

Abstract
Inoculation of Trypanosoma cruzi, Tulahuén strain, into C57BL/6 and BALB/c mice led to an acute infection characterized by marked parasitaemia, myocardial inflammation and thymocyte depletion. While C57BL/6 mice showed a progressive and lethal disease, BALB/c mice partly recovered. To characterize these murine models more effectively, we studied the parasite burden, serum levels of major infection outcome-related cytokines, the in vitro features of T. cruzi infection in peritoneal macrophages and the immunophenotype of thymic cells. The greater disease severity of T. cruzi-infected C57BL/6 mice was not linked to an increased parasite load, as parasitaemia, myocardial parasite nests and amastigote counts in peritoneal macrophages were not different from those in BALB/c mice. Cortical thymocyte loss was accompanied by the presence of apoptotic bodies and fragmented nuclear DNA, whereas fluorocytometric analysis at 17 days postinfection (p.i.) revealed a more pronounced loss of CD4+ CD8+ cells in C57BL/6 mice. This group displayed higher levels of TNF-α on days 14 and 21 p.i., in the presence of lower IL-1β and IL-10 concentrations by days 14 and 21, and days 7 and 14 p.i., respectively. Day-21 evaluation showed higher concentrations of nitrate and TNF-α soluble receptors in C57BL/6 mice with no differences in IFN-γ levels, with respect to the BALB/c group. Increased morbidity of C57BL/6 T. cruzi-infected mice does not seem to result from an aggravated infection but from an unbalanced relationship between pro- and anti-inflammatory mediators.
Keywords: apoptosis susceptibility, cytokine imbalances, T. cruzi, thymus atrophy
INTRODUCTION
Trypanosoma cruzi, which causes Chagas’ disease, is an intracellular protozoan parasite that infects about 20 million people in Latin America with sporadic cases being reported occasionally in the United States [1].
Despite extensive experimental investigations, the mechanism used by the host to control acute T. cruzi infection has not been elucidated fully. Differences in susceptibility to infection may reflect quantitative and/or qualitative dissimilarities in the immune responses against T. cruzi. Given that a variety of immune responses can be mobilized to restrain parasite infection, understanding the precise mechanisms of protection and how they fail in certain circumstances is not only of intrinsic interest, but also essential for the development of control strategies. Experimental evidence indicates that protective immunity to T. cruzi is mediated by the macrophage-activating microbicidal capacity of interferon gamma (IFN-γ) [2–4]. Tumour necrosis factor alpha (TNF-α) was also shown to be effective in controlling acute infection [5], partly by enhancing the in vitro nitric oxide (NO)-dependent trypanocidal activity of IFN-γ activated macrophages [6–8]. However, the presence of NO and TNF-α may at the same time be detrimental, given their participation in tissue injury [9, 10].
To limit the potential damage produced by the accompanying inflammatory reaction or excessive cytokine production, the immune system has evolved a series of immunoregulatory responses, i.e. down-regulatory cytokines. Among these, IL-10 inhibits T cell-dependent IFN-γ production as well as NO and cytokine synthesis by macrophages [11]. In some experimental T. cruzi infections IL-10 inhibits the trypanocidal activity and the TNF-α production displayed by IFN-γ-activated macrophages [12, 13]. TNF-α exerts different biological activities depending on whether it binds to TNFRI (55 kDa) or TNFRII (75 kDa). Both receptors can be cleaved proteolytically and are released into the circulation as soluble forms, thus modulating or blocking the biological effects of TNF-α[14, 15].
Preliminary studies in which C57BL/6 and BALB/c mice were infected with the Tulahuén strain of T. cruzi revealed an acute disease accompanied by thymocyte depletion, with C57BL/6 mice showing progressive and lethal disease and BALB/c mice exhibiting partial recovery. To gain insight into the immunopathological mechanism underlying such divergent disease outcomes, the following parameters were investigated in acutely T. cruzi-infected C57BL/6 and BALB/c mice: (1) the parasite burden and extent of inflammatory infiltrates in myocardial tissue specimens, (2) the in vitro replication of T. cruzi in peritoneal macrophages and the subsequent production of TNF-α, (3) the immunophenotype of thymic cells and a possible apoptotic phenomenon involved in thymocyte depletion and (4) the levels of TNF-α, IL-1β, IFN-γ, NO, IL-10, soluble TNFRI and TNFRII, which could potentially be correlated with the thymic alterations and/or systemic repercussion.
Finally, it is known that the synthesis of macrophage derived proinflammatory cytokines can be triggered by a glycosylphosphatidylinositol (GPI)-anchor structure of T. cruzi[16], a glycolipid reminiscent of a lipopolysaccharide (LPS)-like structure of T. cruzi[17]. This finding, together with the stimulant activity of LPS on TNF-α release [18], led us to explore the circulating levels of TNF-α and its soluble receptors when T. cruzi-infected C57BL/6 and BALB/c mice were challenged with LPS.
MATERIALS AND METHODS
Mice and infection
BALB/c and C57BL/6 mice (both sexes and 8–10 weeks of age) were bred in the animal facilities at the School of Medicine of Rosario, following institutional guidelines for their handling. Animals had access to food and water ad libitum and were injected with 100 viable trypomastigotes of the Tulahuén strain of T. cruzi, by the subcutaneous route. Parasites were maintained by serial passages in BALB/c suckling mice.
Monitoring of acute infection
Bloodstream forms of T. cruzi were assessed under standardized conditions, by direct microscopic observation of 5 μl of heparinized tail venous blood, at 7, 14 and 21 days postinfection (p.i.). Data were expressed as number of parasites/50 fields. Mice were also weighed every other day following infection to monitor the systemic repercussion of the acute disease.
In vitro infection of peritoneal macrophages
Peritoneal macrophages (PM) were obtained from 60- to 90-day-old mice of both strains. Cells were centrifuged and resuspended in MEM (Sigma Chemical Co. St Louis, MO, USA) and cultured in 12-well plates, 3 × 105 cells/well (Chamber Slide Nalge Nunc International, Naperville, IL, USA) containing the same medium supplemented with 10% fetal bovine serum (Gibco BRL, Grand Island, NY, USA), gentamycin 0·2% (10 mg/ml, Gibco) 2% penicillin–streptomycin and 2-mercaptoethanol. After 24 h the culture medium was replaced, and cells were exposed for 24 or 48h to T. cruzi trypomastigotes (Tulahuén strain) at 1:1, 0·5:1 or 0·25:1 parasite–host cell ratio. Culture supernatants from macrophage monolayers were obtained 24 and 48 h following parasite exposure for assessment of TNF-α. Supernatant fluids from 4-, 24- and 48-h cultured macrophages were also investigated for the presence of trypomastigotes. Parallel cultures in eight-well microplates were used to count intracellular parasites by indirect immunofluorescence. Briefly, PM infected 24 or 48 h earlier were washed, to remove parasites that might not have entered the cells, and covered with pooled T. cruzi-positive human sera diluted 1:30 in moist chambers for 30 min. After washing with PBS, fluorescein isothiocyanate (FITC)-conjugated goat antihuman IgG (Sanofi Diagnostics Pasteur, Marnes, France) was added to macrophage monolayers and incubated in a moist chamber at room temperature. Autofluorescence was avoided by previous staining with Evans blue. Extensive washing and mounting in phosphate buffered glycerol was completed before examination of 500 cells under an epifluorescence microscope. Immunofluorescence studies in uninfected PM cultured for 24 or 48 h yielded negative results.
Heart histology
Hearts were removed on day 14 p.i., sliced transversally in three sections, and fixed in buffered formalin. Paraffin-embedded 5 μm sections were stained with haematoxylin and eosin. Tissue parasitism was evaluated by counting the number of parasite nests that were visualized in the three sections. Foci of acute myocarditis were classified as follows: small-sized foci, slight infiltration with damage of one or two myocardial fibres; medium-sized foci, aggregated infiltrates compromising three to five muscle fibres; large-sized foci, heavy accumulation of lymphocytes and macrophages with destruction of more than five muscle fibres. The three sections were examined at whole by an experienced pathologist blinded to the study groups.
Thymic histology
Thymuses were removed at 7, 14 and 21 days p.i., fixed either in Bouin or 10% formaldehyde and embedded in paraffin. Sections were stained with haematoxylin–eosin. The score applied to analyse the thymic involution occurring in childhood acute diseases [19] was used to grade thymic alterations.
Thymocyte apoptosis
Assessment of thymocyte apoptosis was carried out by both analysis of DNA fragmentation and the terminal deoxynucleotide transferase (TdT) dUTP-biotin nick-end labelling (TUNEL) assay. For DNA fragmentation analysis, material corresponding to nearly 25% from each organ was incubated in a lysis buffer containing 50 mm Tris (pH 8·0), 50 mm NaCl, 50 mm EDTA, 1% SDS and proteinase K (50 μg/ml) for 18 h at 55°C. After extraction with phenol–chloroform–isoamylic alcohol (24:24:1), the DNA was precipitated by addition of 300 mm sodium acetate and two volumes of ethanol. Samples were resuspended in distilled water, quantified by 260 nm absorbance and 10 μg of such DNA was electrophoresed for 3h at 90V in 2% agarose containing 5μg/ml ethidium bromide. The dead end colorimetric apoptosis detection system (PROMEGA) was used following the manufacturer’s instructions.
Flow cytometry analysis of thymic cell suspensions
For double staining, thymocytes (106) resuspended in flow buffer (RPMI-1640 without phenol red supplemented with 3% fetal bovine serum (FBS), 0·1% sodium azide and 10 mm HEPES) were stained in one step with cytochrome (Cy)-coupled anti-CD4 and phycoerythrin (PE)-coupled anti-CD8a monoclonal antibodies (MoAbs, PharMingen, San Diego, CA, USA). In some experiments, thymocytes were also stained with fluorescein isothiocyanate (FITC)-coupled anti-ThB MoAbs (PharMingen). Acquisition of a minimum of 105 events was done using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Dead cells were gated on the basis of forward- and side-cell scatter. Background staining values obtained with fluorochrome conjugate isotype controls (Pharmingen) were subtracted. Results were analysed by using the Cell Quest software.
Serum TNF-α, IFN-γ, IL-1β, IL-10, TNFRI and TNFRII determinations
Mice were bled by cardiac puncture at 7, 14 and 21 days p.i. Blood was collected in a sterile, endotoxin-free tube containing no anticoagulant and kept refrigerated until centrifugation. Serum was stored frozen at – 20°C until used. Murine cytokines were measured by specific two-site enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s specifications. ELISA kits for TNF-α (detection limit 5·1 pg/ml), IL-1β (detection limit 3 pg/ml), IFN-γ (detection limit 2 pg/ml), IL-10 (detection limit 4 pg/ml), were from R&D (Minneapolis, MN, USA) and for sTNFRI (detection limit 15 pg/ml) and sTNFRII (detection limit 12 pg/ml) from Genzyme (USA). All samples were assayed in duplicate and plates were read on an ELISA reader at 405 nm. To ascertain the relationship between sTNFRI, sTNFRII and TNF-α, the molar ratios between the soluble receptors with this cytokine were also calculated, as described by Truyens et al.[20]: C1 × M2/C2, in which C1 and C2 are the respective concentrations of sTNFR and TNF-α while M1 and M2 are the molecular masses of sTNFR (I and II) and monomeric TNF-α, respectively.
Nitrite evaluation
Endogenously synthesized NO in serum was measured as nitrate at 7, 14 and 21 days p.i. Serum nitrate was assessed by reducing nitrate to nitrite with a nitrate reductase, followed by the Griess reaction [21]. Nitrite concentration was quantified using various NaNO2 concentrations as standard and data were expressed in μm.
Challenge with LPS
Animals were injected at day 21 p.i. with a single intraperitoneal dose (100 μg/mouse) of LPS from Escherichia coli serotype 0111-B4 (Sigma, St Louis, MO, USA). Serum samples were obtained immediately before and 1·5 and 4h following challenge to assess levels of TNF-α and its soluble receptors.
Statistical analysis
Differences in quantitative measurements were assessed by the Kruskal–Wallis non-parametric analysis of variance and Mann–Whitney U-test. Fisher’s exact test and the χ2 test were employed for comparison of categorical variables.
RESULTS
Parasitaemia, survival time, weight loss and acute myocarditis
As stated in Methods, 60-day-old BALB/c and C57BL/6 mice were infected with 100 trypomastigotes and assessed further for the features of acute infection. As depicted in Table 1, the two mouse strains showed no significant differences in parasitaemia on days 7, 14 and 21 p.i. The mortality rate in BALB/c mice was half that in C57BL/6 mice, and mean survival time was a little longer in the BALB/c mice (Table 1). In agreement with these findings body weight on day 17 p.i. onwards of C57BL/6 mice was significantly below that of BALB/c mice, while mice that overcame the acute disease regained normal weight by day 30–40 p.i. (data not shown). No between-strain differences were noted in the number of parasite nests in the heart at 14 days p.i. (Table 1). Because tissue lesions during acute T. cruzi infection are due to parasite-induced cell destruction followed by focal inflammation, we also determined the extent of myocardial inflammatory infiltrates. Both groups presented a similar occurrence of mononuclear inflammatory infiltrates. Although the number of inflammatory infiltrates was slightly higher in the BALB/c group, statistical comparisons revealed no difference with C57BL/6 mice (Table 1), as did the severity (mostly mild) of myocardial inflammation (data not shown).
Table 1.
Parasitaemia, mortality rate and myocarditis in mice acutely infected with T. cruzi
| Variables | BALB/c | C57BL/6 |
|---|---|---|
| Parasitaemia¶ | ||
| Day 7 p.i. | 12 (8–22) | 13 (1–22) |
| Day 14 p.i. | 43 (22–55) | 42 (4–61) |
| Day 21 p.i. | 470 (210–650) | 291 (111–691) |
| Mortality rate† | 13/32 | 26/27§ |
| Mean survival time‡ | 26·3 ± 0·7 | 23·4 ± 0·3* |
| Parasite nests/heart** | 2·2 ± 1·1 | 1·2 ± 0·3 |
| Number of inflammatory foci*** | 11·5 ± 3·1 | 4·7 ± 1·5 |
Heart samples were obtained on day 14 p.i., and fixed in buffered formalin, whereas paraffin-embedded 5 μm sections were stained with haematoxylin-eosin.
Values represent median (rank) of parasites/50 fields of five mice/group (one representative of three independent experiments).
Mice dead/total mice.
Mean ± standard error of the mean (s.e.m.), days;
and
indicate total numbers of parasite nests and inflammatory lesions detected in the three serial sections, respectively. Values represent the mean ± s.e.m. of 8–10 mice/group (one of two independent experiments).
P < 0·003.
P < 0·01.
Characteristics of the in vitro infection
We further analysed the features of the in vitro infection in PM. Table 2 shows data from representative experiments at the 1:1 parasite:host cell ratio. The number of amastigotes/cell 48 h after parasite exposure did not differ between the groups; the same was true for PM 4 h and 24h after challenge with T. cruzi (data not shown). No between-group differences were seen in the rate of infected PM or in the amount of trypomastigotes recovered in culture supernatants, irrespective of the time-point (4 h, 24h or 48h after parasite exposure). TNF-α was not detectable in culture supernatants after the first 24 h, but detectable in 48-h culture supernatants from C57BL/6 mice, and a single BALB/c mouse (P < 0·015).
Table 2.
Characteristics of the T. cruzi in vitro infection in PM
Values are mean ± s.e.m. of four mice/group (one representative of two experiments performed) at the 1:1 parasite:host cell ratio. †
Counts correspond to the number of intracellular parasites/500 cells. ¶
In 48-h culture supernatants, pg/ml.
Statistical difference in relation to BALB/c, P < 0·015.
Effect of infection on thymocyte subpopulations and apoptosis
Preliminary studies in both mouse models at the time of necropsy indicated marked thymic atrophy. We therefore systematically studied the histology of thymuses taken 7, 14 and 21 days following infection. The number of cortical thymocytes began to decrease during the second week following infection, to reach a grade 4 depletion by day 21 p.i., with no differences between groups. Recovering from the acute disease in BALB/c mice coincided with a recuperation of thymic histological architecture (data not shown). Histology revealed the presence of apoptotic bodies, and gel electrophoresis of DNA extracted from infected thymuses revealed fragmentation in a ladder form (Fig. 1b). This observation was confirmed using the TUNEL staining technique (Fig. 1a). Flow cytometry studies were performed next to determine the repercussion of the infection on the thymic populations of both mouse strains. Cell suspensions from thymuses taken at 10 and 17 days p.i. were processed for flow cytometry analysis of CD4− CD8−, CD4+ CD8−, CD8+ CD4− and CD4+ CD8+ (double positive (DP)) subpopulations. Figure 1c shows the results recorded on day 17 p.i., as they were more representative of thymic changes accompanying acute infection. Infected mice showed a marked loss of DP cells which was significantly higher in C57BL/6 than in BALB/c mice. Decreases in DP thymocytes were accompanied by increases in CD4+ CD8− and CD4− CD8+ single-positive thymocytes, which were more pronounced in C57BL/6 mice. The mean (± s.e.m.) percentages of CD4+ CD8− cells were 23·2 ± 2·9 (n = 6) versus 14 ± 1·5 (n = 6) in infected and uninfected BALB/c mice (P < 0·02), whereas in infected and control C57BL/6 mice they were 35 ± 4·8 and 9·9 ± 1 (P < 0·01, n = 6 each), respectively. Similarly, the percentages of CD4− CD8+ thymocytes in the infected and uninfected groups were 7·3 ± 0·9 versus 3·5 ± 0·4 (P < 0·01, BALB/c mice) and 12·2 ± 1·8 versus 3·75 ± 0·4 (P < 0·01, C57BL/6 mice). The percentages of ThB+ cells (which stains cortical thymocytes) were also significantly lower in C57BL/6 than in BALB/c mice (data not shown).
Fig. 1.
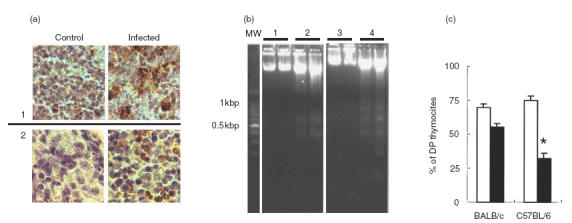
Thymic alterations in T. cruzi-infected BALB/c and C57BL/6 mice. Apoptosis visualization in thymocytes. (a) Sections from thymus medulla (1) and cortex (2) of control and infected mice, stained by TUNEL. (b) Agarose gel electrophoresis of DNA from mouse thymuses. Lanes 1 and 3 correspond to non infected C57BL/6 and BALB/c, respectively. Lanes 2 and 4 correspond to 21-day infected C57BL/6 and BALB/c, respectively. Each lane contains DNA from two different animals loaded independently. (c) Cell cytometric analysis of DP thymocytes from acutely T. cruzi-infected mice; data correspond to mean ± s.e.m. values of six mice/group (one representative of three independent sets of experiments). *P < 0·002. □, Non-infected; ▪, infected.
Levels of circulating cytokines and NO-derived metabolites during acute infection
To ascertain whether differences in infection outcome were associated with a particular pattern of cytokine response, we studied the serum levels of TNF-α and its soluble receptors, IL-1β, IL-10, IFN-γ and NO-derived metabolites. Samples from BALB/c mice correspond to surviving animals. As depicted in Table 3, serum TNF-α concentrations gradually increased in both strains of T. cruzi-infected mice as infection progressed, although levels in C57/BL/6 mice were higher and statistically different from those of the BALB/c group (days 14 and 21 p.i.). In contrast, IL-10 levels, although increased in both strains of mice, were significantly higher in BALB/c mice than in C57BL/6 mice on days 7 and 14 p.i. On day 21, IL-10 levels were similarly increased in both groups. BALB/c mice also contained abundant amounts of IL-1β in their serum 8–12-fold higher than values detected in the C57BL/6 group (days 14 and 21 p.i., respectively). Measurable amounts of IFN-γ in serum were detected only in infected mice and became noticeable by day 14 p.i., with no differences between groups (i.e. day 21 p.i. BALB/c 710 ± 112, n = 12, C57BL 679 ± 136, n = 16, mean ± s.e.m., pg/ml).
Table 3.
Presence of serum cytokines and nitrite in acutely T. cruzi infected mice
| Mediators | BALB/c | C57BL/6 | P-value |
|---|---|---|---|
| TNF-α | |||
| Day 7 p.i. | 30·2 ± 5·9 | 23·8 ± 3 | |
| Day 14 p.i. | 186 ± 29·8 | 269·6 ± 13·3 | P < 0·04 |
| Day 21 p.i. | 448·6 ± 69 | 813 ± 113 | P < 0·02 |
| Uninfected | 15 ± 1·9 | 18 ± 1 | |
| IL-10 | |||
| Day 7 p.i. | 200 ± 14·2 | 64 ± 15·1 | P < 0·005 |
| Day 14 p.i. | 1274 ± 207 | 762 ± 39 | P < 0·05 |
| Day 21 p.i. | 1458 ± 351 | 1577 ± 102 | |
| Uninfected | n.d. | n.d. | |
| IL-1β | |||
| Day 7 p.i. | 112 ± 9·3 | 22·3 ± 2·2 | P < 0·002 |
| Day 14 p.i. | 249·7 ± 38 | 30·5 ± 1·6 | P < 0·002 |
| Day 21 p.i. | 1208 ± 138 | 99·5 ± 7·7 | P < 0·002 |
| Uninfected | n.d. | n.d. | n |
| NO-3 | |||
| Day 7 p.i. | 393 ± 20 | 360 ± 20·7 | |
| Day 14 p.i. | 474 ± 13 | 607 ± 58·8 | |
| Day 21 p.i. | 879 ± 28·5 | 1179 ± 92·7 | P < 0·01 |
| Uninfected | 42 ± 5·6 | 47 ± 6 | |
| sTNFRI | |||
| Day 21 p.i. | 204 ± 22·2 | 433 ± 59·4 | P < 0·01 |
| Uninfected | 10·7 ± 6·5 | 21·5 ± 3·7 | |
| sTNFRI/TNF | |||
| Day 21 p.i. | 0·25 ± 0·02 | 0·31 ± 0·05¶ | |
| Uninfected | 0·43 ± 0·13 | 0·66 ± 0·08 | |
| sTNFRII | |||
| Day 21 p.i. | 1748 ± 156 | 2716 ± 87·5 | P < 0·001 |
| Uninfected | 717 ± 50 | 901 ± 32·7 | P < 0·05 |
| sTNFRII/TNF | |||
| Day 21 p.i. | 1·73 ± 0·29§ | 1·57 ± 0·20§ | |
| Uninfected | 23·4 ± 3·7 | 22·4 ± 2·3 | |
Data are expressed as mean ± s.e.m. (pg/ml and μm for cytokines and nitrate, respectively) of 5–8 mice/group (one representative of three experiments performed); nd, not detectable. Within-strain comparisons: difference from non infected groups,
P < 0·025
P < 0·01.
Regarding NO-derived metabolites, although nitrate levels in infected mice were increased compared to those of control mice at all time-points, no differences between infected BALB/c and C57BL/6 mice were detected on days 7 and 14 p.i. (Table 3). However, on 21 p.i. serum nitrate levels were significantly higher in C57BL/6 mice. Because serum TNF-α concentration differed between BALB/c and C57BL/6, the concentrations of soluble TNF-α receptors (sTNFRI and sTNFRII) were also determined. Measurements at day 21 p.i. showed that C57BL/6 mice had higher levels of sTNFRI and sTNFRII than the BALB/c group. Soluble TNF-α receptors were increased in both groups of infected mice when compared to non-infected animals (Table 3). In the case of the sTNFRII form, comparisons between uninfected mice revealed higher basal levels of this receptor in the C57BL/6 group. Despite increased serum levels of sTNFRI and sTNFRII in C57BL/6 mice, their sTNFRI/TNF and sTNFRII/TNF molar ratios were not different from those of the BALB/c group, mainly because the former group also presented higher TNF-α concentrations in sera.
Response to LPS challenge
To study whether T. cruzi-infected C57BL/6 and BALB/c mice responded differently to LPS, this endotoxin was injected intraperitoneally on day 21 p.i. Serum samples were collected immediately before and 1·5 and 4h following challenge. Data from such experiments are summarized in Fig. 2. In agreement with the results shown in Table 3, prechallenge levels of TNF-α, the concentration of sTNFRI and sTNFRII were lower in BALB/c than C57BL/6 mice (Fig. 2b). Upon LPS exposure, both mouse strains displayed a 12–20-fold increase in TNF-α levels by comparison with non-challenged BALB/c and C57BL/6 counterparts, respectively (Fig. 2a). Both strains of LPS-challenged mice showed a threefold increase in serum sTNFRI concentrations with no difference between them. Challenge with LPS led to a small increase in sTNFRII levels in BALB/c mice, but not in the C57BL/6 mice, whose values remained non-significantly different from those recorded in nonchallenged counterparts (Fig. 2a).
Fig. 2.
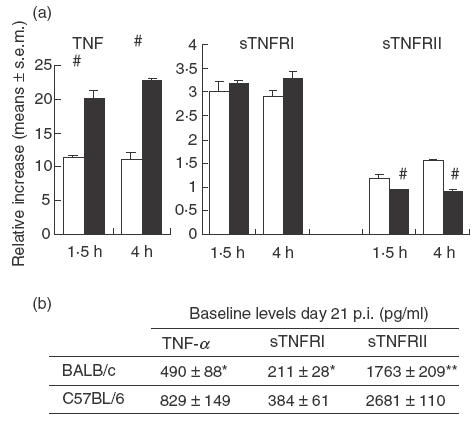
Circulating levels of TNF-α and its soluble receptors in LPS-challenged mice. (a) The relative increase in either TNF-α or its soluble receptors was calculated according the following formula: values in LPS-challenged mice/baseline values at day 21 p.i. (mean ± s.e.m.); #P < 0·01 when compared with BALB/c mice. □, BALB/c; ▪, C57BL/6. (b) Results indicate the mean ± s.e.m. (pg/ml) of 4–5 mice/group (one of two experiments with similar results). Different from C57BL/6 mice, *P < 0·05 and **P < 0·01.
Challenge with LPS in uninfected mice led to a less relevant increase in TNF-α concentrations with no major differences between mouse strains, although 4-h levels tended to be slightly increased in C57BL/6 mice (C57BL 629 ± 162, BALB/c 490 ± 155 pg/ml, mean ± s.e.m. of 4–5 mice/group).
DISCUSSION
The BALB/c and C57BL/6 murine models have been used in recent years in the light of their respective susceptibility and resistance to a series of intracellular pathogens, such as listeriosis [22], leishmaniasis [23] and mycobacteria [24]. With regard to T. cruzi, the same strains have been employed in many experimental studies as a means of investigating the pathogenesis of this trypanosomal infection, with dissimilar results. In comparison to the C57BL background, BALB/c mice seemed to be more susceptible when challenged with the Peru strain of T. cruzi[25]. Other authors have shown, instead, that both mice groups were equally susceptible to the Peruvian strain, with C57BL/10 being more resistant to the Colombian strain [26]. A more recent report indicated an increased susceptibility of C57BL/6 mice to the Tulahuén strain of T. cruzi[13]. It follows that the above-mentioned classification does not necessarily apply to all cases, implying that the interplay between host and parasite genetic differences may ultimately influence the outcome of a mouse infection with T. cruzi.
Beyond such reported dissimilarities, a remarkable finding of the present study was the fact that the greater disease severity of T. cruzi-infected C57BL/6 mice was not linked to an increased parasite load but perhaps to an imbalance in the profile of circulating cytokines and related compounds accompanying infection. Measurements of bloodstream trypomastigotes, parasite nests in myocardial tissues and intracellular growth in PM revealed no differences in the parasite burden between the two mouse strains, indicating that C57BL/6 and BALB/c mice were similarly susceptible in parasitological terms. Nevertheless, C57BL/6 mice developed a progressive disease, characterized by cachexia, severe thymic cell depletion and fatal outcome. Studies carried out in the last decade implicate host mediators triggered by the infection, i.e. TNF, as playing an important role in the wasting associated with chronic infections [27]. In line with findings in which TNF-α was found to exert a detrimental role in this trypanosomiasis [28], we demonstrated that increased TNF-α levels either in serum or in supernatants from T. cruzi-infected macrophages prevailed in C57BL/6 mice, in which the acute disease followed a lethal course. It is known that during experimentally induced and clinical inflammation, circulating soluble TNF receptors protect against excessive levels of TNF [14]. Even though TNF-α concentrations were higher in C57BL/6 mice, this group also displayed comparatively increased values of the soluble receptors making the sTNFRI/TNF and sTNFRII/TNF ratios similar to those recorded in BALB/c mice. Our results seem to be in contrast with data from Truyens et al.[20], who demonstrated that lower sTNFR/TNF ratios prevailed in fatal disease, although comparisons in their study were made between surviving and nonsurviving mice of the same mouse strain. The fact that sTNFR/TNF ratios showed no differences between the infected groups may imply that C57BL/6 mice have an increased cellular susceptibility to the harmful effects of TNF-α, considering the higher weight reductions of these mice. Added to this, IL-10 may play an important immunoregulatory role in this experimental infection in the light of its suppressive effects on proinflammatory cytokine production [29]. The presence of this cytokine in experimentally T. cruzi-infected mice was found to be an essential factor in counterbalancing the TNF-α-driven proinflammatory response [30]. This observation may apply to our situation as C57BL/6 mice exhibited lower levels of IL-10 during the early and intermediate phases of the acute disease.
Our results also indicate that cytokine responses in T. cruzi-infected BALB/c and C57BL/6 mice do not follow a polarized Th1- and Th2-type pattern. Simultaneous increases in IFN-γ and IL-10 are consistent with studies of cytokine responses to intracellular pathogens [31, 32]. These studies have suggested a potential beneficial role of IL-10 for increased host resistance during intracellular infection, despite its anti-inflammatory actions [29]. That the presence of IL-10 is essential for a better resolution of this trypanosomiasis was also illustrated by experiments wherein infection of IL-10-deficient mice resulted in decreased parasitaemia but increased mortality [33, 34].
Although similar in the early phase of acute infection, concentrations of NO-derived metabolites rose more in C57BL/6 mice than in BALB/c mice as the disease progressed. Synthesis of NO, which plays an essential role in resistance to T. cruzi infection [12, 35], can also be detrimental, given its role in inflammation and tissue damage [9]. The results reported here seem to support the latter view, as judged by the disease outcome of C57BL/6 mice. Furthermore, the demonstration that NO production is stimulated by TNF-α and inhibited by IL-10 [36] is in line with present findings.
In general, IL-1 exhibits proinflammatory activity [37], although in infections due to intracellular pathogens, i.e. listeria and leishmania, it may be implicated in immune protection [38, 39]. Furthermore, suppressed T cell responses occurring during experimental T. cruzi infection could be re-established by adding IL-1 to in vitro cultured cells, or by treating animals with IL-1 [40]. Therefore, the higher levels of IL-1β in BALB/c mice may be indicative of an effective immune response providing additional information on the mechanisms involved in protection. Strikingly, BALB/c mice also had increased IL-10 concentrations and this cytokine was shown to inhibit IL-1β synthesis [11], which illustrates the intricate network of regulatory influences surrounding cytokine production and the ultimate fate of the response emerging from such interactions.
Confirming and extending other reports of substantial thymic cell loss during acute T. cruzi infection in mice [41, 42], we now demonstrate that an apoptotic mechanism is involved in such phenomenon. There is evidence that programmed cell death in immune cells can be modulated by cytokines and related compounds. Among them, TNF-α and NO have been proposed as important effector molecules in thymocyte apoptosis [43, 44]. That both mediators may be involved in thymic cell depletion is supported by the demonstration that the higher their levels the more pronounced the relative loss of DP cells. More broadly, thymocyte depletion during T. cruzi infection in the mammalian host may have important consequences for disease outcome. There is a continuous need to replenish mature peripheral T cells that undergo normal turnover throughout life, with 30–50% of the peripheral T cell pool being replaced every 3 days in mice [45]. Thus, thymic disruption could hinder the development of an effective immune response against T. cruzi. On the other hand, thymocyte loss may also affect the autoimmune phenomenon reported during chronic T. cruzi infection [46], because the thymus is important for the generation of T regulatory cells that prevent tissue-specific autoimmunity [47, 48]. In a rat model of T. cruzi infection chronic myocardial lesions, which are thought to have an autoimmune component in its pathogenesis [46], appeared to be intensified by adult thymectomy [49].
Studies in man and in experimental models have demonstrated the presence of TNF in the circulation following the administration of LPS [18]. Because the cell surface of T. cruzi was found to contain a GPI-related glycolipid displaying LPS-like activities [16], we investigated whether changes in serum levels of TNF-α were related to a different T. cruzi infection-associated LPS responsiveness. We found that the LPS-related increase in serum TNF-α levels was much more pronounced in C57BL/6 infected mice. It is now recognized that LPS binds to CD14, a primary receptor for LPS, with Toll-like receptor 4 being involved in CD14-mediated signal transduction [50]. It is therefore tempting to speculate that such an LPS-sensing pathway is facilitated in T. cruzi-infected C57BL/6 mice rendering them more sensitive to LPS-induced TNF-α production.
Present results raise the view that differential outcome of T. cruzi infection may not be linked invariably to a distinct parasite burden, but to a finely tuned balance between responses that, although essential for host resistance, can also contribute to immunopathology.
Acknowledgments
The study was supported by a research grant from FONCYT (BID 1201/OC-AR, 05–06412) and INSERM.
References
- 1.Kirchhoff LV. American trypanosomiasis (Chagas’ disease). A tropical disease now in the United States. N Engl J Med. 1993;329:639–44. doi: 10.1056/NEJM199308263290909. [DOI] [PubMed] [Google Scholar]
- 2.Reed SG. In vivo administration of recombinant IFN-γ induces macrophage activation, and prevents acute disease, immune suppression, and death in experimental Trypanosoma cruzi infections. J Immunol. 1988;140:4342–7. [PubMed] [Google Scholar]
- 3.Torrico F, Heremans H, Rivera MT, Marck EV, Billiau A, Carlier Y. Endogenous IFN-gamma is required for resistance to acute Trypanosoma cruzi infection in mice. J Immunol. 1991;146:3626–32. [PubMed] [Google Scholar]
- 4.Revelli S, Davila H, Ferro ME, et al. Experimental Trypanosoma cruzi infection in the rat. Response to systemic treatment with recombinant rat interferon gamma. Microbiol Immunol. 1995;39:275–82. doi: 10.1111/j.1348-0421.1995.tb02201.x. [DOI] [PubMed] [Google Scholar]
- 5.Santos Lima EC, Garcia I, Vicentelli MH, Vassalli P, Minoprio P. Evidence for a protective role of tumour necrosis factor in the acute phase of Trypanosoma cruzi infection in mice. Infect Immun. 1997;65:457–65. doi: 10.1128/iai.65.2.457-465.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golden JM, Tarleton RL. Trypanosoma cruzi: cytokine effects on macrophage trypanocidal activity. Exp Parasitol. 1991;72:391–402. doi: 10.1016/0014-4894(91)90085-b. [DOI] [PubMed] [Google Scholar]
- 7.Muñoz Fernandez MA, Fernandez M, Fresno M. Synergism between tumour necrosis factor α and interferon γ on macrophage activation against intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur J Immunol. 1992;22:301–7. doi: 10.1002/eji.1830220203. [DOI] [PubMed] [Google Scholar]
- 8.Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FG. Tumour necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun. 1995;63:4862–7. doi: 10.1128/iai.63.12.4862-4867.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–12. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 10.Aggarwal BB, Natarajan K. Tumour necrosis factors: developments during the last decade. Eur Cytokine Netw. 1996;7:93–124. [PubMed] [Google Scholar]
- 11.Moore KW, O’Garra A, Malefyt RW, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11:165–90. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 12.Gazzinelli RT, Oswald IP, Hieny S, James SL, Sher A. The microbicidal activity of interferon-γ-treated macrophages against Trypanosoma cruzi involves an l-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by interleukin-10 and transforming growth factor-β. Eur J Immunol. 1992;22:2501–6. doi: 10.1002/eji.1830221006. [DOI] [PubMed] [Google Scholar]
- 13.Silva JS, Morrisey PJ, Grabstein KH, Mohler KM, Anderson D, Reed SG. Interleukin 10 and interferon γ regulation of experimental Trypanosoma cruzi infection. J Exp Med. 1992;175:169–74. doi: 10.1084/jem.175.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Zee KJ, Kohno T, Fischer E, Rock CS, Moldawer LL, Lowry SF. Tumour necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumour necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci USA. 1992;89:4845–9. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–9. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 16.Ropert C, Gazzinelli RT. Signalling of immune system cells by glysylphosphatidylinositol (GPI) anchor and related structures derived from parasitic protozoa. Curr Opin Microbiol. 2000;3:395–403. doi: 10.1016/s1369-5274(00)00111-9. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg SS, Cordeiro MN, Silva-Pereira AA, Mares-Guia ML. Release of lipopolysaccharide (LPS) from cell surface of Trypanosoma cruzi by EDTA. Int J Parasitol. 1983;13:11–8. doi: 10.1016/s0020-7519(83)80062-9. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez-Ramos JC, Bluethmann H. Molecules and mechanisms operating in septic shock: lessons from knockout mice. Immunol Today. 1997;18:329–34. doi: 10.1016/s0167-5699(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 19.Van Baarlen J, Schuurman HJ, Huber J. Acute thymus involution in infancy and chilhood. A reliable marker for duration of acute illness. Hum Pathol. 1988;19:1155–60. doi: 10.1016/s0046-8177(88)80146-1. [DOI] [PubMed] [Google Scholar]
- 20.Truyens C, Torrico F, Lucas R, De Baetselier P, Buurman WA, Carlier Y. The endogenous balance of soluble tumour necrosis factor receptors and tumour necrosis factor modulates cachexia and mortality in mice acutely infected with Trypanosoma cruzi. Infect Immun. 1999;67:5579–86. doi: 10.1128/iai.67.11.5579-5586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 22.Cheers C, Haigh AM, Kelso A, Metcalf D, Stanley ER, Young AM. Production of colony-stimulating factors (CSFs) during infection: separate determinations of macrophage-, granulocyte-, granulocyte-macrophage, and multi-CSFs. Infect Immun. 1988;56:247–51. doi: 10.1128/iai.56.1.247-251.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinzel FP, Rerko RM, Hujer AM. Underproduction of interleukin-12 in susceptible mice during progressive leishmaniasis is due to decreased CD40 activity. Cell Immunol. 1998;184:129–42. doi: 10.1006/cimm.1998.1267. 10.1006/cimm.1998.1267. [DOI] [PubMed] [Google Scholar]
- 24.Appelberg R, Castro AG, Pedrosa J, Silva RA, Orme IM, Minoprio P. Role of gamma interferon and tumour necrosis factor alpha during T-cell-independent and -dependent phases of Mycobacterium avium infection. Infect Immun. 1994;62:3962–71. doi: 10.1128/iai.62.9.3962-3971.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrightsman R, Krassner S, Watson J. Genetic control of responses to Trypanosoma cruzi in mice: multiple genes influencing parasitaemia and survival. Infect Immun. 1982;36:637–44. doi: 10.1128/iai.36.2.637-644.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrade V, Barral-Netto M, Andrade SG. Patterns of resistance of inbred mice to Trypanosoma cruzi are determined by parasite strain. Braz J Med Biol Res. 1985;18:499–506. [PubMed] [Google Scholar]
- 27.Beutler B, Cerami A. The history, properties, and biological effects of cachectin. Biochemistry. 1988;27:7575–82. doi: 10.1021/bi00420a001. [DOI] [PubMed] [Google Scholar]
- 28.Truyens C, Torrico F, Angelo-Barrios A, et al. The cachexia associated with Trypanosoma cruzi acute infection in mice is attenuated by anti-TNF-α, but not by anti-IL-6 or anti-IFN-γ antibodies. Parasite Immunol. 1995;17:561–8. doi: 10.1111/j.1365-3024.1995.tb00999.x. [DOI] [PubMed] [Google Scholar]
- 29.Smith CR, Terminelli C, Kenworthy-Bott L, Calzetta A, Donkin J. The cooperative effects of TNF-α and IFN-γ are determining factors in the ability of IL-10 to protect mice from lethal endotoxemia. J Leukoc Biol. 1994;55:711–8. doi: 10.1002/jlb.55.6.711. [DOI] [PubMed] [Google Scholar]
- 30.Hölscher C, Mohrs M, Dai WJ, Köhler G, Ryffel B, Schaub GA, Mossmann H, Brombacher F. Tumour necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. 2000;68:4075–83. doi: 10.1128/iai.68.7.4075-4083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bohn E, Heesemann J, Ehlers S, Autenrieth IB. Early gamma interferon mRNA expression is associated with resistance of mice against Yersinia enterocolitica. Infect Immun. 1994;62:3027–32. doi: 10.1128/iai.62.7.3027-3032.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pie S, Bernard PM, Bachi PT, Nauciel C. Gamma interferon and interleukin-10 gene expression in innately susceptible and resistant mice during early phase of Salmonella typhimurium infection. Infect Immun. 1996;64:849–54. doi: 10.1128/iai.64.3.849-854.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrahamsohn IA, Coffman RL. Trypanosoma cruzi-IL-10, TNF, IFN-gamma and IL-12 regulate innate and acquired-immunity to infection. Exp Parasitol. 1996;84:231–44. doi: 10.1006/expr.1996.0109. 10.1006/expr.1996.0109. [DOI] [PubMed] [Google Scholar]
- 34.Hunter CA, Ellis Neyes LA, Slifer T, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. 1997;158:3311–6. [PubMed] [Google Scholar]
- 35.Petray P, Castaños-Velez E, Grinstein S, Örn A, Rottenberg ME. Role of nitric oxide in resistance and histopathology during experimental infection with Trypanosoma cruzi. Immunol Lett. 1995;47:121–6. doi: 10.1016/0165-2478(95)00083-h. [DOI] [PubMed] [Google Scholar]
- 36.Bogdan C, Röllinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12:64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 37.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 38.Mitsuyama M, Igarashi KI, Kawamura I, Ohmori T, Nomoto K. Difference in the induction of macrophage interleukin-1 production between viable and killed cells of Listeria monocytogenes. Infect Immun. 1990;58:1254–60. doi: 10.1128/iai.58.5.1254-1260.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho JL, Badaró R, Schwartz A, et al. Diminished in vitro production of interleukin-1 and tumour necrosis factor-α during acute visceral leishmaniasis and recovery after therapy. J Infect Dis. 1992;165:1094–102. doi: 10.1093/infdis/165.6.1094. [DOI] [PubMed] [Google Scholar]
- 40.Reed SG, Pihl DI, Grabstein KH. Immune deficiency in chronic Trypanosoma cruzi infection. Recombinant IL-1 restores Th function for antibody production. J Immunol. 1989;142:2067–71. [PubMed] [Google Scholar]
- 41.Savino W, Leite de Moraes MC, Joskowicz MH, Dardenne M. Studies on the thymus in Chagas’disease. I. Changes in the thymic microenvironment in mice acutely infected with Trypanosoma cruzi. Eur J Immunol. 1989;19:1727–33. doi: 10.1002/eji.1830190930. [DOI] [PubMed] [Google Scholar]
- 42.Antunez MI, Feisntein RE, Cardoni RL, Grönvik KO. Trypanosoma cruzi. T cell subpopulations in the Peyer’s patches of BALB/c infected mice. Exp Parasitol. 1997;87:58–64. doi: 10.1006/expr.1997.4171. 10.1006/expr.1997.4171. [DOI] [PubMed] [Google Scholar]
- 43.Hernández Caselles T, Stutman O. Immune functions of tumour necrosis factor. I. Tumour necrosis factor induces apoptosis of mouse thymocytes and can also stimulate or inhibit IL-6-induced proliferation depending on the concentration of mitogenic stimulation. J Immunol. 1993;151:3999–4012. [PubMed] [Google Scholar]
- 44.Fehsel K, Kroncke KD, Meyer KL, Huber H, Wahn V, Kolb-Vachofen V. Nitric oxide induces apoptosis in mouse thymocytes. J Immunol. 1995;155:2858–65. [PubMed] [Google Scholar]
- 45.Rocha B, Freitas AA, Coutinho RA. Population dynamics of T-lymphocytes. Renewal rate and expansion on the peripheral lymphoid organs. J Immunol. 1983;131:2158–64. [PubMed] [Google Scholar]
- 46.Soares MBP, Ribeiro Dos Santos R. Immunopathology of cardiomyopathy in the experimental Chagas’disease. Mem Inst Oswaldo Cruz. 1999;94(Suppl. 1):257–62. doi: 10.1590/s0074-02761999000700043. [DOI] [PubMed] [Google Scholar]
- 47.Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–8. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- 48.Seddon B, Mason D. The third function of the thymus. Immunol Today. 2000;21:95–9. doi: 10.1016/s0167-5699(99)01559-5. [DOI] [PubMed] [Google Scholar]
- 49.Bottasso OA, Revelli SS, Davila H, et al. Enhanced myocardial lesions in chronically Trypanosoma cruzi-infected rats subjected to adult thymectomy. Immunol Lett. 1993;37:175–80. doi: 10.1016/0165-2478(93)90028-z. [DOI] [PubMed] [Google Scholar]
- 50.Beutler B. Tlr4. central component of the sole mammalian LPS sensor. Curr Opin Immunol. 2000;12:20–6. doi: 10.1016/s0952-7915(99)00046-1. [DOI] [PubMed] [Google Scholar]