

Abstract
To investigate whether tumour necrosis factor α (TNFα) plays a role in the pathogenesis of hepatitis C virus-associated mixed cryoglobulinaemia (HCV-MC), we measured soluble TNFα and its soluble p55 (sTNFR1) and p75 (sTNFR2) receptors in the serum of patients with HCV-MC. TNFα, sTNFR1 and sTNFR2 were measured in the serum of 32 patients with HCV-MC, 18 patients with hepatitis C without MC (HCV) and 18 healthy volunteers, using specific immunoassays. Correlations between clinical and biological parameters and the concentrations of TNFα and sTNFRs were established by studying detailed clinical records of the 32 HCV-MC patients. Although higher, TNFα levels were not significantly different in HCV-MC patients compared with healthy or HCV controls. sTNFR1 and sTNFR2, however, were significantly higher in HCV-MC compared with controls or with HCV patients, and higher concentrations of sTNFR1 and sTNFR2 were observed in patients with severe visceral vasculitis, compared with patients with limited purpura. sTNFR1 concentrations positively correlated with fibrinogen levels but TNFα, sTNFR1 and sTNFR2 did not correlate with other biological parameters such as rheumatoid factor concentrations, CH50 or C4 values. These data suggest a role for TNFα in the pathogenesis of the immune complex-mediated vasculitis associated with HCV-MC.
Keywords: mixed cryoglobulinaemia, tumour necrosis factor α, soluble TNF receptors, hepatitis C, vasculitis
INTRODUCTION
Tumour necrosis factor α (TNFα) and interleukin (IL)-1α/β are the two most important proinflammatory cytokines in vivo [1–3]. Both cytokines (TNFα and IL-1α) exist as cell-associated, biologically-active precursors acting through juxtacrine interactions on target cells during cell-to-cell contacts [4–7], as well as soluble mature molecules acting through autocrine and paracrine interactions. TNFα and IL-1α/β act on endothelial cells (EC) to induce a proinflammatory state characterized by vascular thrombosis, leucocyte adhesion and tissular infiltration, which has been observed in several models of vasculitis [1–3]. TNFα acts on cells through fixation to two specific membrane receptors, namely p55 TNFR (TNFR1) and p75 TNFR (TNFR2) [2,8]. These two receptors belong to a large family of receptors including CD40, Fas and the nerve growth factor receptor [9]. The TNFR1 mediates the majority of the effects of the soluble 17 kD form of TNFα, while TNFR2 appears to be a specific ligand for the 23 kD precursor membrane forms of TNFα[8,10]. After cell stimulation by various stimuli, including TNFα itself, these two receptors can be proteolitically cleaved into two soluble forms, sTNFR1 and sTNFR2, which can be detected at high concentrations for a prolonged period of time in the circulation of patients with various inflammatory diseases [8,11–13.
Type II and type III mixed cryoglobulinaemia (MC) are associated with autoimmune or chronic infectious diseases [14,15]. MC have been recognized to be strongly associated with chronic hepatitis C virus (HCV) infection [16–19]. MC associated with HCV (HCV-MC) is more likely to be observed in female patients over 50 years-old with cirrhosis on liver biopsy, although this last point is controversial [20,21]. Chronic palpable purpura, leg ulcer, peripheral neuropathy and renal glomerulopathy are well known clinical manifestations of MC [14,15]. HCV-MC can induce cutaneous vasculitis revealed by chronic purpura, but also severe visceral vasculitis reproducing polyarteritis nodosa [22–24]. MC clinical manifestations are due to an immune complex-mediated vasculitis affecting small- and medium-sized vessels [14,15]. Since immune complex-induced vasculitis is characterized by endothelial inflammatory lesions in which TNFα seems to be an important mediator, we investigated whether TNFα and sTNFRs were increased in the serum of patients with HCV-MC, and suggest a role for the cytokine in this disease.
PATIENTS AND METHODS
Patients
Thirty-two patients (aged 36 to 81 years) with HCV-MC were included in this retrospective study. Control groups were composed of 18 patients with HCV chronic infection without MC (HCV, aged 26 to 76) and 18 healthy volunteers (aged 25 to 71). All the patients and the subjects belonging to the control groups were negative for the human immunodeficiency virus. Chronic HCV infection was defined as follows: alanine amino-transferase values more than twice the upper normal limit for more than 6 months, anti-HCV antibodies detected by third generation ELISA, HCV-RNA detected by polymerase chain reaction amplification techniques using specific primers, and histological lesions compatible with chronic hepatitis C on liver biopsy. Patients were considered as having HCV-MC if they had evidence of HCV infection associated with clinical symptoms of vasculitis and the presence of serum cryoprecipitates and complement consumption. All patients with HCV-MC were untreated and clinically active at the time of blood collection. Detailed clinical records were obtained for 32 patients with HCV-MC. Clinical manifestations were classified and scored as follows: low-grade severity vasculitis limited to chronic palpable cutaneous purpura and leukocytoclastic vasculitis on skin biopsy, in the absence of high blood pressure, proteinuria and elevated creatininemia, with normal neurological examination and electromyography; high grade severity vasculitis consisting in extensive cutaneous purpura associated with severe visceral involvement affecting either the kidney (high blood pressure with systolic > 160 mmHg or diastolic > 95 mmHg and/or creatininaemia > 120 μmol/l and/or proteinuria > 1 g/24 h and histological findings related to cryoglobulinemia on renal biopsy), or the peripheral nerves (clinical symptoms and abnormal electromyography associated with vasculitis on neuromuscular biopsy), or the digestive tract, or the central nervous system (ischemic clinical manifestations associated with vasculitis on biopsy and/or microaneurisms/non-athreromatous occlusions on aortoarteriography).
Several biological parameters were studied, including rheumatoid factor (kit for quantitative determination using haemagglutination purchased from Laboratoire Fumouze, Levallois-Perret, France), cryoglobulin, haemolytic complement CH50, complement C4 fraction, C-reactive protein (CRP) and fibrinogen concentrations.
Detection, isolation and characterization of cryoglobulin
Cryoglobulin was precipitated from serum isolated at 37°C and incubated at 4°C for up to 7 days in the presence of 0·1 g/l sodium azide. The immunoglobulin composition of the washed cryoprecipitates was determined using a previously described immunoblotting method [25].
TNFα and soluble TNFRs measurements
Blood was obtained by venipuncture and serum aliquoted and stored at – 80°C until assayed. TNFα, sTNFR1 and sTNFR2 were measured using specific ELISAs from R & D Systems (Abingdon, UK). These assays are sandwich immunoassays using a capture anti-TNFα, anti-sTNFR1 or sTNFR2 MoAbs and polyclonal antibodies, which each did not cross-react with the other molecules measured. The detection limit is less than 5 pg/ml, and intra- and inter-assay variations are less than 10% for TNFα. For both sTNFR1 and sTNFR2 assays, the detection limit is 25 pg/ml, and intra- and interassay variations are less than 8%. These three assays are not influenced by the presence of rheumatoid factors, even at high concentrations, as pointed out by the manufacturer and tested in our laboratory.
Soluble forms of endothelial leucocyte adhesion molecule (sELAM), intercellular adhesion molecule-1 (sICAM-1) and vascular cell adhesion molecule-1 (sVCAM-1) were measured in HCV-MC sera using a specific ELISA from R & D Systems.
Statistical analysis
Analysis was done using the SPSS Base 8.0.1 statistical package (SPSS Inc., Chicago, IL, USA). Values of TNFα and sTNFRs are presented as the mean ± s.e.m. Differences between TNFα and sTNFRs concentrations in the different groups were tested using the Tukey’s test. Correlations between variables were analysed using Pearson’s or Spearman’s rank correlations. Level of significance was fixed at 0·05 for all statistical tests. Tukey’s box-plot representations were used in some of the figures. Briefly, the horizontal line in the middle of a box marks the median of the samples, while the hinges of each box represent the 25th and 75th percentiles (thereby including 50% of the data within a box). The vertical lines extending above and below each box show the range of values that fall within 1·5 s.d. of the hinges. Values between 1·5 and 3 s.d. outside the hinges are considered outside values.
RESULTS
Patients characteristics
Characteristics of the different patient groups are summarized in Tables 1, 2 and 3. The HCV control group (Table 1) consisted of six females and 12 males, with a mean age of 51 ± 15, mean ASAT and ALAT concentrations of 80 ± 10 IU/l and 110 ± 16 IU/l, respectively, and a mean Knodell index of 9 ± 1. The HCV-MC group with vasculitis limited to the skin (Table 2) consisted of 10 females and four males, with a mean age of 66 ± 10, mean ASAT and ALAT concentrations of 71 ± 21 IU/l and 80 ± 25 IU/l, respectively, and a mean Knodell index of 7·4 ± 1. The HCV-MC group with severe visceral vasculitis (Table 3) consisted of 10 females and eight males, with a mean age of 66 ± 11, ASAT concentration (57 ± 11 IU/l), ALAT concentration (68 ± 15) and a mean Knodell index at 7 ± 0·7.
Table 1.
Characteristics of control patients with hepatitis C without mixed c ryoglobulinemia
| Patients, sex, age | ASAT (N: 6–53 IU/l) | ALAT (N: 7–40 IU/l) | HCV RNA | Knodell index |
|---|---|---|---|---|
| 1. F, 6 9 | 79 | 50 | + | 6 |
| 2. F, 76 | 144 | 112 | + | 11 |
| 3. F, 58 | 110 | 180 | + | 14 |
| 4. M, 52 | 102 | 100 | + | 7 |
| 5. M, 45 | 87 | 100 | + | 8 |
| 6. M, 71 | 22 | 17 | + | 9 |
| 7. M, 66 | 96 | 74 | + | 7 |
| 8. M, 27 | 46 | 98 | + | 7 |
| 9. M, 34 | 35 | 85 | + | 8 |
| 10. F, 56 | 184 | 179 | + | 12 |
| 11. M, 34 | 30 | 65 | + | 8 |
| 12. M, 48 | 120 | 170 | + | 13 |
| 13. M, 31 | 24 | 38 | + | 9 |
| 14. M, 55 | 33 | 38 | + | 3 |
| 15. M, 56 | 89 | 135 | + | 13 |
| 16. M, 58 | 95 | 186 | + | 12 |
| 17. F, 26 | 63 | 178 | + | 10 |
| 18. F, 63 | 77 | 85 | + | 8 |
Table 2.
Characteristics of patients with low grade severity forms of hepatitis C-associated mixed cryoglobulinemia vasculitis
| Patients, sex, age | ASAT IU/1 | ALAT IU/l | HCV RNA | Knodell index | Cryo type | Clinical symptoms |
|---|---|---|---|---|---|---|
| 1. F, 67 | 15 | 16 | + | 7 | II | S, J |
| IgMκ | ||||||
| 2. F, 69 | 31 | 22 | + | 7 | III | S, J |
| 3. F, 48 | 29 | 83 | + | 6 | II | S |
| IgMκ | ||||||
| 4. M, 71 | 293 | 184 | + | 14 | II | S |
| IgMκ | ||||||
| 5. M, 78 | 26 | 33 | + | 2 | II | S |
| IgMκ | ||||||
| 6. F, 46 | 100 | 80 | + | 12 | II | S |
| IgMκ | ||||||
| 7. F, 60 | 35 | 47 | + | ND | II | S |
| IgMκ | ||||||
| 8. F, 71 | 25 | 34 | + | 5 | III | S |
| 9. F, 80 | 10 | 18 | + | ND | II | S |
| IgMκ | ||||||
| 10. F, 71 | 60 | 42 | + | 6 | II | S |
| IgMκ | ||||||
| 11. F, 67 | 142 | 128 | + | 7 | II | S, J |
| IgMκ | ||||||
| 12. M, 63 | 166 | 360 | + | 10 | II | S |
| IgMκ | ||||||
| 13. M, 67 | 55 | 38 | + | 7 | III | S |
| 14. F, 61 | 24 | 52 | + | 6 | II | S, J |
| IgMκ |
S, skin; J, joints.
Table 3.
Characteristics of patients with highly severe forms of vasculitis due to hepatitis C-associated mixed cryoglobulinemia
| Patients, sex, age | ASAT IU/1 | ALAT IU/l | HCV RNA | Knodell index | Cryo type | Clinical symptoms |
|---|---|---|---|---|---|---|
| 1. M, 61 | 11 | 20 | + | 5 | III | S, J, DT, PN, K |
| 2. M, 63 | 34 | 17 | + | 11 | II | S, J, K (MPGN) |
| IgMκ | ||||||
| 3. F, 36 | 21 | 16 | + | 2 | II | S, PN, CNS, K |
| IgMκ | ||||||
| 4. M, 60 | 9 | 50 | + | 7 | II | S, J, PN, GS |
| IgMκ | ||||||
| 5. F, 72 | 60 | 75 | + | ND | II | S, J, K (MPGN) |
| IgMκ | ||||||
| 6. M, 71 | 90 | 120 | + | 6 | II | S, J, PN, GS |
| IgMλ | ||||||
| 7. F, 81 | 24 | 32 | + | 6 | II | S, PN, GS |
| IgMκ | ||||||
| 8. M, 58 | 73 | 60 | + | 8 | II | S, PN |
| IgMλ | ||||||
| 9. F, 66 | 77 | 62 | + | 6 | II | S, J, PN |
| IgMκ | ||||||
| 10. F, 74 | 70 | 40 | + | 10 | II | S, J, PN |
| IgMκ | ||||||
| 11. F, 59 | 52 | 65 | + | 7 | III | S, PN |
| 12. M, 81 | 30 | 41 | + | ND | II | S, PN, CNS |
| IgMκ | ||||||
| 13. F, 70 | 100 | 120 | + | ND | II | S, PN, GS |
| IgMκ | ||||||
| 14. M, 71 | 22 | 40 | + | 5 | II | S, DT, K (MPGN) |
| IgMκ | ||||||
| 15. F, 62 | 68 | 85 | + | 10 | II | S, J, PN |
| IgMκ | ||||||
| 16. M, 72 | 28 | 35 | + | 5 | III | S, PN, GS |
| 17. F, 54 | 200 | 300 | + | 11 | II | S, PN |
| IgMκ | ||||||
| 18. F, 76 | 40 | 46 | + | 5 | II | S, J, PN |
| IgMκ |
S: skin, J: joints, DT: digestive tract, K: kidney, MPGN: mem-branoproliferative glomerulonephritis, PN: peripheral nerve, CNS: central nervous system, GS: general symptoms (fever, weight loss).
Increased sTNFR1 and sTNFR2 concentrations in patients with HCV-MC
Although higher, concentrations of TNFα were not significantly different in HCV-MC patients than in healthy controls (25 ± 10 pg/ml versus 5 ± 1·5 pg/ml, P < 0·2, Fig. 1a), or in HCV controls (25 ± 10 pg/ml versus 7·4 ± 3·5 pg/ml, P < 0·3). In addition, TNFα concentrations in HCV patients were not significantly different from healthy controls (7·4 ± 3·5 pg/ml versus 5 ± 1·5 pg/ml, P < 0·9). On the other hand, sTNFR1 (2219 ± 248 pg/ml versus 584 ± 36 pg/ml, P < 0·0001) and sTNFR2 (6987 ± 980 pg/ml versus 2603 ± 136 pg/ml, P < 0·001) were found to be elevated in HCV-MC patients compared with healthy controls (Fig. 1b,c). Similarly, when compared with patients with HCV without MC, sTNFR1 (2219 ± 248 pg/ml versus 1459 ± 98 pg/ml, P < 0·03) and sTNFR2 (6987 ± 980 pg/ml versus 3940 ± 275 pg/ml, P < 0·03) concentrations were found to be significantly higher in HCV-MC patients (Fig. 1b,c).
Fig. 1.
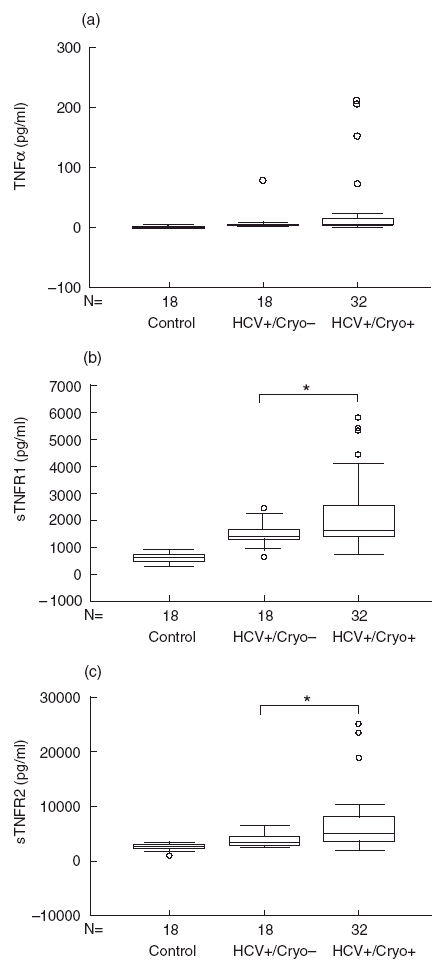
Elevated TNFα, sTNFR1 and sTNFR2 concentrations in patients with HCV-MC. TNFα (a), sTNFR1 (b) and sTNFR2 (c) were measured in patients with HCV-MC (HCV+/Cryo+) and compared with healthy controls (control) and with patients with HCV infection without MC (HCV+/Cryo–). In the box-plot representation, horizontal lines represent the median for each sample, open circles represent outside values (< 0·05, compared with respective control).
Higher sTNFR1 and sTNFR2 concentrations in patients with severe vasculitis
Correlations were studied between TNFα, sTNFR1 and sTNFR2 concentrations and the severity of HCV-MC vasculitis. Fourteen patients belonged to the low-grade severity group consisting of limited cutaneous vasculitis, and 18 patients belonged to the high-grade severity group consisting of severe extracutaneous visceral vasculitis (Tables 2 and 3). TNFα concentrations were not significantly different in patients with severe vasculitis compared with those with limited cutaneous diseases (25 ± 14 pg/ml versus 26 ± 15 pg/ml, data not shown). On the contrary, sTNFR1 concentrations were significantly higher in the case of high-grade severity vasculitis (2625 ± 382 pg/ml versus 1698 ± 850 pg/ml in low- severity vasculitis, P < 0·04, Fig. 2a). sTNFR1 concentrations in the severe vasculitis group were also significantly higher than in the HCV or the healthy control groups (2625 ± 382 pg/ml versus 1459 ± 98, P < 0·03 and 2625 ± 382 pg/ml versus 584 ± 36 pg/ml, P < 0·0001, Fig. 2a). Notably, sTNFR1 concentrations in the low-severity group were significantly higher than in the healthy controls (1698 ± 850 pg/ml versus 584 ± 36 pg/ml, P < 0·01) but not the HCV patients (1698 ± 850 pg/ml versus 1459 ± 98 pg/ml, P < 0·9). Concentrations of sTNFR2 were also found to be significantly higher in patients with severe vasculitis compared with those with low-severity forms (8772 ± 1560 pg/ml versus 4693 ± 649, P < 0·02, Fig. 2b), the HCV patients (8772 ± 1560 pg/ml versus 3940 ± 275 pg/ml, P < 0·001) and the healthy controls (8772 ± 1560 pg/ml versus 2603 ± 136 pg/ml, P < 0·0001). sTNFR2 in the low-severity vasculitis group was not significantly different from that in both the HCV and healthy control groups (4693 ± 649 versus 3940 ± 275 pg/ml, P = 0·9 and 4693 ± 649 versus 2603 ± 136 pg/ml, P = 0·4, respectively).
Fig. 2.
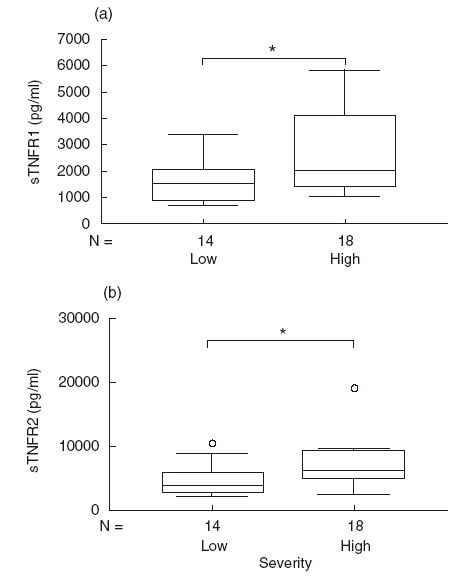
Higher sTNFR1 and sTNFR2 concentrations in HCV-MC patients with severe vasculitis. sTNFR1 (a) and sTNFR2 (b) concentrations were measured in HCV-MC patients with severe vasculitis defined by visceral involvement in comparison with low-severity vasculitis limited to skin involvement (*P < 0·05).
Correlations between sTNFR1 and sTNFR2 and biological parameters in patients with MC
Since this was a major concern in our study, correlations were studied between TNFα, sTNR1, sTNFR2 and the concentrations of rheumatoid factors in the patients with MC. No correlation was observed in any case (P = 0·4, P = 0·7 and P = 0·3, respectively, n = 31, using either Spearman’s or Pearson’s test). No correlations between TNFα and the concentrations of various biological parameters, including CRP (P = 0·7, n = 21), fibrinogen (P = 0·8, n = 21), CH50 (P = 0·2, n = 26), C4 (P = 0·3, n = 27) or cryoglobulin (P = 0·8, n = 28), were observed (data not shown). Similarly, no correlation was observed between sTNFR1 or sTNFR2 concentrations and CH50 (P = 0·5 and P = 0·6, n = 26), C4 (P = 0·9 and P = 0·3, n = 27) or CRP (P = 0·9 and P = 0·6, n = 21, data not shown) levels. On the contrary, a weak significant correlation was observed between sTNFR1 and fibrinogen concentrations (r = 0·5, P < 0·03, n = 21, Fig. 3a), and an almost significant correlation between sTNFR2 and fibrinogen (r = 0·41, P < 0·06, Fig. 3b).
Fig. 3.
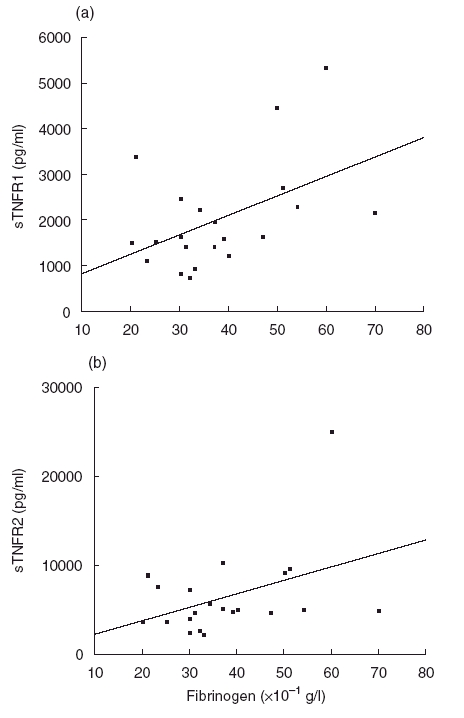
Correlations between sTNFR1 (a), sTNFR2 (b) and fibrinogen concentrations in HCV-MC patients (r = 0·5, P < 0·05 and r = 0·41, P < 0·06, respectively, n = 21).
Correlation between sTNFR1 and sTNFR2 and soluble adhesion molecule concentrations
When studied in all three groups, concentrations of TNFα significantly correlated with both sTNFR1 and sTNFR2 (r = 0·634 and r = 0·579, respectively, P < 0·0001, n = 68, data not shown). Similarly, in HCV-MC patients, a positive correlation was observed between sTNFR1 and TNFα concentrations (r = 0·51, P < 0·003, n = 32) as well as sTNFR2 and TNFα concentrations (r = 0·37, P < 0·05, n = 32, data not shown). In addition, a close correlation was observed between concentrations of sTNFR1 and sTNFR2 (r = 0·78, P < 0·0001, Fig. 4a). In a previous report, we observed that soluble adhesion molecules, especially sVCAM-1, are interesting markers in HCV-MC and are associated with the severity of the disease [26]. We therefore studied correlations between TNFα and sTNFRs levels, and sELAM, sICAM-1 or sVCAM-1 concentrations, in HCV-MC patients. A positive correlation was observed between sELAM concentrations and TNFα (r = 0·46, P < 0·01, data not shown) and sTNFR1 (r = 0·4, P < 0·03), but not sTNFR2 (r = 0·3, P < 0·2, data not shown). A positive correlation was also observed between sVCAM-1 concentrations and sTNFR2 (r = 0·44, P < 0·02, Fig. 4b), but not with sTNFR1 (r = 0·35, P < 0·06) or TNFα (r = 0·26, P < 0·4). No correlation was observed between sICAM-1 concentrations and TNFα, sTNFR1 and sTNFR2 concentrations (data not shown).
Fig. 4.
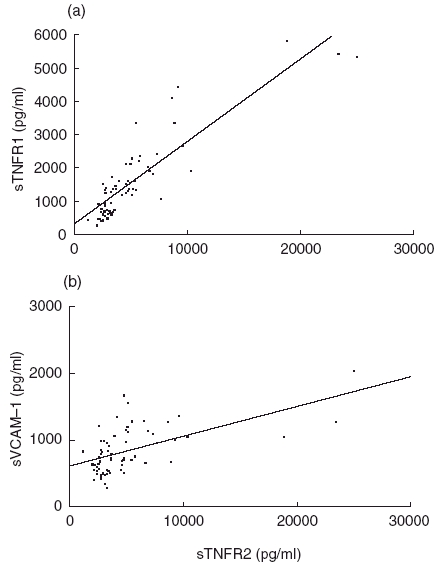
Correlations between sTNFR2 and sTNFR1 (a) and between sTNFR2 and sVCAM-1 (b) concentrations in HCV-MC patients (r = 0·78, P < 0·0001 and r = 0·44, P < 0·02, respectively).
Evolution of sTNFR1 and sTNFR2 in HCV-MC patients over time
For nine patients with HCV-MC, we were able to study sTNFR1 and sTNFR2 concentrations over time. sTNFR1 concentrations remained elevated but appeared largely to parallel clinical status during treatment (Fig. 5, presenting four patients, one with a low-severity form and three with severe forms of the disease). sTNFR2 concentrations paralleled sTNFR1 concentrations but appeared to follow clinical status less closely than sTNFR1 (data not shown).
Fig. 5.
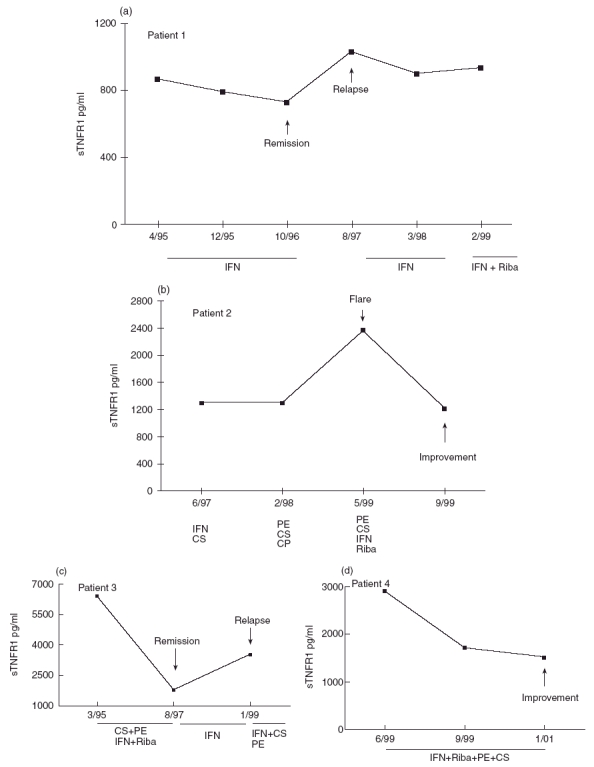
Evolution of sTNFR1 concentrations over time in four HCV-MC patients. Patient 1(a) suffered from a low-severity form of the disease (patient 3 in Table 2). Patients 2(b), 3(c) and 4(d) (patients 10, 1 and 18, respectively, in Table 3) suffered from severe visceral forms of the disease. IFN, interferon; Riba, ribavirin; PE, plasma exchanges; CS, corticosteroids; CP, cyclophosphamide.
DISCUSSION
MC, a major extrahepatic manifestation of chronic HCV infection [16–19], is a cause of immune complex-induced vasculitis affecting small- and medium-sized vessels, characterized by EC inflammation, leucocyte infiltration and vascular thrombosis [14,15]. Since TNFα is a major proinflammatory cytokine known to induce EC inflammation through fixation to its specific TNFR1 and TNFR2 [1,2,8], we asked whether TNFα is involved in HCV-MC pathogenesis. In this study, we found that TNFα levels were higher in patients with HCV-MC, but not significantly different from those in HCV control patients. Concentrations of sTNFR1 and sTNFR2, however, were increased in the serum of patients with HCV-MC compared with healthy volunteers. When compared with patients with chronic hepatitis C, who have previously been shown to have increased serum sTNFRs concentrations [27], sTNFR1 and sTNFR2 were higher in HCV-MC patients. These differences could not be due to variable severity of the underlying liver diseases, since mean Knodell index was higher in the HCV control group than in the HCV-MC patients. In addition, sTNFR1 and sTNFR2 concentrations were found to be significantly higher in cases of severe vasculitis with visceral involvement than in patients with limited cutaneous symptoms. Finally, in a few patients in which a follow-up was possible, sTNFR1 concentrations appear grossly to parallel the clinical status during treatment of HCV-MC, whereas sTNFR2 appears to be a less reliable marker.
A major concern in this study was the presence of rheumatoid factor in the serum of HCV-MC patients. As reported by the manufacturer and tested by us, the assays were not influenced by the presence of serum rheumatoid factor. In addition, neither TNFα nor sTNFRs concentrations correlated with the concentrations of rheumatoid factor, indicating that elevated TNFα and sTNFRs concentrations were not related to the concentrations of rheumatoid factor. A weak positive association has been observed between fibrinogen levels and sTNFR1 concentrations, but other important biological parameters such as CH50, C4 or CRP concentrations were not associated with TNFα or sTNFRs concentrations. In addition, concentrations of TNFα correlated with those of sTNFR1 and sTNFR2. Furthermore, TNFα and sTNFR1 concentrations correlated with concentrations of sELAM, whereas concentrations of sTNFR2 correlated with those of sVCAM-1, two leuko-endothelial adhesion molecules which appear to be involved in HCV-MC pathogenesis [26]. On the contrary, no correlation was found between TNFα, sTNFRs and concentrations of sICAM-1, a molecule which does not seem to be an interesting marker in this disease [26].
Together, these data provide indirect evidence for the involvement of TNFα in the pathogenesis of HCV-MC, although TNFα concentration did not appear to be the most reliable serum marker of its own involvement. Indeed, the assay used in this study is able to detect the 17 kD soluble mature form of TNFα, but not the 26 kD transmembrane precursor form. The latter is, however, biologically active through fixation to TNFR2, which is the main TNF receptor on EC and may play an important local role in HCV-MC pathogenesis [10,28]. A similar observation has been made in the case of soluble IL-1β and membrane-associated IL-1α in a model of infectious vasculitis [7]. Therefore, sTNFRs appears to be a more reliable serum marker of TNFα involvement in HCV-MC, a finding previously reported in other diseases [11–13].
sTNFR1 and sTNFR2 have been found to be increased in numerous other immune complex-mediated human diseases, notably in rheumatoid arthritis [12] and in systemic lupus erythematosus [13]. sTNFRs elevated concentrations reflect the role of TNFα in these two diseases. In rheumatoid arthritis, TNFα has been consistently found in the inflamed joints, and anti-TNF MoAbs have been successfully used to treat patients [29,30]. In animal models of systemic lupus erythematosus, TNFα has been found in the injured kidney and TNFα injections aggravated the disease [31,32]. TNFα involvement has been unequivocally demonstrated in numerous other animal models of immune complex-mediated diseases [33]. Complement fragments such as C5a and C3a, which play a central role during immune complex-mediated injury, as well as immune complexes themselves, have been shown to be potent TNFα and IL-1 inducers from mononuclear cells in vitro [34–37]. In HCV-MC, immune complex deposition on vessels may activate the complement cascade, inducing C3a and C5a production. Through its chemotactic functions on leucocytes, C5a may induce neutrophil and/or monocyte migration to the site of injury [38]. Monocytes activated by both C5a, C3a and immune complexes produce TNFα, which in turn stimulates EC adhesion molecule and tissue factor expression through interaction with its TNFR1 and TNFR2 [28,39]. In accordance with this hypothesis, we recently observed that leuco-endothelial soluble adhesion molecule concentrations are increased in patients with MC and correlated with vasculitis, notably sVCAM-1, which is known to be central in mononuclear cell adhesion to EC [26].
The physiological properties of sTNFRs in vivo appear to be mainly inhibitory of TNFα functions [8]. Interferon α has been used with success to treat essential MC [40] as well as HCV-MC [18,40,41], and these beneficial effects are probably mostly due to its antiviral effect. However, interferon α has also been shown to be a potent inducer of sTNFR1 by monocytes [42] and may thus exert direct anti-inflammatory effects in MC.
In conclusion, this is the first report showing that TNFα, sTNFR1 and sTNFR2 are increased in the serum of patients with HCV-MC, and that sTNFR1 and sTNFR2 levels are associated with the severity of vasculitis, thus suggesting a potential role for TNFα in this disease.
Acknowledgments
The authors are grateful to Laurence Pierret for her assistance in statistical analysis and to Anne Morel-Montero for helpful discussions.
REFERENCES
- 1.Beutler B, Cerami A. The biology of cachectin/TNF, a primary mediator of the host response. Ann Rev Immunol. 1989;7:625–55. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Natarajan K. Tumor necrosis factors: developments during the last decade. Eur Cytokine Netw. 1996;7:93–124. [PubMed] [Google Scholar]
- 3.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 4.Kriegler M, Perez C, DeFay K, et al. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:4 5–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 5.Perez C, Albert I, DeFay K, et al. A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell. 1990;63:251–8. doi: 10.1016/0092-8674(90)90158-b. [DOI] [PubMed] [Google Scholar]
- 6.Kaplanski G, Farnarier C, Kaplanski S, et al. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood. 1994;84:4242–8. [PubMed] [Google Scholar]
- 7.Kaplanski G, Teysseire N, Farnarier C, et al. IL-6 and IL-8 production from endothelial cells stimulated by infection with Rickettsia conorii is via a cell-associated IL-1α dependent pathway. J Clin Invest. 1995;96:2839–44. doi: 10.1172/JCI118354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–3. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 9.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 10.Grell M, Douni E, Wajant H, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:783–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 11.Van Zee KJ, Kohno T, Fischer E, et al. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor α in vitro and in vivo. Proc Natl Acad Sci USA. 1992;89:4845–9. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrera P, Boerbooms AMT, Janssen EM, et al. Circulating soluble tumor necrosis factor receptors, tumor necrosis factor α, and interleukin-6 levels in rheumatoid arthritis. Longitudinal evaluation during methotrexate and azathioprine therapy. Arthritis Rheum. 1993;36:1070–9. doi: 10.1002/art.1780360807. [DOI] [PubMed] [Google Scholar]
- 13.Aderka D, Wysenbeek A, Engelmann H, et al. Correlation between serum levels of soluble tumor necrosis factor receptor and disease activity in systemic lupus erythematosus. Arthritis Rheum. 1993;36:1111–20. doi: 10.1002/art.1780360812. [DOI] [PubMed] [Google Scholar]
- 14.Brouet JC, Clauvel JP, Danon F, et al. Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med. 1974;57:775–88. doi: 10.1016/0002-9343(74)90852-3. [DOI] [PubMed] [Google Scholar]
- 15.Gorevic PD, Kassab HJ, Levo Y, et al. Mixed cryoglobulinemia: clinical aspects and long-term follow-up of 40 patients. Am J Med. 1980;69:287–308. doi: 10.1016/0002-9343(80)90390-3. [DOI] [PubMed] [Google Scholar]
- 16.Ferri C, Greco F, Longombardo G, et al. Antibodies to hepatitis C virus in patients with essential mixed cryoglobulinemia. Arthritis Rheum. 1991;34:1606–10. doi: 10.1002/art.1780341221. [DOI] [PubMed] [Google Scholar]
- 17.Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med. 1992;327:1490–5. doi: 10.1056/NEJM199211193272104. [DOI] [PubMed] [Google Scholar]
- 18.Misiani R, Bellavita P, Fenili D, et al. Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med. 1992;117:573–7. doi: 10.7326/0003-4819-117-7-573. [DOI] [PubMed] [Google Scholar]
- 19.Cacoub P, Lunel-Fabiani F, Musset L, et al. Mixed cryoglobulinemia and hepatitis C virus. Am J Med. 1994;96:124–32. doi: 10.1016/0002-9343(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 20.Cacoub P, Poynard T, Ghillani P, et al. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC Group Multidepartment Virus C. Arthritis Rheum. 1999;42:2204–12. doi: 10.1002/1529-0131(199910)42:10<2204::AID-ANR24>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Lunel F, Musset L, Cacoub P, et al. Cryoglobulinemia in chronic liver diseases: role of hepatitis C virus and liver damage. Gastroenterology. 1994;106:1291–300. doi: 10.1016/0016-5085(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 22.Durand JM, Kaplanski G, Richard MA, et al. Cutaneous vasculitis in patients infected with hepatitis C virus RNA in the skin by polymerase chain reaction. Br J Dermatol. 1993;128:359–60. doi: 10.1111/j.1365-2133.1993.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 23.Cacoub P, Lunel-Fabiani F, Le Thi Huong Du. Polyarteritis nodosa and hepatitis C virus infection. Ann Intern Med. 1992;117:605. doi: 10.7326/0003-4819-116-7-605_2. [DOI] [PubMed] [Google Scholar]
- 24.Cacoub P, Maisonobe T, Thibault V, et al. Systemic vasculitis in patients with hepatitis C. J Rheumatol. 2001;28:109–18. [PubMed] [Google Scholar]
- 25.Musset L, Diemert MC, Taibi F, et al. Characterization of cryoglobulins by immunoblot. Clin Chem. 1992;38:798–802. [PubMed] [Google Scholar]
- 26.Kaplanski G, Cacoub P, Farnarier C, et al. Soluble vascular adhesion molecules are increased in patients with mixed cryoglobulinemia [abstract] Arthritis Rheum. 1996;39(Suppl.):725. [Google Scholar]
- 27.Tilg H, Vogel W, Wiedermann CJ, et al. Circulating interleukin-1 and tumor necrosis factor antagonists in liver disease. Hepatology. 1993;18:1132–8. [PubMed] [Google Scholar]
- 28.Paleolog EM, Delasalle SAJ, Buurman WA, et al. Functional activities of receptors for tumor necrosis factor-α on human vascular endothelial cells. Blood. 1994;84:2578–90. [PubMed] [Google Scholar]
- 29.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 31.Boswell JM, Yui MA, Burt DW, et al. Increased tumor necrosis factor and IL-1β gene expression in the kidneys of mice with lupus nephritis. J Immunol. 1988;141:3050–4. [PubMed] [Google Scholar]
- 32.Tomosugi NI, Cashman S, Hay H, et al. Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor and IL-1. J Immunol. 1989;142:3083–90. [PubMed] [Google Scholar]
- 33.Mulligan MS, Ward PA. Immune complex-induced lung and dermal vascular injury. Differing requirements for tumor necrosis factor-α and IL-1. J Immunol. 1992;149:331–9. [PubMed] [Google Scholar]
- 34.Okusawa S, Dinarello CA, Yancey KB, et al. C5a induction of human interleukin 1. Synergistic effect with endotoxin or interferon γ. J Immunol. 1987;139:2635–40. [PubMed] [Google Scholar]
- 35.Okusawa S, Yancey KB, Van Der Meer JWM, et al. C5a stimulates secretion of tumor necrosis factor from human mononuclear cells in vitro. Comparison with secretion of interleukin1β and interleukin 1α. J Exp Med. 1988;168:443–8. doi: 10.1084/jem.168.1.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takabayashi T, Vannier E, Clarck BD, et al. A new biologic role for C3a and C3a desArg. Regulation of TNF-α and IL-1β synthesis. J Immunol. 1996;156:3455–60. [PubMed] [Google Scholar]
- 37.Vissers MCM, Fantone JC, Wiggins R, et al. Glomerular basement membrane-containing immune complexes stimulate tumor necrosis factor and interleukin-1 production by human monocytes. Am J Med. 1989;134:1–6. [PMC free article] [PubMed] [Google Scholar]
- 38.Marder SR, Chenoweth DE, Goldstein IM, et al. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a desArg. J Immunol. 1985;134:3325–31. [PubMed] [Google Scholar]
- 39.Mackay F, Loetscher H, Stueber D, et al. Tumor necrosis factor α (TNF-α) -induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med. 1993;177:1277–86. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casato M, Legana B, Antonelli G, et al. Long-term results of therapy with interferon-α for type II essential mixed cryoglobulinemia. Blood. 1991;78:3142–7. [PubMed] [Google Scholar]
- 41.Durand JM, Kaplanski G, Lefèvre P, et al. Effects of interferon-alpha 2b on cryoglobulinemia related to hepatitis C virus infection. J Infect Dis. 1992;165:778–9. doi: 10.1093/infdis/165.4.778. [DOI] [PubMed] [Google Scholar]
- 42.Tilg H, Vogel W, Dinarello CA. Interferon-α induces circulating tumor necrosis factor receptor p55 in humans. Blood. 1995;85:433–5. [PubMed] [Google Scholar]