

Abstract
The CC chemokine eotaxin is a potent eosinophil-specific chemoattractant that is crucial for allergic inflammation. Allergen-induced tumour necrosis factor (TNF) has been shown to induce eotaxin synthesis in eosinophils. Nuclear factor-kappaB (NF-κB) and mitogen-activated protein kinases (MAPK) have been found to play an essential role for the eotaxin-mediated eosinophilia. We investigated the modulation of NF-κB and MAPK activation in TNF-induced eotaxin release of human eosinophils. Human blood eosinophils were purified from fresh buffy coat using magnetic cell sorting. NF-κB pathway-related genes were evaluated by cDNA expression array system. Degradation of IκBα and phosphorylation of MAPK were detected by Western blot. Activation of NF-κB was determined by electrophoretic mobility shift assay. Eotaxin released into the eosinophil culture medium was measured by ELISA. TNF was found to up-regulate the gene expression of NF-κB and IκBα in eosinophils. TNF-induced IκBα degradation was inhibited by the proteasome inhibitor N-cbz-Leu-Leu-leucinal (MG-132) and a non-steroidal anti-inflammatory drug sodium salicylate (NaSal). Using EMSA, both MG-132 and NaSal were found to suppress the TNF-induced NF-κB activation in eosinophils. Furthermore, TNF was shown to induce phosphorylation of p38 MAPK time-dependently but not extracellular signal-regulated kinases (ERK). Inhibition of NF-κB activation and p38 MAPK activity decreased the TNF-induced release of eotaxin from eosinophils. These results indicate that NF-κB and p38 MAPK play an important role in TNF-activated signalling pathway regulating eotaxin release by eosinophils. They have also provided a biochemical basis for the potential of using specific inhibitors of NF-κB and p38 MAPK for treating allergic inflammation.
Keywords: eosinophils, eotaxin, NF-κB, p38 MAPK, TNF
INTRODUCTION
Eosinophil plays a pivotal role in the inflammatory reactions of allergic diseases, including asthma, allergic rhinitis and atopic dermatitis [1]. The eosinophils are recruited to the site of inflammation by locally released chemotactic factors. The CC chemokine eotaxin, synthesized by the airway epithelium, alveolar marcophages, bronchial smooth muscle and eosinophils [2,3], is a potent and specific stimulator of eosinophil chemotaxis [4]. Eotaxin binds exclusively to CC chemokine receptor-3 (CCR3) on eosinophils to mediate the biological effects. CCR3 is expressed in high numbers of 4 × 104–4 × 105 receptors per eosinophil [2]. Elevated plasma eotaxin has been found in patients with acute asthma [5], and eosinophil-derived eotaxin is believed to be responsible for the local accumulation of eosinophils at the site of inflammation [3].
The proinflammatory cytokine tumour necrosis factor (TNF) is released in the early stage of allergic inflammation [6]. Endogenously released TNF has been shown to contribute to allergen-induced airway eosinophilia and hyperresponsiveness partly through the initiation of eosinophil degranulation [7,8]. Previous reports have shown that TNF could induce eotaxin synthesis in human lung epithelial cells [9], endothelial cells [10], monocytic cells [11] and dermal fibroblasts [12]. A more recent report indicated that interleukin (IL)-5 down-regulates TNF-induced eotaxin mRNA and protein expression in eosinophils [13]. These evidences suggested that eotaxin may induce eosinophil accumulation through paracrine and/or autocrine mechanisms. However, little is known about the intracellular mechanisms regulating eosinophil-derived eotaxin release in inflammatory processes.
The human eotaxin gene is located on chromosome 17 and comprises three exons and two introns. The 5′-flanking region of the gene contains regulatory elements including binding sites for activator protein-1 (AP-1), nuclear factor-kappaB (NF-κB) [14]. In quiescent cells, NF-κB is sequestered in the cytoplasmic compartment, bound to a group of inhibitory proteins, IκB-α, IκB-β or IκB-ɛ[15]. Upon exposure to inflammatory stimuli, such as bacterial endotoxin and TNF, the inhibitory units including IκB-α are phosphorylated and degraded. This allows the translocation of NF-κB to the nucleus, where it acts as a regulatory element by DNA binding for gene transcription [16]. Recent studies have suggested that activation of NF-κB may be crucial for increased expression of many inflammatory genes of chronic airway inflammation in asthma [17] and regulation of eosinophil apoptosis [18]. A non-steroidal anti-inflammatory drug (NSAID), sodium salicylate (NaSal), was shown to inhibit TNF-induced IκB-α degradation through the activation of p38 mitogen-activated protein kinase (MAPK) in COS-1 cells [19].
MAPK are serine and threonine kinases that can be activated by phosphorylation in kinase cascades. Three distinct MAPKs have been identified in mammalian cells. The p42/p44 extracellular signal-regulated kinase (ERK) are activated by growth factors such as IL-5 [20]; p38 MAPK is activated by osmotic stress, UV irradiation and proinflammatory cytokines including TNF [21]; and c-Jun NH2-terminal protein kinase (JNK)/stress-activated protein kinase (SAPK) is potently activated by irradiation and other environmental stresses such as hyperosmolarity [22]. We have reported previously that dexamethasone-induced apoptosis and activation of JNK and p38 MAPK activity in eosinophils are regulated by caspases [23]. The p38 MAPK has been shown to regulate TNF-induced chemokine ‘regulated upon activation normal T-cell expressed and secreted’ (RANTES) by human bronchical epithelial cells [24].
In the present study, we investigated the effect of TNF on IκB-α pathway-related gene expression, and the modulation of NF-κB and MAPK activity on the regulation of the release of eotaxin and CCR3 expression by eosinophils. Elucidation of the crucial signal transduction mechanisms that control eosinophil-derived eotaxin synthesis and eosinophil recruitment to inflammatory site is fundamental to our understanding of allergic inflammatory disease and will provide better rationale for the design of drug therapy for chemokine-mediated allergic inflammation.
MATERIALS AND METHODS
Isolation of human blood eosinophils from buffy coat and eosinophil culture
Fresh human buffy coat obtained from the Hong Kong Red Cross Blood Transfusion Service was diluted 1:2 with phosphate-buffered saline (PBS) and centrifuged using an isotonic Percoll solution (Amersham and Pharmacia Biotech, Uppsala, Sweden, density 1·082 g/ml) at 4°C for 30 min at 1000 g. The eosinophil-rich granulocyte fraction was collected and washed twice with PBS containing 2% fetal calf serum. The cells were then incubated with anti-CD16 magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) at 4°C for 45 min and CD16-positive neutrophils were depleted by passing through an LS+ column (Miltenyi Biotec) within a magnetic field. With this preparation, the drop-through fraction contained eosinophils with a purity of at least 98%, as assessed by Hemacolor rapid blood smear stain (E. Merck Diagnostica, Darmstadt, Germany). The isolated eosinophils were cultured in RPMI 1640 medium (Gibco Laboratories, NY, USA) supplemented with 10% defined fetal bovine serum (Gibco) and 20 mm Hepes (Gibco).
NF-κB proteasome inhibitor N-cbz-Leu-Leu-leucinal MG-132 (Calbiochem) [25] was dissolved in dimethyl sulphoxide (DMSO) and added to eosinophil culture at a concentration of 20 μm for 1h. In all studies, the concentration of DMSO was 0·2% (v/v).
Endotoxin-free solutions
Cell culture medium was purchased from Gibco, free of detectable lipopolysaccharide (LPS) (<0·1EU/ml). All other solutions were prepared using pyrogen-free water and sterile polypropylene plastic ware. No solutions contained detectable LPS, as determined by the Limulus amebocyte lysate assay (sensitivity limit 12 pg/ml; Associates of Cape Cod, MA, USA).
cDNA expression array analysis
Total RNA was isolated from eosinophils (3 × 106) treated with or without TNF (20 ng/ml) for overnight using Tri reagent (Molecular Research Center Inc, OH, USA). cDNA expression array analysis was performed using human NF-κB Pathway-1 GEArrayTM Kit (SupperArray Inc., Bethesda, MD, USA) according to the manufacturer’s instruction. Briefly, total RNA was reverse transcribed with reverse transcriptase and GEAprimer mix in the presence of biotin-16-dUTP. The biotinylated cDNA probes were denatured and added to the hybridyzation solution. Two indentical GEArrayTM membranes dotted with duplicate cDNAs of 23 human NF-κB pathway-related genes were prehybridized at 68°C for 2h and then hybridized overnight with the cDNA probes. Membranes were washed, blocked and incubated with alkaline phosphatase-conjugated streptavidin. The labelled biotin on the membrane was detected by chemiluminescent method and the luminescence intensities of hybridized cDNA probes were analysed using Bio-rad Quantity OneTM software (Bio-Rad Laboratories, CA, USA). The ratio of gene expression levels between control and TNF-treated human eosinophils was calculated by the fold difference in intensity using β-actin as internal control.
Western blot analysis
Eosinophils (1 × 106) with different treatment were lysed with Laemmli sample buffer (Bio-Rad, CA, USA). Whole-cell lysates were heated at 95°C for 5 min followed by centrifuge at 10 000 g for 10 min to remove cell debris. Equal amounts of proteins were subjected to sodium dodecyl sulphate-12·5% polyacrylamide gel electrophoresis and then proteins were transferred to PVDF membrane (Amersham and Pharmacia Biotech). Membranes were blocked with 5% non-fat milk in Tris-buffered saline Tween 20 (TBST: 10 mm Tris-HCl pH 7·4], 150 mm NaCl, 0·05% Tween 20). The anti-IκB-α, antiphospho-p42/44 MAPK, antiphospho-p38 MAPK and antiphosphorylated ATF-2 antibody (New England Biolabs, MA, USA) were used at dilutions of 1:500, 1:1,000, 1:1000 and 1:1500 in TBST, respectively. Antibody–antigen complexes were detected using ECL chemiluminescent detection system according to the manufacture’s instruction (Amersham and Pharmacia Biotech).
Electrophoretic mobility shift assay (EMSA)
Eosinophils were cultured in the presence or absence of MG-132 or NaSal for 1h before stimulation with TNF-α for 12 h. After cells were harvested, nuclear protein was extracted with cell lysis buffer 50 mm KCl, 0·5% NP-40, 25 mm HEPES, pH 7·8, 100 mm DTT, proteinase inhibitor (1 mm PMSF, 10 mg/ml leupeptin, 20 mg/ml aprotinin)] and then nuclear protein extraction buffer 500 mm KCl, 10% glycerol, 100 mm DTT, proteinase inhibitor (same as lysis buffer)] according to a previous method [26]. The binding reactions were performed with a biotin end-labelled NF-κB oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) (Research Genetics, AL, USA) using LightShiftTM chemiluminescent EMSA kit (Pierce Chemical Co., IL, USA). Biotin-labelled NF-κB oligonucleotide was incubated with the nuclear extract of eosinophils for 20 min at room temperature. These DNA–protein complexes were resolved on a 6% polyacrylamide gel electrophoresis and transferred to a Hybound-N+ membrane (Amersham and Pharmacia Biotech). The biotin end-labelled DNA was detected using a streptavidin–horseradish peroxidase conjugate and a chemiluminescent substrate according to the manufacture’s instructions.
Enzyme-linked immunosorbent assay (ELISA) for eotaxin
Concentration of eotaxin in the culture medium of eosinophils was detected using human eotaxin ELISA kit (R&D systems Inc., MN, USA) according to the manufacturer’s instruction. The detection limit of eotaxin is 5 pg/ml.
Statistical analysis
The differences of means between groups were assessed by the non-parametric Mann–Whitney rank sum test. A probability (P) < 0·05 was considered significantly different. A non-parametric Kruskal–Wallis test was used to assess the differences between several groups. All analyses were performed using the Statistical Package for the Social Sciences (SPSS) statistical software for Windows, version 10·0.0.
RESULTS
Effect of TNF on the expression profile of NF-κB pathway-related genes in human eosinophils
Figure 1 shows the effect of TNF on gene expression profiles related to the NF-κB pathway in eosinophils. As shown in Table 1a, treatment of TNF increased the expression of mRNA levels of NF-κB (p105) and its inhibitory protein IκB-α about three- to twofold compared with control eosinophils. A group of genes encoding proinflammatory cytokines such as TNF, granulocyte and macrophage colony-stimulating factor (GM-CSF), granulocytes colony stimulating factor (G-CSF), monocyte chemotactic protein-1 (MCP-1), interleukin (IL)-8 and IL-6 were up-regulated by TNF in eosinophils (Table 1a). Table 1b shows genes related to NF-κB pathway with similar expression level in eosinophils before or after treatment of TNF, and Table 1c shows undetectable genes in control and TNF-treated eosinophils.
Figure 1.
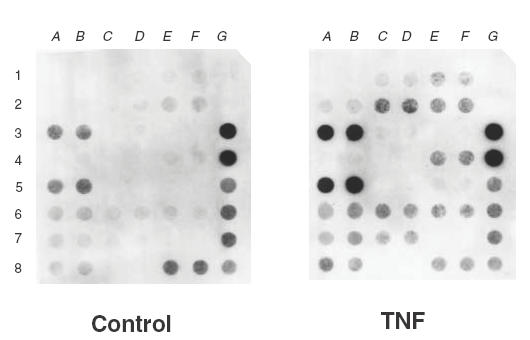
Expression profile of NF-κB pathway-related genes in human eosinophils detected by cDNA expression array system. RNA was extracted from human blood eosinophils after overnight incubation with or without TNF (20 ng/ml). Total RNA was reverse-transcribed and labelled with biotin, and gene expressions were detected using the Nonrad-GEArrayTM kit. Negative control genes: pUC18 DNA (1G, 2G); positive control genes: α-actin (3G, 4G) and glyceraldehyde-3-phosphate dehydrogenase (5G, 6G, 7G, 8E, 8F, 8G).
Table 1.
Differential expression of genes related to the human NF-κB pathway in control or TNF-treated eosinophils
| Location | Genes | Ratio in gene expression level (TNF/control) |
|---|---|---|
| (a) Genes up-regulated by TNF in eosinophils | ||
| (6C) (6D) | NF-κB (p105) | 3·38 |
| (3A) (3B) | IκBα | 2·33 |
| (7C) (7D) | TNF-α | 4·1 |
| (2C) (2D) | GM-CSF | 3·28 |
| (6A) (6B) | MCP-1 | 2·57 |
| (5A) (5B) | IL-8 | 2·1 |
| (4E) (4F) | IL-6 | 1·95 |
| (6E) (6F) | mitogen-activated protein kinase kinase kinase 14 (MAP3K14) | 1·61 |
| (2A) (2B) | G-CSF | 0→ 10·5%* |
| (5E) (5F) | IRF-1 | 0→ 7·14%* |
| (b) Genes similarly expressed in control and TNF-treated eosinophils | ||
| (4A) (4B) | IκB kinase gamma subunit | 0·92 |
| (7A) (7B) | P-selectin | 1·1 |
| (8A) (8B) | Tumour necrosis factor alpha-induced protein 1 | 1·1 |
| (1C) (1D) | c-rel | 0·85 |
| (3C) (3D) | IκB kinase alpha subunit | 0·91 |
| (2E) (2F) | Intracellular adhesion molecule 1 | 1·01 |
| (c) Undetectable genes in control and TNF-treated eosinophils | ||
| (1A) (1B) | c-myc | |
| (3E) (3F) | IκB kinase beta subunit | |
| (4C) (4D) | IL-2 | |
| (5C) (5D) | Inducible nitric oxide synthase | |
| (7E) (7F) | TNF-α | |
| (8C) (8D) | Vascular cell adhesion molecule 1 | |
The ratio of gene expression level between control and TNF treated eosinophils was assessed by the fold difference in luminescence intensity using Human NF-κB Pathway-1 Nonrad-GEArrayTM kit (Super Array).
*The gene expression of G-CSF and interferon regulatory factor-1 (IRF-1) were undetectable in control eosinophils. The expression levels of G-CSF and IRF-1 in TNF-stimulated eosinophils are shown as the percentage of intensity comparing with α-actin internal control.
Effect of MG-132 and NaSal on TNF-induced IκB-α degradation in eosinophils
Incubation of eosinophils with TNF (20 ng/ml) for 5 min was associated with a rapid loss of IκB-α from the cytoplasm. Preincubating eosinophils for 1h with the NF-κB proteasome inhibitor MG-132 (20 μm) partially, and NaSal (20 mm) completely, inhibited TNF-induced IκB-α degradation (Fig. 2). MG-132 or NaSal only did not have any effect on IκB-α protein level (data not shown).
Fig. 2.
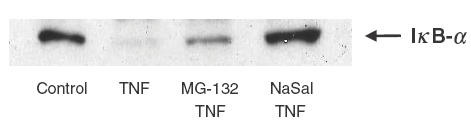
Effect of MG132 and NaSal on TNF-induced-IκB-α degradation in human eosinophils. Eosinophils (1 × 106) were preincubated with or without MG132 (20 μm) or NaSal (20 mm) for 1 h and then stimulated with TNF 20 ng/ml for 5min. Cells were lysed and the amount of IκB-α was analysed using Western blot. This result is representative of essentially identical results of triplicate experiments.
Effect of MG-132 and NaSal on NF-κB activation
The nuclear proteins of eosinophils after different treatments were analysed using the EMSA to determine DNA binding activity of NF-κB. As shown in Fig. 3, the untreated eosinophils (lane 1) exhibited a low NF-κB activity because of the low intensity of band shift. After treatment with TNF (20 ng/ml), a higher NF-κB activation was observed by higher intensity of DNA band shift (lane 2). MG-132 (lane 3) and NaSal (lane 4) were found to suppress the TNF-induced NF-κB activation. The addition of excess unlabelled NF-κB DNA probe in lane 6 did not show any band shift, therefore confirming the specificity of the NK-κB band shift.
Fig. 3.
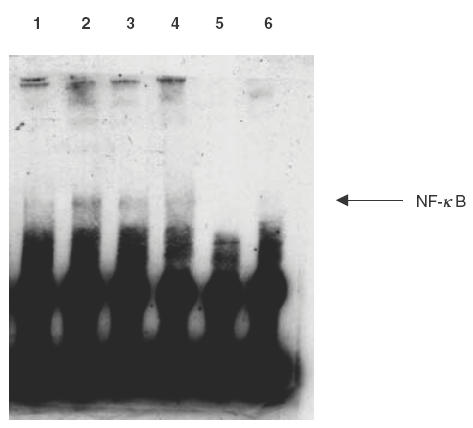
EMSA for NF-κB translocation and DNA binding. Eosinophils (5 × 106) were pretreated with or without MG132 (20 μm) or NaSal (20 mm) for 1h followed by stimulation with TNF-α (20 ng/ml) for 12 h. Nuclear protein was isolated, and EMSA was performed with a biotin end-labelled NF-κB oligonucleotide using LightShift™ chemiluminescent EMSA kit (Pierce). Lane 1–4 were as labelled. Lane 5 had only biotinated NF-κB DNA probe without protein added and lane 6 had biotinated NF-κB DNA probe, excess unlabelled NF-kB DNA probe and TNF-treated protein extract. This result is representative of essentially identical results of triplicate experiments. 1: no treatment; 2: TNF (20ng/ml); 3: MG132 (20μm) and TNF (20ng/ml); 4: NaSal (20μm) and TNF (20ng/ml); 5: negative control (biotinylated NF-κB probe only); 6: biotinylated NF-κB DNA probe, TNF-treated protein extract and excess unlabelled NF-κB DNA probe.
Effect of TNF and SB 203580 on the activation of p38 MAPK in human eosinophils
As shown in Fig. 4a, TNF (20 ng/ml) could induce a rapid phosphorylation of p38 MAPK in human eosinophils in a time-dependent manner. This result is in accord with a previous study [27]. The effect became significant after 2 min of stimulation. However, no phosphorylation of ERK was detected in the same eosinophil cell lysate (Fig. 4b). Pretreatment with specific p38 MAPK inhibitor SB 203580 (1, 5, 10, 20 μm) for 1h shows a dose-dependent inhibition of TNF-induced p38 MAPK activity assessed by the phosphorylation of substrate ATF-2 in eosinophils (Fig. 4c).
Fig. 4.
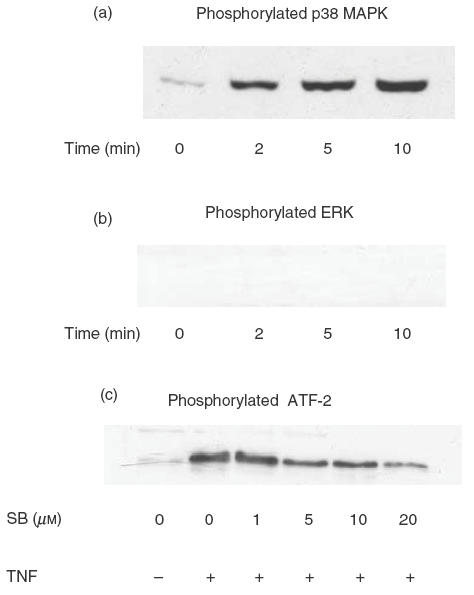
Effect of TNF on the activation of p38 MAPK and ERK. Eosinophils (1 × 106) were stimulated for various times with TNF (20 ng/ml) followed by detection of (a) phosphorylated-p38 MAPK and (b) phosphorylated-ERK in the cell lysate by Western blot. In (c), eosinophils (1 × 106) were treated without or with SB 203580 (SB) (1, 5, 10, 20 μm) for 1 h followed by stimulation with TNF (20 ng/ml) for 5 min. p38 MAPK activity was assessed by the expression of phosphorylated ATF-2 by Western blot. This result is representative of essentially identical results of triplicate experiments.
Effect of MG-132, NaSal and SB 203580 on TNF-induced eotaxin release from human eosinophils
As shown in Fig. 5, eosinophils (1 × 106) cultured in vitro could release eotaxin spontaneously. Addition of TNF (20 ng) significantly augmented the eotaxin release from eosinophils in a time-dependent manner from 1 to 12 h compared with non-treated cells (P < 0·05). For investigating if NF-κB and p38 MAPK were involved in TNF-induced eotaxin release from eosinophils, proteasome inhibitor MG-132 (20 μm), p38 MAPK inhibitor SB 203580 (20 μm) and NaSal (20 mm) were preincubated with eosinophils for 1h before the stimulation with TNF for various times as indicated in Fig. 5. Both MG-132 and NaSal could inhibit significantly TNF-induced eotaxin release at 1h and 2h incubation of eosinophils in vitro (P < 0·05). The inhibitory effect of eotaxin release was not significant at 12 h between MG-132 or NaSal pretreated group and TNF-treated group. However, p38 MAPK inhibitor SB 203580 was shown to inhibit significantly the eotaxin release at all indicated time points (1 h, 2h and 12 h, P < 0·05) and the inhibitory effect is greater than MG-132 and NaSal. Using the non-parametric Kruskal–Wallis test, a non-parametric method equivalent to one-way anova, we found that the asymmetrical significances are 0·082, 0·033 and 0·034 for 1h, 2h and 12h treatment, respectively. These indicate that there were significant difference for eotaxin release between all treated groups in the case of 2 and 12 h. However, inhibitor SB 203580, MG-132 and NaSal alone did not show any significant effect on the spontaneous eotaxin release from eosinophils (data not shown).
Fig. 5.
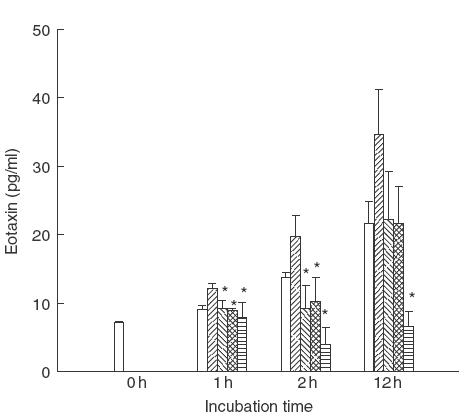
Effect of MG-132, NaSal and SB 203580 on TNF-induced eotaxin release from human eosinophils. Eosinophils (1 × 106) were pretreated with or without MG132 (20 μm), NaSal (20 mm) or SB 203580 (20 μm) for 1 h followed by stimulation with TNF (20 ng/ml) for the time indicated. Eotaxin released in the culture medium from eosinophils was determined by ELISA. Results are expressed as the arithmetic mean ± s.d. from triplicate experiments. Addition of TNF (20 ng) augmented significantly the eotaxin release from eosinophils in a time-dependent manner from 1h to 12 h comparing with non-treated cells (P < 0·05). The difference between TNF-treated groups and other treated groups were assessed by Mann–Whitney rank sum test. *P < 0·05. □, Medium;  , TNF;
, TNF;  , MG-132 and TNF;
, MG-132 and TNF;  , NaSal and TNF;
, NaSal and TNF;  SB 203580 and TNF.
SB 203580 and TNF.
DISCUSSION
Eosinophils have been shown to express TNF receptors [1] and TNF has been found to stimulate IκBα degradation and dissociation of NF-κB in many other cells [16]. A number of proinflammatory cytokines such as IL-1, IL-2, IL-6, IL-8, GM-CSF and RANTES are regulated at the level of gene transcription by NF-κB in many inflammatory diseases [15,28]. The essential role of NF-κB has been implicated in the induction of eosinophilia in allergic airway inflammation using NF-κB p50 subunit deficient mice [29]. However, the detailed intracellular mechanism for the regulation of NF-κB-dependent eosinophilia has not been well understood. A previous report has shown that translocation of NF-κB could induce eotaxin expression by binding to promoter containing NF-κB binding element of eotaxin gene in humans [30]. It was revealed recently that eotaxin protein is preformed and stored in specific granules of normal eosinophils and secrected upon appropriate stimuli such as C5a and ionomycin [3]. In our study, a panel of genes related to the NF-κB pathway was investigated using a cDNA expression array system. TNF could up-regulate the gene expression of NF-κB and its inhibitory protein IκBα in eosinophils (Fig. 1, Table 1a). The expression of a group of proinflammatory cytokine genes including TNF, IL-6, IL-8, GM-CSF, G-CSF and MCP-1 were also increased by treating eosinophils with TNF. Using Western blot analysis and EMSA, we further illustrated that TNF could induce IκB-α degradation and NF-κB activation which could be reversed by NF-κB proteasome inhibitor MG-132. MG-132 was also found to inhibit TNF-induced eotaxin release from human eosinophils. These suggest the important role of NF-κB activation in TNF-induced eotaxin release from eosinophils. Besides, the up-regulation of NF-κB and its inhibitory protein IκBα at gene level may indicate that TNF not only triggers but also maintains its effect on NF-κB mediated signal transduction in eosinophils.
Even though TNF-induced stimulation of I-κB phosphorylation is a signal for the degradation of I-κB, phosphorylation of I-κB is not sufficient to activate NF-κB [31,32]. Moreover, the mechanisms involved in NF-κB translocation have not been defined [17]. In the study of the phosphorylation of IκB-α, no significant change in the level of phospho-IκB-α was detected upon treatment with TNF or MG-132 in our study (data not shown). The non-steroidal anti-inflammatory drug NaSal has been shown to inhibit NF-κB activation in kidney cells [19], neurones [33], monocytic cells [34] and neutrophils [35]. In our study, NaSal was also shown to inhibit TNF-induced IκB-α degradation and NF-κB band shift in eosinophils. NaSal also significantly inhibited TNF-induced eotaxin release up to 2 h incubation in vitro; however, the inhibition by MG-132 and NaSal was not significant on eosinophils when cultured with TNF for 12 h. It may suggest that the initial release of eotaxin is MG-132 and NaSal-dependent, whereas subsequent release may not be NF-κB-dependent. We adopted the concentrations of MG-132 and NaSal from previous studies [19,25] for demonstrating the effective NF-κB inhibition without toxicity to the cells. MG-132 (>20 μm) and NaSal (>20 mm) did not significantly increase their inhibitory effect on IκB-α degradation (data not shown).
The present report shows that TNF could induce phosphorylation of p38 MAPK in eosinophils rapidly and time-dependently (Fig. 4a) but not ERK (Fig. 4b). The specific p38 MAPK inhibitor SB 203580 (20 μm) could largely inhibit TNF-activated p38 MAPK activity in eosinophils (Fig. 4c). The inhibition of p38 MAPK activity decreased the release of eotaxin in eosinophils as shown in Fig. 5. This indicated the important role of the activation of p38 MAPK in the mechanism of TNF-induced eotaxin release from eosinophils. It supports the recent report showing that eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 MAPK [36]. Since p38 MAPK is required for NF-κB-dependent gene expression by the modulation of DNA binding of TATA-binding protein (TBP) to the TATA box [37], it is reasonable that the inhibition of p38 MAPK can down-regulate NF-κB-dependent eotaxin expression in eosinophils. Therefore, it may be possible that the inhibition of p38 MAPK by SB 203580 can block TNF-induced eosinophil-derived eotaxin by indirect inhibiting NF-κB activity and subsequently suppress the eotaxin-induced eosinophil degranulation and chemotaxis. Besides detecting the eotaxin protein released to the culture medium, the mRNA expression of eotaxin in eosinophils was also investigated using fluorescence quantitative RT-PCR (PE Applied Biosystems, CA, USA). The inductive effect of TNF and the inhibitory effect of MG-132, NaSal and SB 203580 at the mRNA level of eotaxin were consistent with the result of ELISA in Fig. 5 (data not shown).
Although NF-κB activation and chemokine production occur in a variety of cell types, their regulation may be different between nonimmune cells and leucocytes. Therefore, cell- and gene-specific regulations of NF-κB need further exploration. Moreover, whether the activity of NF-κB is useful as a marker for the severity of inflammation in certain diseases and assessing the efficacy of therapeutic interventions also requires further investigation.
CCR3 is expressed in high numbers on eosinophils [38]. Blocking of CCR3 has been shown to inhibit eosinophil response to eotaxin, RANTES, and monocyte chemotactic protein-2, -3 and -4 by more than 95% [39]. To supplement our current finding about eotaxin release, we used RT-PCR to detect CCR3 mRNA expression simutaneously with eotaxin release in eosinophils. We found that the expressions of CCR3 mRNA were similar in groups treated with TNF, MG-132, NaSal and SB 203580 (data not shown). In contrast to the regulatory mechanism of eotaxin release, TNF, MG-132, SB 203580 and NaSal did not show any effect on CCR3 expression in eosinophils.
In view of the above findings, we conclude that NF-κB and p38 MAPK are two critical molecules involved in the mechanism of TNF-induced eotaxin release from eosinophils. Blocking of the activation of NF-κB and phosphorylation of p38 MAPK are closely associated with the decreased autocrine release of eotaxin from eosinophils without any significant regulatory effect on CCR3 expression. Specific inhibitors of NF-κB and p38 MAPK can be beneficial in elucidating the complex regulatory mechanism in inflammatory response, and may be clinically useful in treating inflammatory diseases.
Acknowledgments
This study was supported by a Chinese University of Hong Kong Direct Grant for Research and a donation from Zindart (De Zhen) Foundation Ltd, Hong Kong.
REFERENCES
- 1.Walsh GM. Advances in the immunobiology of eosinophils and their role in disease. Crit Rev Clin Lab Sci. 1999;36:453–96. doi: 10.1080/10408369991239277. [DOI] [PubMed] [Google Scholar]
- 2.Rankin SM, Conroy DM, Williams TJ. Eotaxin and eosinophil recruitment: implications for human disease. Mol Med Today. 2000;6:20–7. doi: 10.1016/s1357-4310(99)01635-4. [DOI] [PubMed] [Google Scholar]
- 3.Nakajima T, Yamada H, Iikura M, et al. Intracellular localization and release of eotaxin from normal eosinophils. FEBS Lett. 1998;434:226–30. doi: 10.1016/s0014-5793(98)00863-1. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–56. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 5.Lilly CM, Woodruff PG, Camargo CA, Jr, et al. Elevated plasma eotaxin levels in patients with acute asthma. J Allergy Clin Immunol. 1999;104:786–90. doi: 10.1016/s0091-6749(99)70288-5. [DOI] [PubMed] [Google Scholar]
- 6.Barnes P, Chung KF, Page CP. Inflammatory mediators of asthma: an update. Pharmacol Rev. 1998;50:515–96. [PubMed] [Google Scholar]
- 7.Fujisawa T, Abu-Ghazaleh R, Kita H, Sanderson CJ, Gleich GJ. Regulatory effect of cytokines on eosinophil degranulation. J Immunol. 1990;144:642–6. [PubMed] [Google Scholar]
- 8.Whitcomb EA, Wesolek JH, Pincus SH. Modulation of human peripheral blood eosinophil function by tumor necrosis factor-alpha. Int Arch Allergy Appl Immunol. 1989;89:250–5. doi: 10.1159/000234955. [DOI] [PubMed] [Google Scholar]
- 9.Lilly CM, Nakamura H, Kesselman H, et al. Expression of eotaxin by human lung epithelial cells: induction by cytokines and inhibition by glucocorticoids. J Clin Invest. 1997;99:1767–73. doi: 10.1172/JCI119341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsukura S, Stellato C, Plitt JR, et al. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol. 1999;163:6876–83. [PubMed] [Google Scholar]
- 11.Nakamura H, Haley KJ, Nakamura T, Luster AD, Lilly CM. Differential regulation of eotaxin expression by TNF-alpha and PMA in human monocytic U-937 cells. Am J Physiol. 1998;275:L601–10. doi: 10.1152/ajplung.1998.275.3.L601. [DOI] [PubMed] [Google Scholar]
- 12.Bartels J, Schluter C, Richter E, et al. Human dermal fibroblasts express eotaxin: molecular cloning, mRNA expression, and identification of eotaxin sequence variants. Biochem Biophys Res Commun. 1996;225:1045–51. doi: 10.1006/bbrc.1996.1292. [DOI] [PubMed] [Google Scholar]
- 13.Han SJ, Kim JH, Noh YJ, et al. Interleukin (IL)-5 downregulates tumor necrosis factor (TNF)-induced eotaxin messenger RNA (mRNA) expression in eosinophils. Induction of eotaxin mRNA by TNF and IL-5 in eosinophils. Am J Respir Cell Mol Biol. 1999;21:303–10. doi: 10.1165/ajrcmb.21.3.3467. [DOI] [PubMed] [Google Scholar]
- 14.Corrigan CJ. Eotaxin and asthma: some answers, more questions. Clin Exp Immunol. 1999;116:1–3. doi: 10.1046/j.1365-2249.1999.00740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 16.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 17.Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med. 1998;158:1585–92. doi: 10.1164/ajrccm.158.5.9706116. [DOI] [PubMed] [Google Scholar]
- 18.Ward C, Chilvers ER, Lawson MF, et al. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. 1999;274:4309–18. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- 19.Schwenger P, Alpert D, Skolnik EY, Vilcek J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol Cell Biol. 1998;18:78–84. doi: 10.1128/mcb.18.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Groot RP, Coffer PJ, Koenderman L. Regulation of proliferation, differentiation and survival by the IL-3/IL-5/GM-CSF receptor family. Cell Signal. 1998;10:619–28. doi: 10.1016/s0898-6568(98)00023-0. [DOI] [PubMed] [Google Scholar]
- 21.Raingeaud J, Gupta S, Rogers JS, et al. Proinflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–6. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 22.Derijard B, Hibi M, Wu IH, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–37. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JP, Wong CK CWK. L. Role of caspase in dexamethasone-induced apoptosis and activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in human eosinophils. Clin Exp Immunol. 2000;122:20–7. doi: 10.1046/j.1365-2249.2000.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto S, Matsumoto K, Gon Y, et al. p38 MAP kinase regulates TNF alpha-, IL-1 alpha- and PAF-induced RANTES and GM-CSF production by human bronchial epithelial cells. Clin Exp Allergy. 2000;30:48–55. doi: 10.1046/j.1365-2222.2000.00641.x. 10.1046/j.1365-2222.2000.00641.x. [DOI] [PubMed] [Google Scholar]
- 25.Fiedler MA, Wernke-Dollries K, Stark JM. Inhibition of TNF-alpha-induced NF-kappaB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol. 1998;19:259–68. doi: 10.1165/ajrcmb.19.2.3149. [DOI] [PubMed] [Google Scholar]
- 26.Leonard SS, Wang S, Shi X, Jordan BS, Castranova V, Dubick MA. Wood smoke particles generate free radicals and cause lipid peroxidation, DNA damage, NFκB activation and TNF-α release in macrophages. Toxicology. 2000;150:147–57. doi: 10.1016/s0300-483x(00)00256-0. 10.1016/s0300-483x(00)00256-0. [DOI] [PubMed] [Google Scholar]
- 27.Bracke M, van de Graaf E, Lammers JW, Coffer PJ, Koenderman L. In vivo priming of FcalphaR functioning on eosinophils of allergic asthmatics. J Leukoc Biol. 2000;68:655–61. [PubMed] [Google Scholar]
- 28.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Cohn L, Zhang DH, Homer R, Ray A, Ray P. Essential role of nuclear factor kappaB in the induction of eosinophilia in allergic airway inflammation. J Exp Med. 1998;188:1739–50. doi: 10.1084/jem.188.9.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hein H, Schluter C, Kulke R, Christophers E, Schroder JM, Bartels J. Genomic organization, sequence, and transcriptional regulation of the human eotaxin gene. Biochem Biophys Res Commun. 1997;237:537–42. doi: 10.1006/bbrc.1997.7169. 10.1006/bbrc.1997.7169. [DOI] [PubMed] [Google Scholar]
- 31.Alkalay I, Yaron A, Hatzubai A, et al. In vivo stimulation of I kappa B phosphorylation is not sufficient to activate NF-kappa B. Mol Cell Biol. 1995;15:1294–301. doi: 10.1128/mcb.15.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Tumor necrosis factor alpha-induced phosphorylation of I kappa B alpha is a signal for its degradation but not dissociation from NF-kappa B. Proc Natl Acad Sci USA. 1994;91:12740–4. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grilli M, Pizzi M, Memorandum M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383–5. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 34.Oeth P, Mackman N. Salicylates inhibit lipopolysaccharide-induced transcriptional activation of the tissue factor gene in human monocytic cells. Blood. 1995;86:4144–52. [PubMed] [Google Scholar]
- 35.Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–9. [PubMed] [Google Scholar]
- 36.Kampen GT, Stafford S, Adachi T, et al. Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood. 2000;95:1911–7. [PubMed] [Google Scholar]
- 37.Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-kappaB-dependent gene expression. The role of TATA-binding protein (TBP) J Biol Chem. 1999;274:30858–63. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- 38.Daugherty BL, Siciliano SJ, DeMartino JA, Malkowitz L, Sirotina A, Springer MS. Cloning, expression, and characterization of the human eosinophil eotaxin receptor. J Exp Med. 1996;183:2349–54. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heath H, Qin S, Rao P, et al. Chemokine receptor usage by human eosinophils. The importance of CCR3 demonstrated using an antagonistic monoclonal antibody. J Clin Invest. 1997;99:178–84. doi: 10.1172/JCI119145. [DOI] [PMC free article] [PubMed] [Google Scholar]