are now in the process of increasing the yield of the fusion protein in order to carry out further characterization including protection assays with the wild virus. Once this has been achieved, these molecules, either in their present form or modified, might justifiably be considered for clinical use.
Acknowledgments
This work was funded by the Department of Biotechnology, Government of India. We are grateful to the British Council for facilitating the collaboration between AIIMS and PICR. We thank Dr P.L. Stern and Dr R.E. Hawkins for their generous laboratory support.
References
- 1.WHO. World Survey of Rabies 30 (for the year 1994) Geneva: World Heath Organization; 1994. WHO/EMC/ZOO/96.3. [Google Scholar]
- 2.Hemachudha T, Phuapradit P. Rabies. Curr Op Neurol. 1997;10:60–7. doi: 10.1097/00019052-199706000-00016. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Fact sheet N99. Geneva: World Heath Organization; At: http://www.who.int/inf-fs/en/fact099.htm. [Google Scholar]
- 4.Production of ARS. Health Information of India 1995 & 96. New Delhi: Ministry of Health and Family Welfare, Government of India; 1996. p. 240. ISSN 0971-0159. [Google Scholar]
- 5.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 6.Parmley SF, Smith GP. Antibody selectable filamentous fd phage vectors: affinity purification of target genes. Gene. 1988;73:305–18. doi: 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- 7.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–55. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 8.Featherstone F. Bispecific antibodies: the new magic bullets. Lancet. 1996;348:536. doi: 10.1016/s0140-6736(05)64681-8. [DOI] [PubMed] [Google Scholar]
- 9.van de Winkel JGJ, Bast B, de Gast GC. Immunotherapeutic potential of bispecific antibodies. Immunol Today. 1997;18:562–4. doi: 10.1016/s0167-5699(97)01167-5. [DOI] [PubMed] [Google Scholar]
- 10.Vaughan TJ, Osbourn JK, Tempest PR. Human antibodies by design. Nature Biotechnol. 1998;16:535–9. doi: 10.1038/nbt0698-535. [DOI] [PubMed] [Google Scholar]
- 11.Mack M, Gruber R, Schmidt S, Riethmuller G, Kufer P. Biologic properties of a bispecific single chain antibody directed against 17–1A (EpCAM) and CD3: Tumor cell-dependent T cell stimulation and cytotoxic activity. J Immunol. 1997;158:3965–70. [PubMed] [Google Scholar]
- 12.Wiktor TJ. WHO Monograph Series No 23. In: Kaplan MM, Koprowski H, editors. Laboratory Techniques in Rabies. 3. Geneva: WHO; 1973. p. 96. [Google Scholar]
- 13.Dietzschold B. Techniques for the purification of rabies virus, its subunits and recombinant virus. In: Meslin F-X, Kaplan MM, Koprowski H, editors. Laboratory Techniques in Rabies. 4. Geneva: WHO; 1996. pp. 175–180. [Google Scholar]
- 14.Lowry D, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;195:265–75. [PubMed] [Google Scholar]
- 15.Hoogenboom HR, Griffiths AD, Johnson KS, Chiswell DJ, Hudson P, Winter G. Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucl Acids Res. 1991;19:4133–7. doi: 10.1093/nar/19.15.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization: human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch EF, Maniatis T, editors. Molecular cloning, A laboratory manual. 2. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 18.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci. 1979;76:4350. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fitzgerald EA. Potency test for antirabies serum and immunoglobulin. In: Meslin F-X, Kaplan MM, Koprowski H, editors. Laboratory Techniques in Rabies. 4. Geneva: WHO; 1996. pp. 417–422. [Google Scholar]
- 20.Reed LV, Muench H. A simple matter of estimating fifty percent end points. Am J Hyg. 1938;27:493. [Google Scholar]
- 21.Dietschold B, Rupprecht CE, Fu ZF, Koprowski H. Rhabdoviruses. In: Fields BN, Knipe DM, Howley PM, et al., editors. Virology. 2. Philadelphia: Lippincott-Raven Publishers; 1996. pp. 1137–59. [Google Scholar]
- 22.Tordo N. Characteristics and molecular biology of the rabies virus. In: Meslin F-X, Kaplan MM, Koprowski H, editors. Laboratory Techniques in Rabies. 4. Geneva: WHO; 1996. pp. 28–51. [Google Scholar]
- 23.Tordo N, Poch O. Structure of rabies virus. In: Campbell JB, Charlton KM, editors. Rabies. Boston: Kluwer Academic Publishers; 1988. pp. 25–45. [Google Scholar]
- 24.Crise B, Ruusala A, Zagouras P, Shaw A, Rose JK. Oligomerization of glycolipid-anchored and soluble forms of the vesicular stomatitis virus glycoprotein. J Virol. 1989;63:5328–33. doi: 10.1128/jvi.63.12.5328-5333.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doms RW, Keller DS, Helenius A, Balch WE. Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J Cell Biol. 1987;105:1957–69. doi: 10.1083/jcb.105.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikematsu H, Harindranath N, Ueki Y, Notkins AL, Casali P. Clonal analysis of a human antibody response. II. Sequences of the VH genes of human IgM, IgG, and IgA to rabies virus reveal preferential utilization of VHIII segments and somatic hypermutation. J Immunol. 1993;150:1325–37. [PMC free article] [PubMed] [Google Scholar]
- 27.Ikematsu W, Kobarg J, Ikematsu H, Ichiyoshi Y, Casali P. Clonal analysis of a human antibody response. III. Nucleotide sequences of monoclonal IgM, IgG, and IgA to rabies virus reveal restricted V kappa gene utilization, junctional V kappa J kappa and V lambda J lambda diversity, and somatic hypermutation. J Immunol. 1998;161:2895–905. [PubMed] [Google Scholar]
- 28.Griffiths AD, Williams SC, Hartley O, et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994;13:3245–60. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coll JL. The glycoprotein G of rhabdoviruses. Arch Virol. 1995;140:827–51. doi: 10.1007/BF01314961. [DOI] [PubMed] [Google Scholar]
- 30.Perelson AS, Oster GF. Theoretical studies of clonal selection: minimal antibody repertoire size and reliability of self non-self discrimination. J Theor Biol. 1979;81:645–70. doi: 10.1016/0022-5193(79)90275-3. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths AD, Malmqvist M, Marks JD, et al. Human anti-self antibodies with high specificity from phage display libraries. EMBO J. 1993;12:725–34. doi: 10.1002/j.1460-2075.1993.tb05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietzschold B, Kao M, Zheng YM, Chen ZY, Maul G, Fu ZF, Rupprecht CE, Koprowski H. Delineation of putative mechanisms involved in antibody mediated clearance of rabies virus from the central nervous system. Proc Natl Acad Sci USA. 1992;89:7252–6. doi: 10.1073/pnas.89.15.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koprowski H, Van Der Scheer J, Black J. Use of hyperimmune anti-rabies serum concentrates in experimental rabies. Am J Med. 1994;8:412–20. doi: 10.1016/0002-9343(50)90224-5. [DOI] [PubMed] [Google Scholar]
- 34.Hooper DC, Morimoto K, Bette M, Weihe E, Koprowski H, Dietzschold B. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J Virol. 1998;72:3711–935. doi: 10.1128/jvi.72.5.3711-3719.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]