

Abstract
Human HLA class I deficiency is a rare disease which, in most of the patients described to date, results from a defect in subunit 1 or 2 of the peptide transporter associated with antigen processing (TAP). The clinical features of TAP deficiency include a chronic inflammation of the respiratory tract and/or granulomatous skin lesions. In this report, we describe two adult siblings with an HLA class I deficiency. One individual had only spontaneously-healing skin granulomatous lesions, while the second did not display any of the symptoms associated with HLA class I deficiency and could be considered to be healthy. We show that the patients display a homozygous TAP2 mutation which blocks the maturation of HLA class I molecules. Cell surface expression of these molecules is strongly reduced, but three times higher than on cells from other previously described TAP-deficient individuals. This higher expression results, at least in part, from the presence of HLA-B7 molecules which are probably empty of peptide. The numbers of CD8+ αβ T cells are almost normal in these patients. The anti-EBV T-cell response of one patient is mediated by HLA-B7 restricted CD8+ αβ T lymphocytes recognizing the BMRF1 nuclear EBV antigen, demonstrating that CD8+ αβ T cells can participate in anti-viral responses. This study shows that TAP deficiency can remain totally asymptomatic for several decades, and suggests that in some cases, TAP-independent immune responses provide efficient protection from most of the common intracellular pathogens.
Keywords: bare lymphocyte syndrome, peptide transporter, deficiency, granuloma, Epstein-Barr virus
INTRODUCTION
Sixteen HLA class I deficient patients have been described to date [1,2] and may be classified into two groups. In the first group, three individuals, two of whom were related, showed a transcription defect in both the HLA class I and β2-microglobulin (β2m) genes, and a 10-fold reduction in the expression of HLA class I molecules. This type of defect is partially overcome by inflammatory cytokines and, probably for this reason, is asymptomatic. The second group concerns patients presenting a 100-fold reduction in the cell surface expression of HLA class I molecules due to mutations in subunits 1 or 2 of the transporter associated with antigen processing (TAP). As a result of this deficiency, antigenic peptides derived from the degradation of proteins in the cytosol cannot be transported into the lumen of the endoplasmic reticulum where they would normally associate with neosynthesized HLA class I molecules. Thus, the presentation of most intracellular antigens by HLA class I molecules is blocked. Paradoxically, these patients do not develop either recurrent or severe viral infections or neoplastic diseases as would have been expected in HLA class I deficient individuals. Disease generally appears late in childhood and consists of chronic bacterial infections of the upper respiratory tract. Pathogenesis is likely to be mediated by neutrophils, which attack the lung tissues, due to sustained chemoattraction or activation resulting from inefficient pathogen clearance. Autoreactive activated NK cells may also be involved in the progressive degradation of lung tissues. In several cases, severe chronic cutaneous granulomatous lesions have been described [3]. The origin of these lesions is poorly understood, since no particular pathogens could be identified in the skin lesions [2]. NK and γδ T cells, which may be present in the granulomatous lesions and are potentially autoreactive, could be involved in this last aspect of the clinical features associated with TAP deficiency.
In the present study, we describe two adult siblings who display a strong reduction in the level of cell surface expression of HLA class I molecules. One patient is asymptomatic, while the second only had minor skin lesions which spontaneously healed. We analysed the genetic defect and T-cell responses in one of these patients.
METHODS
Patients
Patient 1, a 46-year-old man, was initially hospitalized for intracranial hypertension related to a congenital abnormality (cyst of the septum pelucidum), which was treated by placing a ventriculo-peritoneal shunt. Six months later, he had unexplained complications, which subsequently proved to be due to obstruction of the shunt. His medical history was unremarkable except for a chronic eruption on one leg, which appeared at the age of 43. When the patient was seen for the first time aged 46, the eruption consisted of erythematous and brownish confluent lupoid papules and plaques. At that time, histological observations did not reveal granulomatous lesions. However, 6 months later, a second cutaneous biopsy showed multiple epithelioid granulomas in the dermis, rich in T cells but devoid of CD56+ cells. No micro-organisms were detected and, in particular, the patient displayed no evidence of tuberculosis or sarcoidosis. An anti-tuberculosis treatment was given for 9 months. The lesion healed 4 months after the end of the treatment. Using serological techniques, no HLA class I molecules could be detected on the blood cells.
Patient 2 was a totally asymptomatic 30-year-old woman, whose HLA class I deficiency was detected during family screening.
Cells
Daudi and HeLa cell lines, and the HB122 hybridoma producing the anti-HLA-A3 MoAb GAP-A3, were obtained from ATCC (Manassas, VA, USA). The ST-EMO (HLA-A*0301, –B*1516, –Cw*1402, TAP2–/TAP2–) cell line has been described previously [4] and IHW9055 (HLA-A*0301, –B*1402, –Cw*0802), IHW9001 (HLA–A*2402, –B*0702, –Cw*0702), IHW9026 (HLA–A*2601, –B*3801, –Cw*1203) are HLA homozygous Epstein-Barr virus transformed B (EBV-B) lymphoid cell lines of the XIIth International Histocompatibility Workshop cell line panel.
EBV-B cell lines were raised from the PBMCs of both patients and their parents, but one of the two patients’ cell lines gave a high ratio of dead cells. Consequently, data were obtained using only the cell line HA-L (patient 2, TAP2–/TAP2–) and HA-T cells (TAP2+/TAP2–) from one parent.
Cytometry
Cells were labelled with W6/32 (pan-anti-HLA class I, Dako, Trappes, France), B1G6 (anti-β2m, Beckman Coulter, Villepinte, France), GAP-A3 (anti-HLA-A3), 126.39 (anti-HLA-Bw6, kindly provided by Dr K. Gelsthorpe, The National Blood Service, Sheffield, UK) and BB7.1 (anti-HLA-B7, Silenus, Melbourne, Australia) MoAbs, or an IgG isotype control. Next, the cells were stained with FITC- or phycoerythrin-conjugated goat anti-mouse Ig Abs (Silenus and Dako, respectively) and analysed on a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) using Cellquest software. The expression of HLA class I molecules was assessed by the mean fluorescence intensity.
Complementation assay
EBV-B cell lines were infected with recombinant vaccinia viruses expressing either the TAP1 or the TAP2 subunit [5]. The expression of HLA class I molecules on infected cells was quantified as described above.
Peptides
Synthetic peptides derived from BMFR1 EBV antigen (KVKQAFNPL, IPSHSSNTAL) were purchased from Sigma-Genosys (Cambridge, UK). The sequences of these peptides were chosen for their ability to bind HLA-B7 molecules, as deduced using the programme of HLA peptide binding predictions at http://bimas.dcrt.nih.gov/molbio/hla_bind/.
Mutation analysis
Total RNA from EBV-B cells was prepared with an RNeasy extraction kit (Qiagen, Les Ullis, France). RNA was reverse transcribed with AMV reverse transcriptase (Eurogentec, Seraing, Belgium) using random hexanucleotides (Roche Diagnostics, Meylan, France) as primers. TAP2 cDNA fragments were amplified, using different sets of specific oligonucleotides (Eurogentec), which allowed the amplification of approximately 600 bp fragments distributed along the entire coding sequence of the mRNA. Amplification was performed in an OmniGene Hybaid thermocycler (Ashford, UK) under the following conditions: 94°C, 30s; 56°C, 30 s; 72°C, 1 s, 30–35 cycles. The mutation was identified by direct sequencing of amplified cDNA and genomic fragments using a BigDye Terminator sequencing kit (Perkin-Elmer France, Les Ullis, France). The sequence of TAP2 mRNA in the region of the mutation was determined by RT-PCR using the oligonucleotides ATTCCAAGACGTCTCCTTTG and CCTGAGAAGAGGGCCCAGT specific for sequences in exons VIII and XII, respectively. The amplified cDNA was purified on an agarose gel and sequenced using the primer TGGCTTCCCTTCTCCCCT. Finally, the sequence of the mutated TAP2 gene was checked by PCR using oligonucleotides 478 (CCCCCTCGAGAATCTGTACCAGCCCACAGG) and 479 (CCCCGAATTCGGAGAACAGCACAGGCTCCT), hybridizing with sequences in exons IX and X. Sequencing reactions were carried out with the primer CCAGCCCACAGGGGGACAGGTG.
Metabolic labelling and immunoprecipitation
The biosynthesis of HLA class I molecules was analysed as previously described [4]. Briefly, EBV-B cells were pre-incubated for 60 min in methionine- and cysteine-free RPMI 1640 medium (ICN Pharmaceuticals, Orsay, France). The cells were then centrifuged, resuspended in 0·5 ml of the same medium supplemented with 250 mCi S35 methionine and cysteine (Promix; Pharmacia-Amersham, Courtaboeuf, France) and incubated for 30 min. After washing, the cells were incubated for different times in RPMI 1640 complete medium. All the following steps were performed at 0°C. The cells were recovered by centrifugation and lysed in 1% Triton X100, 150 mm NaCl, 1 mm Tris, pH 8, and the nuclei were pelleted. Cleared lysates were first incubated with protein A-sepharose (Pharmacia-Amersham) and 5 μl non-immune murine serum, after which non-adsorbed proteins were incubated for 2 h with 5 μg W6/32; immune complexes were recovered on protein A-sepharose. Immuno-adsorbed proteins were then either treated with Endoglycosidase h (New England Biolabs, Beverly, MA, USA) according to the manufacturer’s recommendations, or left untreated. Samples in both cases were separated by SDS PAGE. The gels were fixed, incubated with Amplify (Pharmacia-Amersham) and dried, and the bands were revealed by autofluorography.
Anti-EBV T-cell responses
Only cells from patient 2 were available for this test. An anti-EBV T-cell line was raised by stimulating PBMCs from this patient with irradiated autologous EBV-B cells, as previously described [6]. The cells were depleted of CD4+ T cells using anti-CD4 magnetic beads (Dynabeads, Compiègne, France), and of γδ T cells using a pan-anti-γδ MoAb (IMMU510, Beckman Coulter) and anti-mouse Ig Dynabeads. Clones were obtained by seeding CD8+ T cells at 0·3 cells/well [7].
The T cells were tested for cytotoxicity against autologous, HLA-matched (IHW9055 and IHW9001), HLA mismatched (IHW9026) and HLA class I deficient (Daudi) cell lines using a standard chromium release assay.
To identify the EBV antigen(s), 1·5 × 104 COS cells were co-transfected with 100 ng of an expression vector coding for an EBV protein, and 100 ng of an expression vector coding for one of the HLA class I molecules. Transfected COS cells were tested 48 h after transfection in a CTL stimulation assay by adding 103, 104 or 105 polyclonal T cells, or 5 × 103 cloned T cells. Culture supernatant fluids were harvested 6 h later and tested for TNFα content by measuring their cytotoxicity to WEHI 164 clone 13 in a colorimetric assay [8]. Nine EBV lysis proteins (BZLF1, BMLF1, BRLF1, BHLF1, BLLF1, BCRF1, BMRF1, BALF2 and BALF4), and all of the latent EBV proteins (EBNA1, 2, 3A, 3B, 3C, LP, LMP1 and LMP2) [7,9], were tested.
RESULTS
HLA class I phenotype and genotype of the patients
Molecular typing confirmed the patients to be HLA homozygous: HLA–A*03011, –B*07021, –Cw*0702, DRB1*15, DQB1*06. Serological typing of HLA class I molecules was unsuccessful, suggesting a low expression of these molecules. The levels of HLA class I expression on PBMCs and EBV-B cells from the two patients, one parent (HAT) and one previously described TAP-deficient individual (EMO) [4], were compared by flow cytometry. HLA class I expression, as assayed with the anti-β2m or pan-anti-HLA class I MoAbs B1G6 and W6/32, was 10 and 15 times lower on PBMCs and EBV-B cells from patient HAL than on the same cells of the parent (Fig. 1a,b), but three times higher than on cells from the TAP-deficient patient EMO. The expression of HLA class I molecules on PBMCs or EBV-B cells from the second patient appeared to be 20% less than on the same cells from HAL (not shown).
Fig. 1.
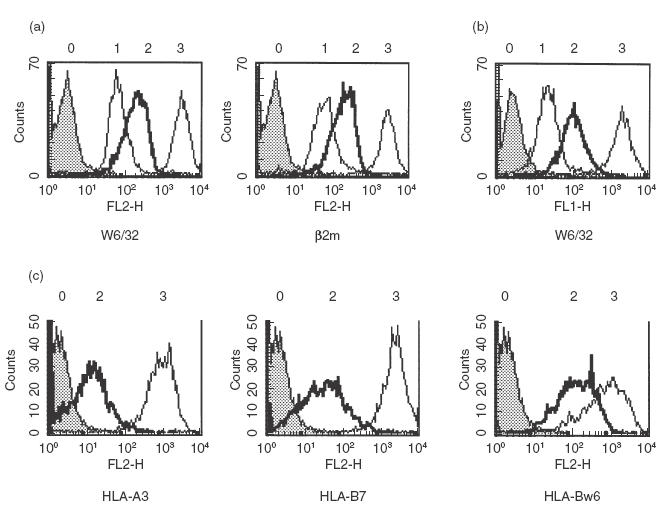
Cell surface expression of HLA class I molecules. (a) PBMCs from one patient (HAL), a parent (HAT) and a previously described TAP2-deficient patient (EMO) were labelled with the MoAb W6/32 (pan-anti-β2m-associated HLA class I heavy chain complex) an anti-β2m MoAb and analysed by cytofluorimetry. (b) Corresponding EBV-B cells were stained with W6/32 and analysed by cytofluorimetry. (c) Expression of HLA-A3 and HLA-B molecules on HA-L and HA-T EBV-B cells was investigated using anti-HLA-A3 (GAP-A3), -HLA-B7 (BB7·1), or -HLA-Bw6 (126.39) MoAbs. 0, control IgG; 1, 2, 3, cells from EMO, HAL and HAT, respectively.
HLA-B molecules can be divided serologically into two groups. Thus, ST-EMO cells are HLA–Bw4+, while HA-L cells are HLA–Bw6+. When an anti-HLA-Bw6 MoAb was used to stain HA-L cells, the expression of HLA-B7 molecules appeared to be reduced by 83% of the level on normal cells, demonstrating that cell surface levels of HLA-B7 molecules are moderately low on TAP-deficient cells (Fig. 1c). In contrast, the expression of these molecules as assessed by staining with an HLA-B7-specific MoAb, BB7.1, appeared to be reduced by 98–99% of normal levels, probably because the HLA-B7 molecules are empty, or loaded with a limited repertoire of peptides and consequently poorly recognized by the BB7.1 MoAb. A similar reduction of 98–99% was also observed when an anti-HLA-A3-specific MoAb was used to stain HA-L or ST-EMO cells, or an anti-HLA-Bw4 MoAb used to stain ST-EMO cells (data not shown).
Blood cell populations were investigated by flow cytometry (Table 1). The numbers of NK cells were found to be normal, and CD8+ αβ T cells were present in both patients. The ratios of CD8+ to CD4+ αβ T cells in PBMCs from the patients were comparable with those in PBMCs from the parents. Apart from an expansion of γδ T-cell numbers in patient 1, no significant differences in the numbers of peripheral T and NK cells were observed between the patients and their parents.
Table 1.
Analysis of blood cell populations from patients 1 and 2, and the parents (3, 4)
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Lymphocytes (per mm3) | 3000 | 3700 | 1300 | 2184 |
| Lymphocyte subpopulations (% lymphocytes) | ||||
| NK | 13 | 29 | 16 | 24 |
| TCR αβ | 70 | 59 | 67 | 57 |
| TCR γδ | 17 | 3 | 1 | 1 |
| TCR αβ CD8+ | 14 | 12 | 20 | 11 |
| TCR αβ CD4+ | 56 | 38 | 49 | 40 |
| TCR αβ CD4−CD8− | 1 | 1 | 1 | 1 |
| CD8/CD4 (αβ) | 0·19 | 0·24 | 0·29 | 0·21 |
Identification of a mutation in the ATP-binding cassette region of the TAP2 subunit
Since the HLA homozygosity suggested that the defect was linked to HLA, and since the TAP genes are located near the HLA class II genes, a diagnosis of TAP deficiency was highly probable. A complementation test was performed by infecting the EBV-B HA-L cells and, as a control, TAP2– ST-EMO cells, with recombinant vaccinia viruses expressing the TAP1 or TAP2 subunit. Infection of EBV-B cells with TAP2-, but not TAP1-recombinant viruses restored a cell surface expression of HLA class I molecules similar to levels seen on TAP+ cells (Fig. 2a), demonstrating that the cell lines had a TAP2 defect. A peptide transport defect could be confirmed by incubating HA-L cells at 27°C in the absence or presence of peptides of the BMRF1 EBV antigen predicted to bind HLA-B7 molecules. The cells were then stained with either an anti-HLA-B7 or an anti-HLA-Bw6 MoAb. Overnight incubation of the cells at 27°C alone induced a twofold increase in cell surface HLA-B molecules, detected by both MoAbs (data not shown). When the cells were incubated with the HLA-B7-specific peptides, a further 1·5-fold increase in cell surface HLA-Bw6 reactive molecules was revealed, while the anti-HLA-B7 MoAb demonstrated a sixfold increase in cell surface HLA-B7 antigen (data not shown). The ratio of mean fluorescence staining of the cells incubated in the presence of peptides and labelled with anti-HLA-B7- or the anti-HLA-Bw6-specific MoAb was similar to that seen with the hemizygous cell line HA-T (Fig. 2b). These data strongly suggest that most of the HLA-B7 molecules expressed on the plasma membrane of HA-L cells are empty, but can be loaded with exogenous peptides.
Fig. 2.
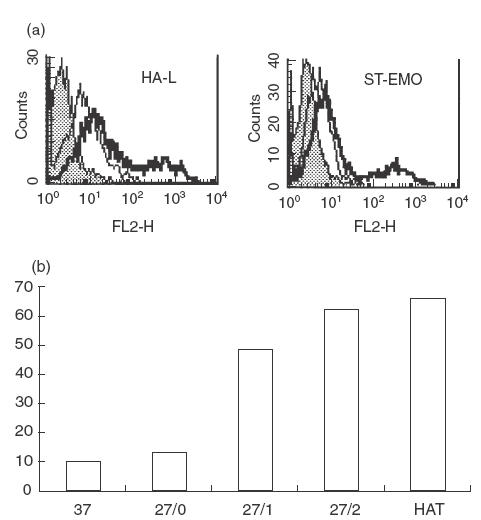
Defect in the transport of peptides. (a) EBV-B cells HA-L and ST-EMO were infected with recombinant vaccinia viruses expressing the TAP1 (thin line) or TAP2 subunit (bold line). Cells were stained with the MoAb W6/32, or an isotype control (grey histogram). (b) HA-L cells were incubated at 27°C in the presence (27/1, 27/2) or absence (27/0) of exogenous peptide 1(IPSHSSNTAL) or peptide 2 (KVKQAFNPL). Controls included cells incubated at 37°C (37) and a hemizygous cell line (HAT). Cells were stained with the anti-HLA-Bw6 MoAb 126.39 or the anti-HLA-B7 MoAb BB7.1. Ratios of the mean fluorescence intensity (expressed in percent) after staining with BB7.1 compared with 126.39 were determined under the different conditions.
To determine the nature of the mutation, RNA extracted from the EBV-B cell lines HA-L (patient 2) and HA-T (one parent) was reverse-transcribed and overlapping fragments covering the entire coding sequence of TAP2 mRNA were amplified. The quantity of amplification products was reproducibly about 4–10 times less when using cDNA from HA-L compared with that obtained after amplification of TAP2 cDNA from the hemizygous HA-T cell line. In control experiments using TAP1-specific oligonucleotides, no differences in the amounts of TAP1 mRNA were observed between the two cell lines (data not shown). The sequence of the 560 nucleotides upstream from the transcription start point of the TAP2 gene was identical in both cell lines, suggesting that the mutation was located in the transcribed part of the gene and not in the promoter (data not shown).
Sequencing of the amplified TAP2 cDNA fragments allowed identification of a point mutation at nucleotide 1635 in the coding sequence of TAP2 mRNA from HA-L cells, where a G was missing (Fig. 3). Since the electropherogram obtained using mRNA from HA-T cells (TAP2+/TAP2–) showed only the normal sequence, we concluded that the quantity of mRNA derived from the mutated allele was low compared with that derived from the functional allele. In contrast, sequencing genomic DNA from the same cell lines revealed a G to A substitution at the end of intron IX of the TAP2 gene in HA-L cells, and an overlay of the normal and mutated allelic nucleotide in HA-T cells (represented by N,Fig. 3 right bottom panel). The G to A substitution is located in the acceptor splicing site of intron IX of the TAP2 gene which, combined with the presence of a G in the first position of exon X, causes a one base shift of the splicing site (Fig. 3) and explains the missing G in the sequence of the mutated mRNA. Additional experiments demonstrated that the lower quantity of mutated TAP2 mRNA is indeed a consequence of the mutation (data not shown).
Fig. 3.
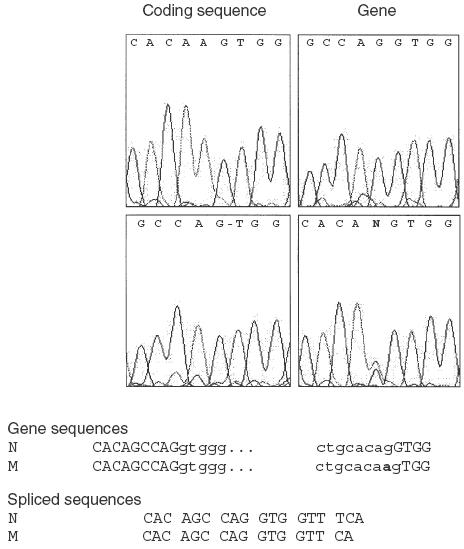
Characterization of the mutation. Electropherograms of the mutated region in the coding and gene sequences of patient 2 and one parent are shown. At the position of the mutation in intron IX, two overlapping bases can be read in the electropherogram from the parent (N, for G and A), while a missing base in the coding sequence from the patient is indicated by a –. Nucleotide sequences of the gene and coding regions of the normal (N) and mutated (M) alleles are given. The mutation is indicated in bold, and exon nucleotides are represented in block characters.
One consequence of the mutation is a predicted inhibition of the maturation of HLA class I heavy chains. Thus, HA-L (TAP2–/TAP2–) and HA-T (TAP2+TAP2–) cells were pulse-labelled for 30 min and cold-chased for various times, after which the cells were lysed and HLA class I molecules immunoprecipitated. The immunoprecipitates were either treated with endoglycosidase h (EndoH) or left untreated, and the proteins analysed by gel electrophoresis and autofluorography. Immediately after pulse labelling, most HLA class I heavy chains from the TAP2+/TAP2– cells were EndoH sensitive, but nearly all were resistant after 1 h of cold-chase. Conversely, all HLA class I heavy chains from the TAP2– homozygous cells remained EndoH sensitive (Fig. 4). Thus, the maturation of HLA class I molecules is blocked in HA-L cells, as in other TAP-deficient cells.
Fig. 4.
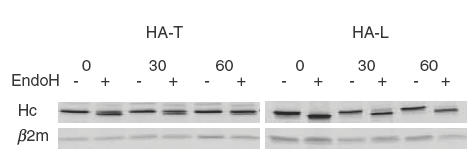
Absence of maturation of the HLA class I heavy chain in HA-L cells. EBV-B cells HA-L (TAP2–/TAP2–) or HA-T (TAP2+/TAP2–) were pulse-labelled for 30 min with 35S-methionine and -cysteine and chased for 0, 30 or 60 min. Cell membranes were solubilized and HLA class I molecules immunoprecipitated with the MoAb W6/32. The immunoprecipitates were treated (+) or not (–) with EndoH and separated by SDS-PAGE. Hc, HLA class I heavy chain; β2m, coimmunoprecipitated β2m.
Anti-EBV T-cell response
The involvement of CD8+ αβ cytotoxic T cells in anti-viral responses was demonstrated by stimulating PBMCs from patient 2 with autologous EBV-B cells. CD4+ and γδ T cells were depleted with magnetic beads, and the remaining T cells were tested for cytotoxicity against autologous and HLA-matched or mismatched EBV-B cell lines (Fig. 5a). The results showed that the anti-EBV T cells killed both autologous EBV-B cells and normal HLA–B*0702/HLA–C*0702 cells (IHW9001), but not HLA–A*0301 expressing cells (TAP2– ST-EMO and TAP+ IHW 9055 cells), the HLA mismatched cell line IHW9026 or HLA class I-deficient Daudi cells. Additional experiments were performed to characterize the EBV antigen(s). COS cells were transiently co-transfected with cDNA encoding different EBV antigens, and cDNA encoding either HLA–B*0702 or HLA–Cw*07; the transfected cells were incubated with the polyclonal T cells and T-cell activation was monitored by quantification of TNFα release. Among all HLA/EBV combinations, only the co-transfection HLA–B*0702/BMRF1 cDNAs induced the release of TNFα (data not shown). This reactivity was confirmed using a T-cell clone obtained by limiting dilution from the polyclonal T-cell line (Fig. 5b).
Fig. 5.
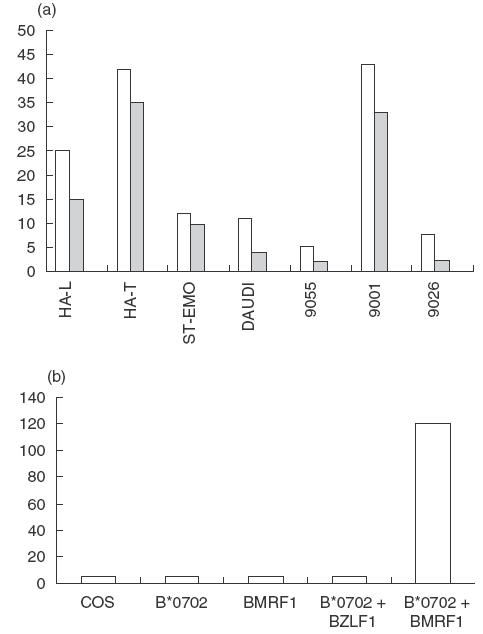
Anti-EBV T-cell response. (a) An anti-EBV T-cell line was raised by stimulating PBMCs from patient HAL with autologous EBV-B cells. The T cells were tested for cytotoxicity towards autologous EBV-B cells (HA-L, TAP2–/TAP2–, A*03011, –B*07021, –Cw0702), hemizygous cells (HA-T, TAP2+/TAP2–), ST-EMO cells (TAP2–/TAP2–, HLA-A*0301), and the TAP2+ cell lines IHW 9055 (HLA-A*0301), IHW 9001 (HLA-B*0702, HLA-Cw*0701), IHW 9026 (HLA-mismatched) and Daudi (β2m-deficient). (□) 10/1; ( ) 3/1; (▪) 0/1. (b) Antigen specificity of an anti-EBV T-cell clone. COS cells co-transfected with HLA-B*0702 and BMRF1 cDNAs were incubated for 48 h with T cells. Controls included COS cells transfected with HLA-B*0702 cDNA alone, or BMRF1 cDNA alone, or HLA-B*0702 and BZLF1 cDNA, an irrelevant EBV protein. The TNFα release (ng/ml) of the T cells was assessed by its cytotoxic effect on WEHI cells after 18h.
) 3/1; (▪) 0/1. (b) Antigen specificity of an anti-EBV T-cell clone. COS cells co-transfected with HLA-B*0702 and BMRF1 cDNAs were incubated for 48 h with T cells. Controls included COS cells transfected with HLA-B*0702 cDNA alone, or BMRF1 cDNA alone, or HLA-B*0702 and BZLF1 cDNA, an irrelevant EBV protein. The TNFα release (ng/ml) of the T cells was assessed by its cytotoxic effect on WEHI cells after 18h.
DISCUSSION
We have identified two HLA class I-deficient siblings with a homozygous mutation in subunit 2 of the TAP. Their TAP2 gene is mutated at the end of intron IX, resulting in a frame shift at codon no. 545, 34 amino acids downstream from the Walker site A of the ATP-binding cassette. Such a mutation destroys the ATP-binding site and the activity of the TAP2 subunit, but should preserve interactions with both the TAP1 subunit, as is seen with the TAP2-deficient cell line BM63.1 [10], and with tapasin.
One of the patients displayed skin lesions reminiscent of those described in several TAP-deficient patients who developed chronic, slowly progressive granulomatous lesions on the legs and face [2,11]. In most cases, these lesions worsen to chronic ulcerations and/or mutilation of the mid-face. The lesions of our patient were considerably less serious and spontaneously healed. Although this has already been reported once [11], our patient lacks the other cardinal feature of this syndrome, namely recurrent bacterial infections of the respiratory tract with subsequent bronchiectasis. The 30-year-old sister has the same deficiency but is totally asymptomatic.
Using a pan anti-HLA class I MoAb, the level of surface expression of HLA class I molecules on the patients’ cells appeared fivefold higher compared with other, previously described, TAP-deficient individuals. Additional experiments suggested that this apparent higher expression may be explained, at least in part, by the presence of HLA-B molecules recognized by anti-HLA Bw6 but not anti-HLA-B7 MoAbs. These molecules, which are not recognized by the BB7.1 MoAb, may be empty since (i) cell surface HLA-B7 molecules could be loaded by adding synthetic peptides predicted to bind HLA-B7 to the culture medium, and (ii) binding of these peptides to HLA-B7 molecules restored recognition by the anti-HLA-B7 MoAb. However, we cannot exclude the possibility that some of the BB7.1– HLA-Bw6+ molecules were already loaded with low affinity peptides which could be displaced by the exogenous peptides. We could not investigate the effect of the TAP2 mutation on the cell surface expression of HLA-C molecules, since no HLA-C specific MoAbs are available.
One remarkable feature of the blood cell populations of our patients was the presence of nearly normal numbers of CD8+ αβ T cells. This could arise from the higher than usual expression of HLA class I molecules compared with other TAP-deficient patients, which might allow the development of larger numbers of such cells at two levels: first, during thymic selection, and secondly, in the homeostatic regulation of blood cell populations, both of which are dependent on HLA class I molecules [12]. Patient 1 had quite high numbers of γδ T cells. This expanded population coincides with the presence of a local skin inflammation and is compatible with the observation that γδ T cells are present in granuloma from TAP-deficient patients [3]. Histological staining of the skin lesion of our patient revealed the presence of T cells, but we were unable to determine whether these cells were αβ or γδ. Interestingly, no CD56+ cells and, consequently, no NK cells were observed in the skin lesion. Whether granuloma disease is related to the expansion of γδ T cells, while a better outcome of this disease, i.e. lack of chronic ulceration, results from the absence of NK cells in skin lesions, nevertheless remains an open question.
We could raise an HLA-B7-restricted αβ CD8+ T-cell line which killed autologous- and HLA-B7-matched EBV-B cells. This cell line was stimulated by transfected cells co-expressing HLA-B7 and EBV BMRF1 molecules. This reactivity could be confirmed using a subclone. BMRF1 is a nuclear protein and is not expected to cross the classical transport pathway of HLA class I molecules from the endoplasmic reticulum to the cell surface. TAP-independent antigen presentation of two viral nuclear proteins in mice has also been reported [13]. These proteins may be transported into endosomal compartments by chaperone molecules such as hsc73 [14,15]. In these compartments, the antigens may be degraded and captured by HLA class I molecules, which are internalized from the cell surface and then recycled back to it [16]. TAP-independent processing of two other EBV proteins, BCRF1 [9] and LMP2 [17], has been described but BMFR1 is, to our knowledge, the first example of a nuclear TAP-independent EBV antigen.
TAP-deficient patients are not abnormally susceptible to viral infections. However, the chronic inflammation of their airways is interpreted as a consequence of an inefficient immune response in the lungs. The absence of a chronic inflammatory syndrome in these new TAP-deficient patients suggests that their immune defense is better, or may operate under different environmental conditions, compared with that of previously described TAP-deficient patients. Alternatively, the lung disease associated with TAP deficiency may be under the influence of other genes. This study thus reveals that TAP deficiency may be completely asymptomatic for decades, and suggests that TAP-deficient individuals may be discovered in other atypical situations.
Acknowledgments
We thank Dr D. Lipsker (Dermatology Clinic, Faculty of Medicine, University of Strasbourg) for critical reading of the manuscript, Professor E. Grosshans (Dermatology Clinic, University of Strasbourg) for histological analyses and Dr K. Gelsthorpe for the gift of the anti-HLA-Bw6 MoAb. Part of this work was supported by the Etablissement Français du Sang-Alsace; J. Zimmer was supported by a grant from the Lions Action ‘Vaincre le Cancer’, Luxembourg.
REFERENCES
- 1.de la Salle H, Donato L, Zimmer J, et al. The genetics of primary immunodeficiency diseases. New York: Oxford University Press; 1999. HLA class I deficiencies; pp. 181–8. [Google Scholar]
- 2.Gadola SD, Moins-Teisserenc HT, Trowsdale J, et al. TAP deficiency syndrome. Clin Exp Immunol. 2000;121:173–8. doi: 10.1046/j.1365-2249.2000.01264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moins-Teisserenc HT, Gadola SD, Cella M, et al. Association of a syndrome resembling Wegener’s granulomatosis with low surface expression of HLA class-I molecules. Lancet. 1999;354:1598–603. doi: 10.1016/s0140-6736(99)04206-3. [DOI] [PubMed] [Google Scholar]
- 4.de la Salle H, Hanau D, Fricker D, et al. Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science. 1994;265:237–41. doi: 10.1126/science.7517574. [DOI] [PubMed] [Google Scholar]
- 5.de la Salle H, Zimmer J, Fricker D, et al. HLA class I deficiencies due to mutation in subunit 1 of the peptide transporter TAP1. J Clin Invest. 1999;103:R9–R13. doi: 10.1172/JCI5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toubert A, Gomard E, Grumet FC, et al. Identification of several functional subgroups of HLA-B27 by restriction of the activity of antiviral T killer lymphocytes. Immunogenetics. 1994;20:513–25. doi: 10.1007/BF00364354. [DOI] [PubMed] [Google Scholar]
- 7.Scotet E, David-Ameline J, Peyrat MA, et al. T cell response to Epstein-Barr virus transactivators in chronic rheumatoid arthritis. J Exp Med. 1996;184:1791–800. doi: 10.1084/jem.184.5.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scotet E, Peyrat MA, Saulquin X, et al. Frequent enrichment for CD8 T cells reactive against common herpes viruses in chronic inflammatory lesions: towards a reassessment of the physiopathological significance of T cell clonal expansions found in autoimmune inflammatory processes. Eur J Immunol. 1999;29:973–85. doi: 10.1002/(SICI)1521-4141(199903)29:03<973::AID-IMMU973>3.0.CO;2-P. 10.1002/(sici)1521-4141(199903)29:03<973::aid-immu973>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Saulquin X, Bodinier M, Peyrat MA, et al. Frequent recognition of BCRF1, a late lytic cycle protein of Epstein-Barr virus, in the HLA-B*2705 context: evidence for a TAP-independent processing. Eur J Immunol. 2001;31:708–15. doi: 10.1002/1521-4141(200103)31:3<708::aid-immu708>3.0.co;2-5. 10.1002/1521-4141(200103)31:3<708::aid-immu708>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Kelly A, Powis SH, Kerr LA, et al. Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature. 1992;355:641–4. doi: 10.1038/355641a0. [DOI] [PubMed] [Google Scholar]
- 11.Willemsen M, De Coninck A, Goossens A, et al. Unusual clinical manifestation of a disfiguring necrobiotic granulomatous disease. J Am Acad Dermatol. 1995;33:887–90. doi: 10.1016/0190-9622(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 12.Cho BK, Rao VP, Ge Q, et al. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192:549–56. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigal L, Rock K. Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J Exp Med. 2000;192:1143–50. doi: 10.1084/jem.192.8.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–34. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schirmbeck R, Bohm W, Reimann J. Stress protein (hsp73)-mediated, TAP-independent processing of endogenous, truncated SV40 large T antigen for Db-restricted peptide presentation. Eur J Immunol. 1997;27:2016–23. doi: 10.1002/eji.1830270828. [DOI] [PubMed] [Google Scholar]
- 16.Gromme M, Uytdehaag FG, Janssen H, et al. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci USA. 1999;96:10326–31. doi: 10.1073/pnas.96.18.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SP, Thomas WA, Blake NW, et al. Transporter (TAP)-independent processing of a multiple membrane-spanning protein, the Epstein-Barr virus latent membrane protein 2. Eur J Immunol. 1996;26:1875–83. doi: 10.1002/eji.1830260831. [DOI] [PubMed] [Google Scholar]