

Abstract
It is generally accepted that multiple sclerosis (MS) is mediated by autoreactive T cells and that myelin basic protein (MBP) is one of the target autoantigens. The T-cell response to MBP has been analysed extensively, largely through the use of T-cell lines (TCL) and T-cell clones (TCC), and to date, three immunodominant regions (13–32, 84–103 and 144–163) have been described. However, given that TCL may represent a skewed pattern of peptide reactivity, we have developed a kinetic response assay in which the proliferation of peripheral blood mononuclear cells (PBMC) from MS patients and healthy individuals was measured directly against a panel of peptides spanning the full length of human MBP. Furthermore, PBMC from each subject were tested three times over the course of 18 months. A high proportion of MS patients exhibited a significant response to eight MBP regions (1–24, 30–54, 75–99, 90–114, 105–129, 120–144, 135–159 and 150–170). TCC were subsequently generated from MS subjects and were used to further define the epitope recognized in each case. Overall, normal individuals recognized significantly fewer peptides. In addition, we noted that the T-cell recognition of any one peptide can fluctuate, appearing at one time point, regressing, and subsequently reappearing at a later date. This study provides new insight into the recognition profile and dynamics of myelin-antigen-specific T cells in MS.
Keywords: multiple sclerosis, myelin basic protein, kinetic response assay, epitope immunodominance, cycling of peptide recognition
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory disease characterized by multiple demyelinating lesions disseminated throughout the CNS white matter, occurring at various sites and times [1, 2]. MS is thought to be mediated by autoreactive T cells and, amongst the myelin components that represent the putative autoantigens, myelin basic protein (MBP) has been the most extensively studied. This is partly due to the finding that MBP is immunogenic and that MBP-specific T lymphocytes have encephalitogenic activity in experimental animals [3–5]. Furthermore, recent evidence arising from a clinical trial using an MBP-TCR antagonist has shown a clear correlation between exacerbation of disease and an increased precursor frequency of MBP-specific T cells [6]. This clearly demonstrates that MBP-specific T lymphocytes can contribute to MS in humans.
It is well established that MBP-specific T cells are present in the peripheral blood of MS patients as well as in healthy individuals. Therefore, the mere presence of autoreactive cells in the periphery is not sufficient for the development of MS. We and others [7–9] have shown that one of the differences between healthy individuals and MS patients lies in the activation state of the autoreactive T cells. That is, MBP-specific T cells in MS patients are more likely to have been activated in vivo and to have differentiated into memory cells compared with control subjects.
Early studies using T-cell lines (TCL) and T-cell clones (TCC) revealed that MS patients recognize three immunodominant regions within human MBP (amino acid (aa) 13–32, aa 84–103 and aa 144–163) [10–14]. This restricted epitope recognition was thought to persist for several months irrespective of clinical disease activity [13]. To date, the profile of MBP reactivity in MS patients and healthy individuals has been defined largely through the use of TCL and TCC. However, recent work showing that some TCC are susceptible to apoptosis in cell culture conditions suggests that surviving MBP-reactive TCL and TCC may represent a skewed repertoire with respect to the pattern of peptide reactivity [15]. Few studies have carried out direct ex vivo analyses involving peripheral blood mononuclear cell (PBMC) cultures to examine (a) whether MS patients respond to a broad spectrum of MBP peptides and (b) how anti-MBP T-cell responses evolve and whether such responses are stable over time. As the use of MBP-reactive TCL and TCC may obscure our understanding of epitope recognition in MS, we have developed a kinetic response assay in which PBMC from patients and controls are cultured with a panel of overlapping peptides spanning the whole length of human MBP. T-cell proliferative responses from each individual were measured over a period of 2 weeks. In addition, PBMC from most of our MS subjects were examined in the kinetic response assay three times over a period of 18 months in order to investigate the stability of the response to MBP in ongoing disease. The epitopes recognized by our study group were further characterized with the aid of TCC generated from patients and controls. The use of TCC allowed mapping of the epitopes contained within regions previously identified in the kinetic response assay.
MATERIALS AND METHODS
Antigens
Human MBP was prepared from brain white matter as described by Deibler et al.[16]; its purity was >95% as assessed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). MBP and Mycobacterium tuberculosis purified protein derivative (PPD) (UK Central Veterinary Laboratory, Surrey) were used in proliferative assays at previously determined optimal concentrations, which were 50 μg/ml for each. A panel of 15-mer overlapping peptides spanning the whole MBP molecule was synthesized using standard F-moc chemistry on an Abimed AMS 422 multiple peptide synthesizer (Abimed, Langenfeld, Germany). Each peptide was displaced by five aa and overlapped by 10 aa. We produced 33 peptides that were pooled into groups of three; pools were tested at the optimum concentration of 50 μg/ml such that in vitro, each peptide was present at a concentration of 16·6 μg/ml.
Patients and control subjects
Eleven patients with clinically definite or laboratory supported definite MS [17], with an age range of 26–49 years, were studied. A clinical history of each patient is provided in Table 1A. Eight of the 11 patients were receiving interferon-β treatment and the remaining three MS patients had received no corticosteroid treatment or interferon-β treatment for at least 3 months prior to the commencement of the study. All patients whose MS was Relapsing-Remitting were in remission at the time of sample collection. One patient (MS 49) had secondary-progressive MS. The control group consisted of 12 healthy individuals with an age range of 27–53 years, and none had received immunosuppressive therapy for at least 3 months prior to the blood sample being obtained.
Table 1(a).
(a) Clinical details of MS patients.
| Patient number | HLA DR type | Age and sex | Disease duration | Clinical status | Treatment at time of assay |
|---|---|---|---|---|---|
| MS10 | DRB1*2,3 | 26 F | 3 years | RR | β-IFN |
| MS17 | DRB1*2,4 | 37 F | 8 years | RR | None |
| MS19 | DRB1*2,4 | 40 F | 6 years | RR | None |
| MS39 | DRB1*2,7 | 39 F | 8 years | RR | β-IFN |
| MS41 | DRB1*2,1 | 48 F | 16 years | RR | β-IFN |
| MS43 | DRB1*2,4 | 38 F | 5 years | RR | β-IFN |
| MS49 | DRB1*2,7 | 37 F | 4 years | SP | β-IFN |
| MS57 | DRB1*2,4 | 49 F | 11 years | RR | β-IFN |
| MS59 | DRB1*2,4 | 47 F | 14 years | RR | β-IFN |
| MS60 | DRB1*2 | 47 F | 19 years | RR | β-IFN |
| MS67 | DRB1*2,13 | 49 F | 6 years | RR | None |
RP, Relapsing-Remitting MS; SP, Secondary Progressive MS.
Our studies had received local ethical approval, and informed written consent was obtained from all patients and healthy individuals involved.
Tissue culture medium
RPMI-1640 medium (Life Technologies, Paisley, UK) supplemented with 20 mm HEPES (Sigma, Poole, UK), penicillin (100 cells/ml), streptomycin sulphate (100 μg/ml) and 4 mml-glutamine (all from Life Technologies), was used as the tissue culture medium. Medium without serum was used for washing lymphoid cells and TCL. For all culture conditions and assays, medium was supplemented with 10% heat-inactivated autologous plasma.
Culture conditions and T-cell proliferative assays
Citrated peripheral blood (50–100 ml) was collected by venepuncture from each subject. The cell culture conditions used throughout this study were largely based on techniques originally described in this laboratory [18]. Peripheral blood mononuclear cells were isolated by density centrifugation on Histopaque-1077 (Sigma) and cultured in 1·5 ml volumes in 24-well tissue culture plates (Nunc International, Costar, Corning Inc. New York, USA), at a concentration of 1 × 106 cells/ml, containing no antigen, PPD, MBP or peptides of MBP. The plates were incubated at 37°C in a humidified atmosphere of 5% CO2/95% air. Between days 5 and 14, duplicate aliquots of 100 μl were withdrawn from each culture, transferred to a 96-well round-bottom microtitre plate and pulsed with 0·4μCi 3H-thymidine (Amersham International, Amersham, UK). After 18 h, cells were harvested onto glass fibre mats (LKB-Wallac, Turku, Finland) using a Mach 111 harvester 96 (Tomtec, Orange, NJ, USA). 3H-thymidine incorporation was determined using a Microbeta liquid scintillation counter (LKB-Wallac). Test wells containing antigen were considered positive when the δcpm was >1000 and the Stimulation Index (SI) > 3; δcpm = cpm antigen containing culture – cpm culture without antigen, and SI = cpm antigen containing culture/cpm culture without antigen.
The T-cell proliferative responses in MS patients were analysed at three separate time points; subjects were re-bled 4–10 months following the first time point, and blood samples for the third time point were collected after a further 3–8 months.
Tissue typing
Genomic DNA was prepared from PBMC of each individual by the salting-out method [19]. Typing for the polymorphism of HLA class II DR-loci was performed by polymerase chain reaction amplification with sequence specific primers, as described previously [20]. The DR type of each individual in our study is presented in Table 1A and B according to the nomenclature of Schreuder et al.[21].
Table 1(b).
(b) The HLA-DR type of healthy individuals
| Control subject number | HLA DR type | Age and sex |
|---|---|---|
| N1 | DRB1*1,2 | 37 F |
| N2 | DRB1*1,2 | 44 F |
| N3 | DRB1*2,3 | 40 M |
| N4 | DRB1*2,3 | 33 M |
| N5 | DRB1*2,7 | 28 F |
| N6 | DRB1*2,7 | 46 M |
| N7 | DRB1*2,4 | 52 F |
| N8 | DRB1*2,3 | 53 F |
| N9 | DRB1*2,3 | 38 F |
| N10 | DRB1*2,7 | 39 M |
| N11 | DRB1*2,11 | 42 F |
| N12 | DRB1*2,4 | 27 F |
The HLA nomenclature used in this table and throughout our study is as defined by Schreuder et al. (Schreuder et al. 1999).
Generation of TCL and TCC
MBP-specific TCL were generated from eight MS patients and one healthy control donor. PBMC from each subject were separated as described above and cultured at 1 × 106 cells/ml in 6-well plates in the presence of MBP (50 μg/ml); an aliquot of PBMC from each subject was frozen and stored for subsequent re-stimulations. Seven days later, the cells were fed with fresh medium containing 20 cells/ml IL-2 (i.e. 2% Lymphocult-HT; Biotest Ltd, Birmingham, UK) and on day 12 of culture, all cells were subjected to the first round of re-stimulation, i.e. cells were re-stimulated with antigen, IL-2 and irradiated (2500 Rad) autologous PBMC as a source of antigen-presenting cells (APC), at a cell ratio of 1 T cell:5 APC. Cells were expanded in IL-2 every 3–4 days and on day 14, were re-stimulated with antigen, IL-2 and PBMC, as described above. On the day of the first re-stimulation, cells were examined for specific proliferation to MBP. Briefly, 2 × 104 T cells and 1 × 105 irradiated autologous PBMC were cultured in triplicate, in 96-well round-bottom plates, in the presence of MBP. Cells were cultured for 2 days and pulsed with 3H-thymidine at 0·4μCi/well during the last 18 h of the culture. Cells were then harvested as described above, and a TCL with a δcpm >1000 and a SI > 3 was considered to be MBP-specific.
Following three re-stimulation/expansion cycles, TCL were cloned using PHA (Sigma) in the presence of autologous irradiated PBMC as APC. T cells were plated under limiting dilution conditions at 0·1 cell/well, 0·3 cell/well and 1 cell/well, and cultured in Terasaki plates (Nunc International) with 1 × 104 irradiated PBMC, 5 μg/ml PHA and 20 cells/ml IL-2. After 10–12 days, growth-positive wells were expanded onto 96-well round-bottom plates using 1 × 105 irradiated PBMC, 5 μg/ml PHA and IL-2. Three days later, wells were fed with fresh medium containing IL-2 and on day 7, the clones were expanded onto 48-well plates using 5 × 105 irradiated PBMC, PHA and IL-2; at this point, clones were tested in proliferation assays for specific responses to MBP. MBP-specific clones were expanded a week later onto 24-well plates using 1 × 106 irradiated PBMC, PHA and IL-2. The clones were maintained in 24-well plates using a 7–10 day re-stimulation/expansion cycle, as described above. The ability of TCC to recognize the panel of MBP peptides was tested by proliferation assays, as described above.
RESULTS
HLA DR type of MS patients and healthy donors
Based on an extensive analysis, conducted in our laboratory, involving 68 MS patients and 40 healthy individuals, we have found that the HLA DR2 type (DRB1*1501,15021; DRB5*0101) was expressed in 56% of MS patients and 25% of healthy individuals [9]. Given this strong DR2 association in MS, we chose to analyse DR2+ve patients; consequently, the control group consisted of 12 DR2+ve healthy individuals (Table 1A and B).
MBP-peptide recognition amongst MS patients and healthy individuals
We have developed a kinetic response assay in which PBMC from MS patients and healthy subjects were tested for their ability to respond to a panel of overlapping 15-mer synthetic peptides spanning the full length of human MBP. The proliferative response of PBMC from each culture was examined at five time points over a period of 2 weeks, and the kinetic profile of the response to MBP and peptides was compared with the response to PPD, the latter representing a secondary response/memory antigen.
All patients and healthy individuals gave a classical secondary kinetic response to PPD (a typical example is shown in Fig. 1). The response to MBP in MS patients and healthy controls peaked later than the response to PPD (Fig. 1). In addition, our results suggest that the response to MBP and its peptides is not modified or reduced by interferon-β treatment, as patients receiving interferon-β responded equally well to MBP and recognized no fewer epitopes than patients on no treatment. This can be seen by comparing T-cell responses between patients MS17 and MS67 (patients receiving no interferon-β) and the remaining patients who were on interferon-β treatment (see Table 2).
Fig.1.
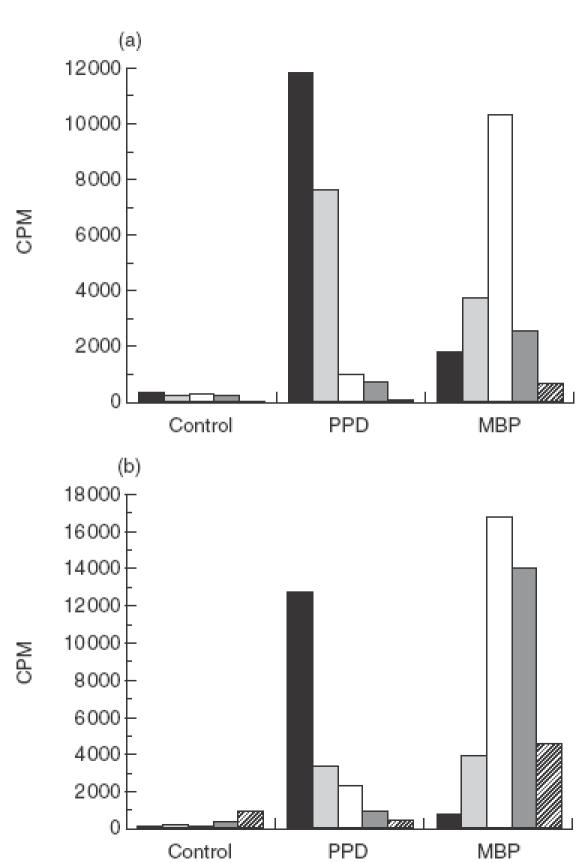
PBMC isolated from an MS patient (a) and normal individual (b) were tested for proliferative responses to PPD and whole MBP, measured by 3H-thymidine uptake. The kinetic profile of the MBP response was compared with that to the recall antigen PPD. (▪) Day 5; ( 00077;) day 7; (□) day 9; (
00077;) day 7; (□) day 9; ( 00036;) day 11; (
00036;) day 11; ( 00004;) day 13.
00004;) day 13.
Table 2.
Peak Stimulation Index (SI) values to MBP and MBP-peptides in MS patients obtained at three separate time points (see Materials and Methods)
| Time point | MBP | 1–24 | 15–39 | 30–54 | 45–69 | 60–84 | 75–99 | 90–114 | 105–129 | 120–144 | 135–159 | 150–170 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS10 | 1 | 14 | 17 | 3 | 15 | 55 | 1 | 1 | 2 | 15 | 1 | 2 | 1 |
| 2 | 4 | 1 | <1 | <1 | <1 | <1 | 9 | 4 | 1 | <1 | 4 | 1 | |
| 3 | 8 | 3 | 1 | 1 | 1 | <1 | 1 | 1 | 2 | 1 | 1 | 0 | |
| MS17 | 1 | 29 | 1 | 1 | 1 | 1·5 | 2 | 1 | <1 | 1 | 1 | 1·5 | 2 |
| 2 | 51 | 1 | <1 | 1 | <1 | <1 | <1 | <1 | <1 | <1 | 1 | 5 | |
| 3 | 51 | 1 | <1 | 2 | 1 | 1 | <1 | 2 | 1 | 1 | 1 | 1 | |
| MS19 | 1 | 1 | 2 | 1 | <1 | <1 | <1 | 1 | <1 | <1 | <1 | <1 | <1 |
| 2 | 1·5 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | |
| MS39 | 1 | 39 | 3 | 5 | 1 | 3 | 2 | 2 | 7·5 | 6 | <1 | 2 | 2 |
| 2 | 58 | 1 | <1 | <1 | 2 | 2 | <1 | <1 | <1 | 1 | <1 | <1 | |
| 3 | 50 | 8 | 1 | 1 | 1 | 2 | 1 | 1·5 | 1 | <1 | 3 | <1 | |
| MS41 | 1 | 59 | 2 | 3 | 2 | 2 | 1·5 | 1 | 1 | 2 | 6 | 7 | 3 |
| 2 | 116 | <1 | 1 | <1 | <1 | <1 | 1 | <1 | 1 | <1 | <1 | <1 | |
| 3 | 74 | 2 | 1·5 | 1 | 2 | 2 | 8 | <1 | <1 | <1 | 95 | <1 | |
| MS43 | 1 | 32 | 4 | 22 | 3 | 3 | 3 | 24 | 4 | 2 | 12 | 3 | 4 |
| 2 | 75 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | |
| 3 | 66 | 2 | 2 | 1 | 13 | 2 | 1 | 2 | 2 | 2 | 7 | 1 | |
| MS49 | 1 | 6 | 5 | 9 | 11 | 5 | 4 | 11 | 4 | 35 | 6 | 5 | 31 |
| 2 | 202 | 2 | 8 | 23 | 4 | 3 | 2 | 73 | 2 | 2 | 11 | 34 | |
| 3 | 6 | 13 | 8 | 17 | 1 | 1 | 11 | 11 | 4 | 1 | 7 | 6 | |
| MS57 | 1 | 36 | 8 | <1 | 1·5 | 1 | 1 | 2 | 2 | <1 | <1 | <1 | <1 |
| 2 | 7 | 2 | 2 | 2 | 1 | 1 | <1 | <1 | <1 | <1 | <1 | <1 | |
| 3 | 6 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | |
| MS59 | 1 | 57 | 2 | 1 | 65 | 1 | 1 | 9 | 37 | 1 | 1 | 2 | 14 |
| 2 | 27 | 1 | <1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| 3 | 14 | 1 | 9 | 2 | 1 | 1 | 4 | 2 | 2 | 63 | 137 | 1 | |
| MS60 | 1 | 44 | 3 | 2 | 19 | 8 | 2 | 28 | 9 | 11 | 16 | 16 | 12 |
| 2 | 133 | 1 | <1 | 83 | <1 | <1 | 20 | <1 | 1 | <1 | 2 | <1 | |
| 3 | 23 | 2 | <1 | 13 | <1 | 2 | <1 | <1 | <1 | 10 | 26 | 2 | |
| MS67 | 1 | 49 | 2 | 3 | 2 | 1 | 2 | 2 | 1 | <1 | 1 | 1 | 58 |
The SI values shown here are derived from the day on which the T-cell response peaked. Background cpm was measured for each day and varied between 80 and 700cpm; a positive response (bold) was defined according to SI > 3 and δcpm > 1000. All patients, including MS19, gave a positive response to PPD at each time point (data not shown). It was not possible to collect samples for a third time point from patients MS19 and MS67.
As shown in Table 2, the regions most commonly recognized by MS patients were 75–99, 90–114 and 150–170 (6/11 patients), followed by 1–24, 120–144 and 135–159 (5/11 patients), and 15–39, 30–54 and 105–129 (4/11 patients). Three patients responded to region 45–69, and none of the MS patients responded to 60–84. By contrast, all healthy individuals recognized whole MBP, but only three responded to one or more peptides (individuals N6, N10 and N11; Table 3). Overall, 9/12 healthy individuals did not respond to any of the overlapping peptides, whereas only 1/11 MS patients (MS19) consistently failed to recognize either MBP or the MBP peptides. Notably, this patient was unique in not responding to either MBP or peptides derived from it.
Table 3.
Peak Stimulation Index (SI) values to MBP and MBP-peptides in healthy individuals
| MBP | 1–24 | 15–39 | 30–54 | 45–69 | 60–84 | 75–99 | 90–114 | 105–129 | 120–144 | 135–159 | 150–170 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N1 | 20 | <1 | <1 | <1 | <1 | 0 | 1 | <1 | 1 | <1 | <1 | <1 |
| N2 | 54 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 | <1 |
| N3 | 14 | 2 | 2 | 2 | 1 | 4 | 6 | 1 | 1 | <1 | 2 | 3 |
| N4 | 6 | 1 | 1·5 | <1 | <1 | <1 | 1 | 1 | <1 | <1 | 1 | <1 |
| N5 | 27 | <1 | 1·5 | <1 | <1 | <1 | 1·5 | 1 | <1 | <1 | 1 | <1 |
| N6 | 29 | 5 | 1 | 1 | 3 | 2 | 5 | 6 | <1 | 2 | 3 | 2 |
| N7 | 100 | 2 | <1 | 2 | <1 | <1 | <1 | <1 | 2 | <1 | 2 | 2 |
| N8 | 33 | 3 | <1 | 1·5 | <1 | <1 | 1 | 1 | 1 | 1 | 2·5 | 1·5 |
| N9 | 104 | <1 | <1 | 2 | <1 | 1 | 1 | <1 | 1 | 2 | 1 | <1 |
| N10 | 72 | 3 | <1 | 1 | <1 | 2 | 1·5 | <1 | <1 | 71 | 5 | <1 |
| N11 | 11 | 4 | 1 | 18 | <1 | 12 | 5 | <1 | 2 | 7 | 7 | 8 |
| N12 | 89 | 1 | <1 | <1 | <1 | <1 | 1 | 1 | <1 | <1 | <1 | <1 |
Background cpm was measured for each day and varied between 80 and 700cpm; a positive response (bold) was defined according to SI > 3 and δcpm > 1000.
Overall, the day on which the response to MBP and/or peptides peaked did not differ between healthy individuals and patients, with peak proliferation appearing between days 7–13, as illustrated for patients MS59 and MS60 in Figs 2 and 3, respectively.
Fig.2.
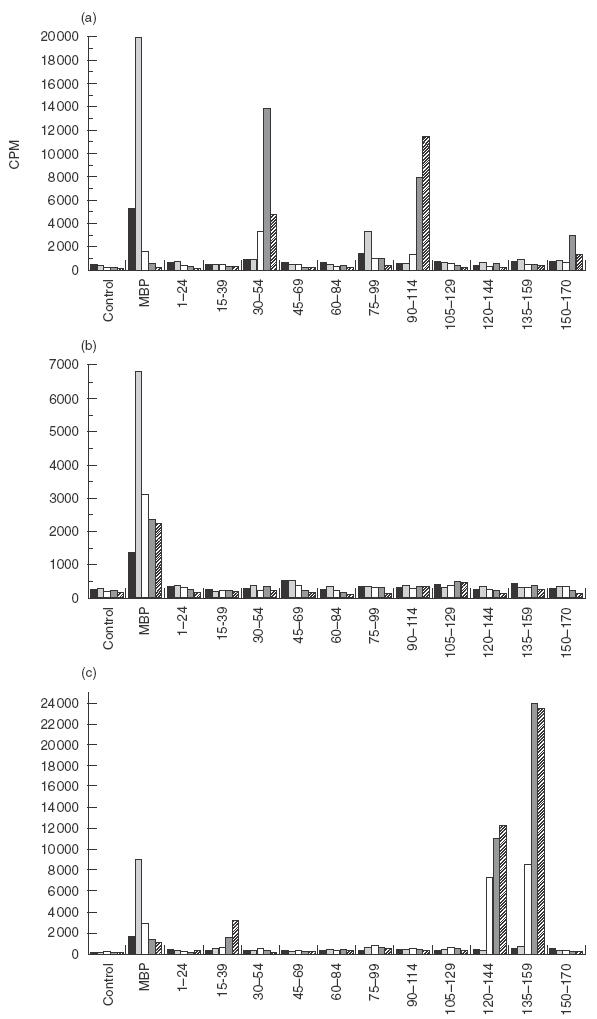
PBMC from an MS patient (MS59) were cultured in the presence of MBP and a panel of peptides spanning the full length of MBP; proliferation was measured by 3H-thymidine uptake. The response observed at (a) the first time point was significantly different from that at (b) the second (after 8 months) and (c) third time points (3 months following the second sample). (▪) Day 5; (00077;) day 7; (□) day 10; (00036;) day 12; (00004;) day 14.
Fig.3.
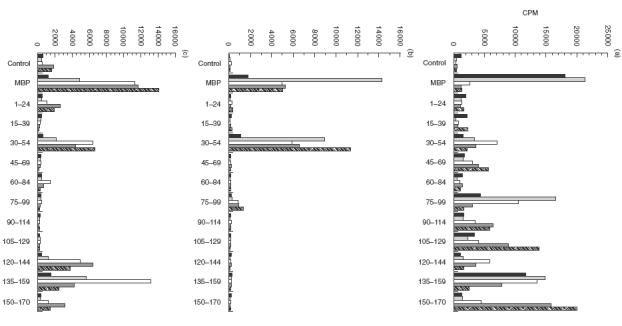
PBMC from patient MS60 were cultured in the presence of MBP and a panel of MBP-peptides, and proliferation was measured by 3H-thymidine uptake. (a) First time point; (b) second time point; (c) third time point. The patient’s broad peptide recognition pattern regressed by the second time point (10 months following the first time point) and reappeared 8 months later (third time point). (▪) Day 5; (blk14;) day 7; (□) day 9; (blk14;) day 11; (00004;) day 13.
MBP-peptide recognition in MS patients changes over time
Having established that patients with MS respond to a broad spectrum of MBP peptides, we next decided to examine whether PBMC recognition in the same individuals was focused and stable over the course of approximately 4–18 months. As shown in Table 2, MS patients did not exhibit the same peptide recognition pattern at each time point. Figure 2 represents an example of an MS patient (MS59) who responded to four MBP peptides (30–54, 75–99, 90–114 and 150–170) on the first time point; no peptide recognition was observed when the patient was recalled 8 months later, but a new pattern had appeared 3 months after that (aa 15–39, 120–144 and 135–159). Figure 3 (MS60) illustrates an example of a patient whose broad epitope response regressed to a focused response over the period of 18 months.
Overall, our results demonstrate that patients with MS do not exhibit fixed patterns of recognition (Table 2). Within each patient, the PBMC response to several peptides can persist, regress and/or shift to new regions of MBP, as exemplified by patients MS59 and MS60.
Fluctuations of peptide recognition in MS patients
When the PBMC response to peptides was analysed at three different time points, it became clear that in certain patients epitope recognition appeared to alternate rather than shift irreversibly to new peptide regions. For example, as shown in Fig. 3 and Table 2, patient MS60 exhibited a cycling pattern of recognition to aa 120–144 and 135–159. Residues 120–144 and 135–159 were amongst many recognized at the first time point tested; the response to these two regions regressed by the second time point and then reappeared at the third time point measured 8 months later. Other examples include the kinetic profiles of patients MS41 and MS49, whereby the recognition of aa 75–99, 105–129 and 135–159 alternated over several time points spanning a period of 18 months.
Mapping the response to MBP
TCC were generated from eight MS patients and one healthy individual, and used to clarify the specificity of the peptide regions identified in the kinetic response assay. The specificity of each TCC was tested by its proliferative response to the panel of 15-mer peptides. The specificity of all TCC is summarized in Table 4.
Table 4.
Summary of the MBP-and peptide-specific T-cell clones
| TCC | MBP region | Specific peptide |
|---|---|---|
| MS39:A7* | 1–24 | 5–19 |
| MS49:D3* | 30–54 | 30–44 |
| MS49:C8* | 30–54 | 30–44 |
| MS49:A8* | 30–54 | 30–44 |
| MS49:B6* | 30–54 | 30–44 |
| MS39:D7* | 60–84 | 60–74 |
| MS43:A7* | 75–99 | 83–99 |
| MS41:B6* | 75–99 | 83–99 |
| MS41:A2* | 75–99 | 83–99 |
| MS41:C6* | 75–99 | 83–99 |
| N5:8 † | 75–99 | 83–99 |
| MS60:A2* | 105–129 | 110–124 |
| MS60:B3* | 105–129 | 110–124 |
| MS60:E1* | 120–144 | 130–144 |
| MS17:A3* | 120–144 | 130–144 |
| MS60:F7* | 135–159 | 140–154 |
| MS60:D1* | 135–159 | 140–154 |
| MS57:A1* | 135–159 | 140–149 |
| MS59:F1* | 135–159 | 140–154 |
| N5:19 † | 135–159 | 140–154 |
| MS43 A3 * | 150–170 | 156–169 |
| MS43:D2† | 150–170 | 156–169 |
A map of the specificity of the peptide regions identified in the kinetic response assay was obtained through the use of TCC generated from MS patients and healthy individuals. Most of the peptides used in screening assays were 15-mer in length. However, a few were 10-mer, and one peptide was 17-mer. The specificity of each TCC was tested at least two times.
TCC derived from MS patients.
TCC derived from healthy individuals.
The TCL and TCC were selected against MBP, and all gave significant proliferative responses (i.e. δcpm > 1000 and SI > 3) to whole MBP and to the identified peptide. In addition, clones were generated to most of the regions which induced responses in Table 2. However, region 90–114, which was recognized by six patients, has produced no clones to date. Despite the fact that none of the patients responded to region 60–84, one yielded a clone to 60–74. The eight patients listed in Table 4 produced clones to one or two of their preferred epitopes from Table 2, but not to all the regions identified. Lastly, the panel of clones specific to pool 135–159 demonstrates that this region includes more than one T-cell epitope (aa 140–154 and aa 140–149).
DISCUSSION
A number of studies have attempted to delineate the immunological differences between healthy individuals and MS patients [22–25], and have analysed the T-cell reactivity to MBP [10, 11, 12, 13, 14, 26]. The body of information arising from these studies has led to a clearer understanding of the cells involved in the pathogenesis of MS, but has also given rise to conflicting data regarding the profile and dynamics of CD4+ T-cell reactivity to MBP. An explanation for the discrepancy between results may lie in the techniques used in the various studies. Without exception, studies which involved freshly-isolated PBMC have measured in vitro proliferative responses to myelin antigens at a single time point only, usually on day 6 of culture [27]. In order to map the epitopes involved in the immune response to MBP in MS patients and healthy individuals, we developed a kinetic response assay in which PBMC taken over a period of 18 months were cultured for 2 weeks with a panel of 15-mer peptides spanning the whole length of human MBP. By analysing T-cell responses at five time points (between days 5–13), we were able to establish that MS patients recognize at least one MBP-derived peptide; we would not have observed this result had we measured proliferation at only one time point because peak proliferation varied amongst MS patients and ranged between days 7–13.
Overall, the majority of patients responded to a broad spectrum of peptides compared with the control group in which only three individuals recognized one or more MBP epitopes. Notably, one of the healthy individuals was a laboratory colleague working with MBP (N10). Furthermore, in certain MS patients, the response pattern suggested cycling, with certain responses waning for a period of time only to reappear later.
We developed the kinetic response assay as a means of measuring the most frequent T-cell responses to the immunodominant regions of MBP. Several studies have employed limiting dilution analysis to examine frequencies of MBP reactive T cells in MS patients and healthy subjects, and have reported comparable frequencies of one in 105–106 T cells [8, 10, 28]. By contrast, T-cell frequencies expressing TCR chain transcripts associated with MBP p85–99 recognition were estimated to be as high as one in 300 [29] in a group of MS patients. If such a high frequency of activated autoreactive T cells was indeed present in MS patients, then our kinetic response assay would be expected to detect a response to the immunodominant epitopes at any stage of the disease and on every time point tested; this was clearly not observed. It could be argued that autoreactive T cells undergo Fas-mediated apoptosis upon antigen stimulation in vitro, thereby reducing the number of T cells measured by limiting dilution analysis. However, in a preliminary study, culture of PBMC with MBP and an anti-Fas ligand antibody (NOK2) failed to increase the anti-MBP response in MS patients (unpublished data). Therefore, assuming that in many patients T-cell frequencies are low, the kinetic assay will yield a response only when a relatively high frequency of MBP-specific T cells is present.
In addition to the three previously defined immunodominant regions (13–32, 84–103 and 144–163), we found at least four more major regions which could be important in the pathogenesis of MS. Amongst these are peptides 5–19 and 30–44. Regions 45–69 and 105–129 were only recognized by MS patients, with peptide 110–124 being defined as an MBP-specific T-cell epitope within the 105–129 region. Only one patient (MS19) consistently failed to respond to MBP and its peptides. Interestingly, however, this patient responded well to an immunodominant epitope within PLP (manuscript in preparation).
Previous studies have indicated that T-cell recognition in MS can display two patterns. Epitope spreading can result in the loss of recognition of certain epitopes [30]. By contrast, molecular studies have revealed the long-term stability of particular T-cell clonotypes in MS [31]. Our results demonstrate that the T-cell response to determinants of MBP is not stable over time but is clearly variable. In addition, progression of MS is not necessarily accompanied by the loss of response to a particular epitope(s) and the concurrent emergence of new self-determinants. This is seen in patient MS49 who experienced a severe relapse in the period between the first and second kinetic response assay, and yet recognition of regions 15–39, 30–54 and 150–170 persisted, while other peptide responses disappeared and new ones appeared. Variability was also observed in patient MS59 in whom, over the course of one year, we could demonstrate the disappearance of an epitope-specific response and the concurrent shift to a new pattern of peptide recognition. The anti-MBP response was not only variable within any one individual, but also appeared to alternate; that is, an anti-peptide response could be present at one time point, regress at a later time point and then subsequently reappear in a process of ‘T-cell cycling’. Notably, amongst the few healthy individuals whose PBMC exhibited a response to peptides, some also demonstrated T-cell cycling (data not shown).
The alternation of peptide recognition over time most probably reflects the frequency of circulating autoreactive T cells at any given time point; when these are low, responses may regress rather than disappear irreversibly. A peptide response will only be detected when the frequency of autoreactive T cells reaches a certain threshold. Our hypothesis on T-cell cycling is partly supported by a recent study on PLP [32] in which the frequencies of circulating autoreactive T cells fluctuated over time in patients with MS as well as in healthy individuals.
Our overall findings therefore differ from other studies examining the long-term dynamics of the T-cell repertoire to myelin autoantigens [30, 31]. While others found that progression of MS involved the sequential appearance and regression of epitope recognition, we demonstrate that in certain MS patients, epitope-specific responses are cyclical, i.e. a specific T-cell response may appear, regress and reappear months later reflecting changes in frequency of T cells. Once a specific T-cell repertoire is established within an individual, it is not necessarily lost during the course of disease. The T-cell epitope cycling that occurs within the repertoire of a particular individual rather emphasizes the complex and dynamic nature of the immune response associated with MS.
Six out of the 11 DR2+ve patients in this study recognized region 75–99, which is in full agreement with the existing reports that 50–60% of DR2+ve patients respond to an epitope within 75–99. It is clear from our work, however, that patients with DR2 recognize many other regions within MBP.
We generated TCL and TCC from MS patients and healthy individuals in order to confirm the response observed to the panel of peptides, and to further define the MBP regions identified in our assays. Clones were selected against eight of the 11 regions spanning the MBP molecule. Interestingly, even though residues 90–114 and, to a lesser extent, 15–39 were recognized frequently by MS patients, TCC specific for these determinants have yet to be developed in vitro. It may be relevant that a proportion of MBP-reactive TCL, specific for region 111–139, were susceptible to apoptosis in vitro following stimulation with MBP [15]. Furthermore, these Fas-sensitive MBP-specific T cells were not deleted in vivo in patients with MS as opposed to healthy individuals [15]. This argument clearly does not hold for all epitopes since we have managed to obtain TCC for many different MBP epitopes. Equally, however, an epitope(s) within the 90–114 region may be ‘cryptic’, with T cells recognizing the synthetic peptide but not the intact MBP protein. However, the higher frequency of 90–114 responses amongst DR2+ve MS patients (6/11) than DR2+ve controls (1/12) argues that they are disease specific. Alternatively, the T cells that recognize the synthetic peptide 90–114 may be cross-reactive with different disease-associated proteins. Taken together, these observations reinforce our view that the combined use of kinetic analysis of fresh PBMC and clonal analysis of established antigen-reactive TCC is required to provide a complete picture of the T-cell repertoire to MBP and/or other myelin antigens.
One of our most interesting observations is that MS patients responded far more frequently to peptide epitopes than healthy controls, despite the fact that MBP-reactive T cells were present in both, perhaps reflecting differences in their activation state [33]. Previous investigations demonstrated that MBP-specific CD4 T cells in MS patients had been activated in vivo, and that these differentiated memory cells were less dependent on CD28-mediated co-stimulation for proliferation [8]. This suggests that, in MS patients, the response to MBP peptides resides predominantly in the memory T-cell compartment, and memory T cells can be clonally expanded following presentation of antigen in conditions of weak co-stimulation. In contrast, the response to MBP in most healthy individuals resides in the naïve T-cell population, and in vitro culture conditions may not provide sufficient co-stimulatory signals for proliferation in response to peptides. Work from this and other laboratories has shown that, in MS patients, T cells recognizing MBP exhibited either the CD45RA or the CD45RO phenotype, whereas in the majority of healthy individuals, the response to MBP resided in the CD45RA T-cell population alone [9, 34]. The demonstration that the MBP response in MS patients resided in part in the CD45RO (memory) T-cell compartment provides an explanation for the peptide reactivity profile seen in this study. It is likely that some peptides can be presented by APC without processing and presentation by activated APC. Peptides may thus be presented by APC bearing low levels of co-stimulatory molecules. It follows then that memory T cells, which are less dependent on co-stimulation when compared with naïve cells, will proliferate preferentially in the presence of peptides. The MBP molecule, on the other hand, would need to be processed by APC, which would then provide the appropriate co-stimulatory signals for the proliferation of resting T cells. This would explain why all individuals respond well to MBP, why in healthy individuals this response is observed in the CD45RA population, and why healthy individuals fail to respond to peptide epitopes in the kinetic response assay.
In summary, we have demonstrated that most MS patients, as opposed to healthy individuals, respond to a broad spectrum of MBP peptides. Furthermore, there is considerable heterogeneity in the pattern of epitope recognition between different patients, and there is no single dominant epitope. The T-cell recognition pattern alternates rather than shifting irreversibly to novel epitopes, and in some cases, can exhibit a cyclical pattern of epitope recognition most likely due to changes in the frequency of circulating autoreactive T cells. These results suggest that antigen-specific therapies based on single peptide epitopes/analogues are unlikely to be effective in complex diseases such as MS, unless they can evoke the ‘bystander suppression’ shown for some (but not all) epitopes within a protein [35]. Given the heterogeneous nature of epitope recognition shown here, it is clear that a cocktail of peptides should be selected for further trials of peptide therapy in MS.
Acknowledgments
The authors wish to acknowledge the Multiple Sclerosis Society of Great Britain and Northern Ireland and the Wellcome Trust for financial support. They would also like to thank the patients and staff at Frenchay and Derriford Hospitals, and the healthy blood donors from their department for their involvement in this study. The assistance of J. Coad, Drs N. Viner and M. Harber is also much appreciated.
References
- 1.McFarlin DE, McFarland HF. Multiple Sclerosis. Part I. N Engl J Med. 1982;307:1183–8. doi: 10.1056/NEJM198211043071905. [DOI] [PubMed] [Google Scholar]
- 2.McFarlin DE, McFarland HF. Multiple Sclerosis. Part II. N Engl J Med. 1982;307:1246–51. doi: 10.1056/NEJM198211113072005. [DOI] [PubMed] [Google Scholar]
- 3.Segal BM, Raine CS, McFarlin DE, Voskuhl RR, McFarland HF. Experimental allergic encephalomyelitis by the peptide encoded by exon 2 of the MBP gene – a peptide implicated in remyelination. J Neuroimmunol. 1994;51:7–19. doi: 10.1016/0165-5728(94)90123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voskuhl RR, McFarlin DE, Stone R, McFarland HF. T lymphocyte recognition of a portion of myelin basic protein encoded by an exon expressed during myelination. J Neuroimmunol. 1993;42:187–92. doi: 10.1016/0165-5728(93)90009-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamvil S, Nelson P, Trotter J, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317:355–8. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- 6.Bielekova B, Goodwin BR, Ichert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis:Results of a phase II clinical trial with an altered peptide ligand. Nature Med. 2000;6:1167–75. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 7.Allegreta M, Nicklas JA, Sea S. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science. 1990;247:718–21. doi: 10.1126/science.1689076. [DOI] [PubMed] [Google Scholar]
- 8.Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. J Clin Invest. 1998;101:725–30. doi: 10.1172/JCI1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponsford M, Mazza G, Coad J, Campbell MJ, Zajicek J, Wraith DC. Differential responses of CD45+ve T cell subsets to MBP in multiple sclerosis. Clin Exp Immunol. 2001;124:315–22. doi: 10.1046/j.1365-2249.2001.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 1990;346:183–7. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]
- 11.Valli A, Sette A, Kappos L, et al. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest. 1993;91:616–28. doi: 10.1172/JCI116242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meinl E, Weber F, Drexler K, et al. Myelin basic protein-specific T lymphocyte repertoire in multiple sclerosis. J Clin Invest. 1993;92:2633–43. doi: 10.1172/JCI116879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvetti M, Ristori G, D’Amato M, et al. Predominant and stable T cell responses to regions of myelin basic protein can be detected in individual patients with multiple sclerosis. Eur J Immunol. 1993;23:1232–9. doi: 10.1002/eji.1830230606. [DOI] [PubMed] [Google Scholar]
- 14.Kozovska M, Zang YCQ, Aebischer I, et al. T cell recognition motifs of an immunodominant peptide of myelin basic protein in patients with multiple sclerosis:structural requirements and clinical implications. Eur J Immunol. 1998;28:1894–901. doi: 10.1002/(SICI)1521-4141(199806)28:06<1894::AID-IMMU1894>3.0.CO;2-W. 10.1002/(sici)1521-4141(199806)28:06<1894::aid-immu1894>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- 15.Zang YCQ, Kozovska MM, Hong J, et al. Impaired apoptotic deletion of myelin basic protein-reactive T cells in patients with multiple sclerosis. Eur J Immunol. 1999;29:1692–700. doi: 10.1002/(SICI)1521-4141(199905)29:05<1692::AID-IMMU1692>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 16.Deibler GE, Martenson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prepara Biochem. 1972;2:139. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 17.Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis – guidelines for research protocols. Ann Neurol. 1983;13:227–31. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 18.Plebanski M, Burtles SS. In vitro primary responses of human T cells to soluble protein antigens. J Imm Meth. 1994;170:15–25. doi: 10.1016/0022-1759(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 19.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acid Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olerup O, Zetterquist H. HLA-DR typing by PCR amlification with sequence-specific primers (PRC-SSP) in 2 hours – an alternative to serological DR typing in clinical practice including donor recipient matching in cadaveric transplantation. Tissue Antigens. 1992;39:225–35. doi: 10.1111/j.1399-0039.1992.tb01940.x. [DOI] [PubMed] [Google Scholar]
- 21.Schreuder GMT, Hurley CK, Marsh SGE, et al. The HLA dictionary 1999. Hum Immunol. 1999;60:1157–81. doi: 10.1016/s0198-8859(99)00111-1. [DOI] [PubMed] [Google Scholar]
- 22.Lisak RP, Zweiman B. In vitro cell-mediated immunity of cerebrospinal-fluid lymphocytes to myelin basic protein in primary demyelinating diseases. N Eng J Med. 1977;297:850–3. doi: 10.1056/NEJM197710202971602. [DOI] [PubMed] [Google Scholar]
- 23.Vandenbark AA, Chou YK, Bourdette D, et al. Human lymphocyte-T response to myelin basic-protein – selection of lymphocyte-T lines from MBP-responsive donors. J Neurosci Res. 1989;23:21–30. doi: 10.1002/jnr.490230104. [DOI] [PubMed] [Google Scholar]
- 24.Johnson D, Hafler DA, Fallis RJ, et al. Cell-mediated-immunity to myelin-associated glycoprotein, proteolipid protein, and myelin basic-protein in multiple-sclerosis. J Neuroimmunol. 1986;13:99–108. doi: 10.1016/0165-5728(86)90053-6. [DOI] [PubMed] [Google Scholar]
- 25.Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CC, Ben Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest. 1993;92:2602–8. doi: 10.1172/JCI116875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tejada-Simon MV, Hong J, Rivera VM, Zhang JZ. Reactivity pattern and cytokine profile of T, cells primed by myelin peptides in multiple sclerosis and healthy individuals. Eur J Immunol. 2001;31:907–17. doi: 10.1002/1521-4141(200103)31:3<907::aid-immu907>3.0.co;2-1. 10.1002/1521-4141(200103)31:3<907::aid-immu907>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 27.Greer JM, Csurhes PA, Cameron KD, McCombe PA, Good MF, Pender MP. Increased immunoreactivity to two overlapping peptides of myelin proteolipid protein in multiple sclerosis. Brain. 1997;120:1447–60. doi: 10.1093/brain/120.8.1447. [DOI] [PubMed] [Google Scholar]
- 28.Pette M, Fujita K, Wilkinson D, et al. Myelin autoreactivity in multiple sclerosis:recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple sclerosis patients and healthy donors. Proc Natl Acad Sci USA. 1990;87:7968–72. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bieganowska K, Ausubel LJ, Modabber Y, Slovik E, Messersmith W, Hafler DA. Direct ex vivo analysis of activated, Fas-Sensitive autoreactive T cells in human autoimmune disease. J Exp Med. 1997;185:1585–94. doi: 10.1084/jem.185.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuohy VK, Yu M, Yin L, Kawczak JA, Kinkel RP. Spontaneous regression of primary autoreactivity during chronic progression of experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 1999;189:1033–42. doi: 10.1084/jem.189.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects. Brain. 2000;123:508–18. doi: 10.1093/brain/123.3.508. [DOI] [PubMed] [Google Scholar]
- 32.Pender MP, Csurhes PA, Greer JM, et al. Surges of increased T cell reactivity to encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects. J Immunol. 2000;165:5322–31. doi: 10.4049/jimmunol.165.9.5322. [DOI] [PubMed] [Google Scholar]
- 33.Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol. 1998;160:1532–8. [PubMed] [Google Scholar]
- 34.Burns J, Batholomew B, Lobo S. Isolation of myelin basic protein-specific T cells predominantly from the memory T-cell compartment in multiple sclerosis. Ann Neurol. 1999;45:33–9. 10.1002/1531-8249(199901)45:1<33::aid-art7>3.3.co;2-7. [PubMed] [Google Scholar]
- 35.Anderton SM, Wraith DC. Hierarchy in the ability of T cell epitopes to induce peripheral tolerance to antigens from myelin. Eur J Immunol. 1998;28:1251–61. doi: 10.1002/(SICI)1521-4141(199804)28:04<1251::AID-IMMU1251>3.0.CO;2-O. 10.1002/(sici)1521-4141(199804)28:04<1251::aid-immu1251>3.3.co;2-f. [DOI] [PubMed] [Google Scholar]