

Abstract
It has been proposed that Trypanosoma cruzi, the aetiologic agent of Chagas' disease, produces mitogenic substances responsible for the polyclonal B-cell activation observed during the acute phase of the infection. Isolation and characterization of the molecules involved in the induction of polyclonal activation observed during infectious diseases have posed a great challenge for the immunologist over the last decade. In this work we report that a 33 kD protein obtained from an alkaline fraction of T. cruzi epimastigotes (FI) stimulates proliferation and promotes differentiation into antibody-secreting cells of normal murine B cells in a T-cell independent manner. By flow cytometry we also found that the 33 kDa protein induces an increase in the expression of MHC class II and B7.2 but not B7.1 molecules on the B-cell surface. Sequencing by mass spectrometry identified the T. cruzi 33 kD protein as hypothetical oxidoreductase, a member of the aldo/ketoreductase family. In this report we demonstrate that this protein is also present in the infective bloodstream trypomastigote form of the parasite and was identified as T. cruzi mitochondrial malate dehydrogenase (mMDH) by enzyme activity and by Western blotting using a specific mMDH polyclonal antiserum. The biologic relevance of mMDH-induced polyclonal activation concerning T. cruzi infection is discussed.
Keywords: Trypanosoma cruzi, polyclonal B-cell activation, Chagas' disease, malate dehydrogenase
INTRODUCTION
Chagas' disease, caused by the haemoflagellate Trypanosoma cruzi, is a widespread tropical disease. This pathology affects nearly 20 million people in Central and South America with a significant impact on public health and important socio-economic implications. Disease pathology is a polemic issue of intensive research and it has been postulated that it is a consequence of the interactions between T. cruzi and the host immune system [1].
During T. cruzi acute experimental infection, all compartments of the immune system are disturbed as illustrated by T and B cell polyclonal activation, cytokine production, thymic atrophy, immunosuppression, and high autoantibody production [2–5]. The majority of the antibodies triggered by the infection are nonspecific and fail to bind parasite antigens [6]. Polyclonal activation has been proposed as the hallmark of immunological disorders during the acute phase of this parasitic infection and regarded as a ruling factor for parasite establishment [7].
This type of response is not a feature distinctive of T. cruzi infection since many viruses, bacteria and parasites [8–10] induce polyclonal activation as an evasive mechanism which hinders pathogen specific immune responses and allows for the chronic persistence of the infectious agent in the host.
In T. cruzi infection polyclonal activation could result from production of cytokines leading to the stimulation of bystander poised B and T cells and/or from the production of mitogenic substances by the microorganism [7].
Among the polyclonal B-cell activators from T. cruzi we could mention lipopolysaccharide-like substances [11], an excretory/ secretory antigen of 24 kD released by the parasite [12], an alkaline antigenic fraction (FI) from the epimastigote form of the parasite [13] and a recently identified proline racemase [14]. We have previously demonstrated that an alkaline fraction (FI) from T. cruzi-epimastigotes induces polyclonal B cell activation by T-cell-independent BCR-directed stimulation [15].
Recently, Reina San Martín et al. [16] proposed a new vaccine strategy advocating the neutralization of mitogens or superantigens that promote polyclonal activation and allowing instead for protective specific immune mechanisms. They claimed that the identification and characterization of the molecules involved in the induction of polyclonal activation evidenced during infection becomes relevant when selecting potential vaccine candidates in the light of immune protection.
Based on this proposal and since FI is a complex antigenic fraction, the aim of the present work was to identify and characterize the polyclonal B-cell activating-protein present in FI. We demonstrated that a 33 kDa protein (p33) from FI stimulates the activation, proliferation and differentiation of normal murine B cells into antibody-secreting plasmocytes, in a T-cell independent fashion. We have also noticed that p33 preferentially induced an increase in costimulatory molecules such as B7.2 and MHC class II on the B cell surface. Enzymatic digestion of the isolated p33 and sequence analysis of purified peptides suggested identity with an oxidoreductase, a member of the aldo/ketoreductase family. This protein was identified as a T. cruzi-mMDH since it was recognized by a specific mMDH polyclonal antibody and exhibited MDH enzyme activity. Futhermore, in this paper we evidence that T. cruzi-mMDH is present in the infective bloodstream trypomastigote form of the parasite.
MATERIALS AND METHODS
Mice
BALB/c 6-week-old mice were purchased from the Comisión Nacional de Energía Atómica (CNEA) (Buenos Aires, Argentine). Argentine animal experimental guidelines were followed.
Trypanosoma cruzi alkaline fraction (FI)
Epimastigote forms from the Tulahuén strain of T. cruzi were harvested from cultures in monophasic medium [17]. The epimastigotes were disrupted by seven cycles of freezing and thawing and then centrifuged at 105 000 g. The supernatant, named F105, was subjected to isoelectric focusing (IEF) on agarose gel at pH gradient 3–10. The unstained gel zone corresponding to pI 7–10 was removed, homogenized, eluted with phosphate buffer saline (PBS) pH 7·2, dialysed for 3 days against PBS and finally used as the antigenic fraction (FI) [13].
Trypanosoma cruzi trypomastigote homogenate
Trypomastigote forms of T. cruzi (Tulahuén strain) were obtained from the supernatant of the Vero cell line 6–8 days after primary infection. Parasites were harvested by centrifugation at low revolutions. The parasites were present in the supernatant. The supernatant was centrifuged at 5000 g and washed three times in ice cold PBS pH 7·2 and kept at –70°C. Then, trypomastigotes were lysed by seven cycles of freezing and thawing and a total homogenate of trypomastigotes was obtained.
Treatment of FI with proteinase K
The treatment of FI with proteinase K was performed under conditions similar to those previously described [18]. FI (1 mg/ml) was treated with 10 μg/ml of proteinase K (Sigma, St Louis) for 2 h at 42°C. After this time, FI treated with proteinase K (FI-pK) was dialysed against PBS overnight at 4°C and then it was sterilized by filtration for further use in cell culture.
Gel filtration chromatography
FI (3 mg/ml) was dialysed against buffer 20 mm sodium phosphate, pH 7·2 overnight at 4°C and a 0·5-ml aliquot was applied to a Superose 12 HR10/30 column (Pharmacia Biotech, Uppsala, Sweden) equilibrated with the same buffer and attached to a fast protein liquid chromatography (FPLC) system (Pharmacia Biotech). Elution (0·5 ml/min) was performed at 4°C and 1 ml fractions were collected and automatically read at A280. Material from each fraction was dialysed against PBS, adjusted to 0·5 mg/ml and sterilized by filtration (0·22 μm filter) for further use in cell culture.
SDS-PAGE
The different fractions eluted from the Superose 12 column were subjected to SDS-PAGE according to Laemmli [19] by using a 5% stacking gel and 12·5% resolving gel in the presence of SDS and β-2 mercaptoethanol and visualized by silver staining.
Isolation of the 33kD T. cruzi protein (p33)
After SDS-PAGE the 33 kD protein was isolated by an electroelution procedure using an electroelution cell following the manufacturer's instructions (Model 422 Electro-Elutor, Biorad, Hercules, CA) [20]. Briefly, FI (≈ 3 mg/ml) was submitted to a preparative SDS-PAGE and the gel zone corresponding to the p33 band was excised. Then, the protein was electrophoretically eluted from the minced gel in 25 mm Tris, 192 mm glycine, 0·1% SDS. The SDS contaminating the isolated p33 was removed by placing the eluted fraction in crushed ice for 4 h, and the crystallized SDS was removed by pelleting it in a bench Microfuge. Then, the sample was extensively dialysed against PBS at 4°C and sterilized for further use in cell culture.
‘In gel’ digestion of proteins, extraction of peptides and mass spectrometry analysis
SDS-PAGE gels were stained with Coomassie brillant blue, and the bands of interest excised and subjected to in situ digestion with trypsin as described [21]. A small aliquot (0·5 μl) of the extract was taken up and analysed by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. Gel extracts were pooled, dried down, resuspended in 10μl of 0·1% TFA and subjected to automated desalting following a procedure previously described [22]. Briefly, the resuspended extracts were injected onto a RP-HPLC nanocolumn (Vydac C18, 300 μm i.d.) at a flow of 3–5 μl/min using a Smart HPLC instrument with automatic fraction collection (Pharmacia Biotech, Uppsala, Sweden) equipped with a flow splitter and working at about 100 μl/min. Peptides were eluted in a single step with methanol/water (7:3) containing 0·1% formic acid, and fractions of about 5 μl were collected. The fraction containing the peptide pool was identified by MALDI-TOF analysis of an 0·5 μl aliquot of each fraction. The remainder of the fraction was used for nanospray-ion trap (nESI-IT) mass spectrometry analysis.
Analysis by MALDI-TOF mass spectrometry were per-formed using a Kompact Probe instrument (Kratos-Shimazdu, Manchester, UK), equipped with an extended flight tube of 1·7 m and delayed extraction, operating in linear mode. 0·5 μl of the fractions to be analysed were applied onto target and dried out. 0·5 μl of saturated α-ciano-4-hydroxycinnamic acid matrix in water:acetonitrile (1:1) containing 0·1% TFA was then added and dried out. Calibration was made externally by using a set of synthetic peptides.
Analysis by nESI-IT were performed using an ion-trap mass spectrometer model LCQ (Finnigan, ThermoQuest, San Jose, CA, USA) equipped with a nanospray interface, exactly as previously reported [23]. De novo sequencing of peptides was performed by doing multiple subfragmentation (MSn) steps [23,24], which helped to determine the nature of the fragmentation series.
Western blot
In order to analyse whether p33 is recognized by a specific polyclonal mMDH rabbit antiserum (kindly supplied by Dr Nowicki [25]) and whether this protein is present in the trypomas-tigote forms of the parasite, p33 and a total homogenate of trypomastigotes were run on SDS-PAGE, electroblotted onto nitrocellulose membranes and incubated with the antiserum mentioned above (1:5000). The Western blots were then incubated with horseradish peroxidase-conjugate antirabbit goat IgG and developed using either H2O2 plus 4-chloro-1-naphtol as the substrate or an ECL kit.
Determination of enzyme activity
In order to retain the possible malate dehydrogenase activity of p33 we purified the protein using a chromatofocusing column. The activity of malate dehydrogenase, in the direction of oxaloacetate reduction, was determined spectrophotometrically at 37°C as the decrease in absorbance at 340 nm, in a reaction mixture containing 100 mm phosphate buffer pH 7.4, 1 mm oxaloacetate, 0.17 mm NADH and the enzyme, which was added last to start the reaction [26].
Isolation of normal murine B cells
A B-cell enriched population from normal spleen cells was obtained by depletion of T cells by magnetic isolation. The cells were prepared by gently teasing splenic material in RPMI, the debris was removed and a cell suspension collected. Then, the spleen cells were depleted of erythrocytes by treatment with Tris-buffered ammonium chloride, washed and resuspended in RPMI containing 10% FBS, 2 mml-glutamine, 50 μmβ2-mercaptoethanol and 20 μg/ml gentamicin. Subsequently, the cell suspension was incubated with anti-Thy 1·2 coated magnetic beads (Dynal, France) at a final concentration of 1 × 107 beads/ml, with a bead to cell ratio of 4:1, for 20 min at 37°C. Thy 1·2 positive cells were collected by magnetic separation. The effectiveness of T-cell depletion was assessed by staining the T cell-depleted spleen mononuclear cells with anti-CD3 FITC-labelled antibodies followed by FACS; contaminating T cells were < 2%. In all cases, cell viability was > 95%, as determined by trypan blue dye exclusion.
Cell proliferation assay
The B-cell enriched population (2 × 105 cells/well) in presence of macrophages was cultured in triplicate at a volume of 200 μl in flat-bottom 96-well tissue culture plates (Corning), for 72 h with 45 μg/ml FI, 20 μg/ml of the different fractions eluted from the Superosa column, different concentrations of p33, 20 μg/ml LPS or medium alone. Cells were harvested and proliferation was assayed by measuring the incorporation of 1 μCi of[3H]TdR/well during the final 18 h of culture, using a beta-scintillation counter. Results are expressed as incorporation of radioactivity (cpm ± s.d.).
Isotype determination
For determination of immunoglobulin synthesis, B cells were incubated with p33 (20 μg/ml) or medium alone at a density of 3 × 106 cells/ml in a volume of 2 ml for 6 days, and supernatants were collected and assayed in a isotype specific ELISA. 96-well ELISA plates were coated with the isotype specific goat-antimouse IgG1, IgG2b, IgG3 and IgM antibodies overnight at 4°C, extensively washed and blocked by the addition of BSA 1% for 1 h at 37 °C. Supernatants of p33 or nonstimulated B cells were incubated for 1 h at 37°C and then overnight at 4°C. After washing with PBS-Tween, peroxidase conjugated antimouse IgG (when the plates were coated with anti-IgG isotype) or antimouse IgM were added and incubated for 1 h at 37°C. The reaction was developed with o-phenylendiamine. The concentrations were measured with reference to standard curves using known amounts of the respective murine immunoglobulin isotypes (Sigma, St Louis).
Flow cytometry assays
The B-cell enriched population (2 × 106), in presence of macrophages, was incubated at 37°C for 48 h with 20 μg/ml of p33 or medium alone. Then, the cells were harvested and the cell viability was measured. Subsequently the cells were washed three times with balanced salt solution (HANK'S) containing 0·5% bovine serum albumin (BSA) and 0·01% NaN3. Cells were then incubated with antimouse CD32/CD16 antibodies for 1 h at 4°C in order to block immunoglobulin nonspecific binding through Fc receptors. After this time, the cells were then stained with PE-labelled antimouse CD19+ and FITC-labelled antimouse B7.1, B7.2 or FITC-labelled antimouse CD19 and PE-labelled antimouse MHC class II molecules (PharMingen, San Diego) antibodies for 30 min at 4°C. After appropriate washes, the stained cells were fixed in 2% formaldehyde and stored at 4°C in the dark until analysed. Two colour-analysis was carried out in a FACS flow cytometer (Ortho Diagnostic System, Raritan, NJ) within the viable cell population. Results were analysed using the WinMDI software.
Statistical analysis
All statistical analysis was performed using either the Student's t-test or the Welch t-test (for two groups). P < 0·05 was considered significant.
RESULTS
FI-mitogenic activity was abolished by proteinase K treatment
Previously we reported that FI, a complex antigenic fraction constituted of several proteins, was able to induce polyclonal normal murine B cell activation [13]. However, we did not demonstrate whether the compound responsible for the mitogenic activity was of a protein nature. To do this, we treated FI with proteinase K and assessed its capacity to induce B-cell proliferation in the presence of accessory cells. As shown in Fig. 1, treatment of FI with proteinase K (FI-pK) abrogated FI-mitogenic activity. We clearly observed that the thymidine uptake (cpm) reached by FI-pK-stimulated B cells was similar to the value obtained by B cells cultured without any stimulus. As a control for a positive proliferative response, B cells were cultured with untreated-FI under similar experimental conditions.
Fig. 1.

Effect of F1 proteinase K-treatment on B cell proliferation. B cells in the presence of macrophages (2 × 105/well) were incubated with medium alone (control, □), 45 μg/ml FI (FI,  ) or FI (45 μg/ml) treated with proteinase K (FI-pK, ▪). After culturing for 3 days, [3H]-thymidine uptake (cpm) was measured (see Materials and Methods). Results represent the mean ± s.d. This set of results is representative of three individual experiments. The asterisk indicates a significant difference (P < 0·005) compared with FI-stimulated B-cells.
) or FI (45 μg/ml) treated with proteinase K (FI-pK, ▪). After culturing for 3 days, [3H]-thymidine uptake (cpm) was measured (see Materials and Methods). Results represent the mean ± s.d. This set of results is representative of three individual experiments. The asterisk indicates a significant difference (P < 0·005) compared with FI-stimulated B-cells.
Figure 1 shows that proteinase K treatment abolishes B cell proliferation, thus suggesting that proliferation is dependent on protein stimulation and not on contaminating polysaccharides.
Identification of a 33kD T. cruzi-protein with B-cell mitogenic activity
In order to identify and purify the T. cruzi protein/s responsible for B cell proliferation and further explore other properties, we first isolated proteins of FI according to their molecular weights. Briefly, FI was submitted to gel filtration chromatography as described in the Material and Methods. According to the absorbance at 280 nm we collected 15 fractions eluted from the column. A protein profile is depicted in Fig. 2a where we can observe the maximal level of absorbance between fractions 8–10.
Fig. 2.
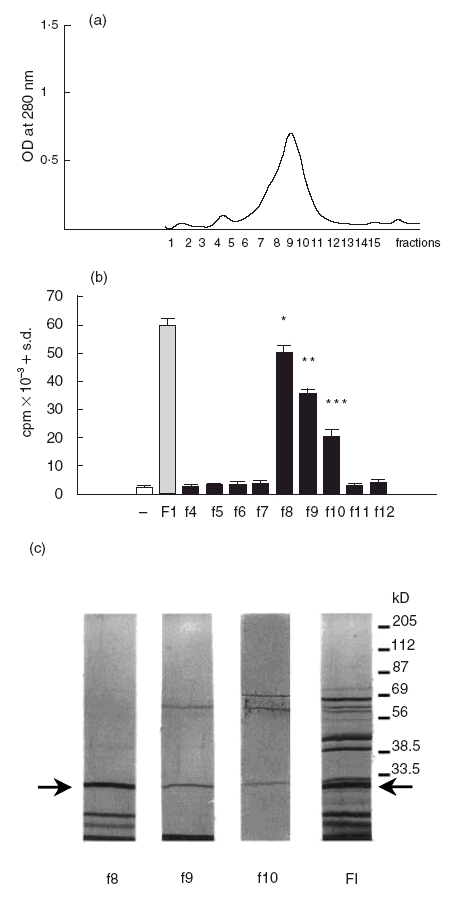
(a) Gel filtration of FI on Superose 12 column. FI was obtained by IEF as described in the Materials and Methods and applied to a Superose 12 HR 10/30 column attached to an FPLC system. Fractions of 1 ml were collected and automatically read at OD 280 nm. (b) Proliferative response of B cells stimulated with fractions eluted from the Superose 12 column. B cells in the presence of macrophages (2 × 105/well) were incubated with 45 μg/ml FIor 20 μg/ml of each fraction eluted from the column (▪) or medium alone (control, □) for 3 days.[3H]-Thymidine uptake (cpm) was measured and the results represent the mean ± s.d. This set of results is representative of three individual experiments. The asterisks indicate significant differences (*P < 0·05, **P < 0·007, ***P < 0·005) compared with unstimulated B-cells. (c) SDS-PAGE of FI and the fractions eluted from the Superose 12 column. FI and the fractions with B-cell mitogenic activity obtained from FI by gel filtration were separated by SDS-PAGE and silver stained. The molecular weights of markers are indicated in the right side of the figure. The arrows indicate the position of p33.
Once each fraction was adjusted to a concentration of 0·5 mg/ml protein, the capacity of these fractions to promote B cell proliferation was evaluated. Normal murine B cells cultured in the presence of accessory cells (T-cell depleted spleen cell population), were stimulated with 20 μg/ml of the different fractions and proliferation was assayed by thymidine uptake. Thus, Fig. 2b shows that B cells exhibit a high response to components in fractions 8–10 and the highest level of proliferative response was evidenced when fraction 8 (f8) was added to the culture, reaching cpm values similar to those with the complete antigenic fraction (FI). No B-cell proliferative response to fractions 4–7 or 11–12 was detected.
In order to explore the protein profile of the fractions with mitogenic activity, samples were resolved by SDS-PAGE and silver stained. Figure 2c shows that the fractions have several proteins with different molecular weights. However, a band of 33 kDa occurs in all fractions with mitogenic activity. We observed that in fraction 8, which displays the highest mitogenic activity, the 33 kDa band is the most prominent protein. In this figure we indicated the presence of the 33 kDa protein in FI.
Since the fractions capable of inducing B-cell proliferation contain proteins beside 33 kDa, we attempted to purify the 33 kDa protein by an electroelution procedure to evaluate its mitogenic capacity. Briefly, FI was separated by SDS-PAGE as described in the Material and Methods and the p33 band was excised and then electrophoretically eluted from the minced gel. To test the purification, the electroeluted material was submitted to SDS-PAGE and silver stained. Under this condition only one protein of 33 kDa was observed (data not shown).
Next we examined whether the 33 kDa purified protein promoted the proliferation of B cells. Normal murine B cells cultured in the presence of macrophages (T-cell depleted spleen cell population) were stimulated with different concentrations of p33 for different periods of time. By kinetic studies we observed that p33 induced a low level of B cell proliferation at 24–48 h and that maximal proliferation was detected between 72 and 96 h (data not shown). As illustrated in Fig. 3, p33 significantly increased the proliferative response of B cells, in a dose-dependent way, as compared to nonstimulated B cells. After 3 days of culture the cpm reached by the cells stimulated with the highest concentration of p33 was seven times higher than the control. The value obtained with p33 was similar to that obtained when the cells were cultured with LPS, a typical murine B-cell mitogen.
Fig. 3.

Proliferative response of B cells stimulated with p33. B cells in the presence of macrophages (2 × 105/well) were incubated with medium alone (□), 20 μg/ml LPSor different concentrations of p33 (▪) for 3 days and the [3H]-Thymidine uptake (cpm) was measured. Results represent the mean ± s.d. This set of results is representative of three individual experiments. The asterisks indicate significant differences (*P < 0·001,**P < 0·007, ***P < 0·005) compared with unstimulated B-cells.
Trypanosoma cruzi p33 activates normal B cells and induces the secretion of different immunoglobulin isotypes
Previously, it has been reported that a number of pathogen agents that nonspecifically stimulate B cells are able to up-regulate the expression of costimulatory molecules as well as MHC class II molecules [27–29].
To evaluate the effects of p33 from T. cruzi on the expression of B7.1, B7.2 and MHC class II molecules, B cells from normal mice were cultured in the presence of macrophages and stimulated with 20 μg/ml of purified p33. On day 2, cells were washed and incubated with PE-labelled anti-CD19 in combination with FITC-labelled antimouse antibodies to B7.1, B7.2 or FITC-labelled anti-CD19 jointly with PE-labelled anti MHC class II molecules. CD19+ cells (B cells) were gated and analysed for the expression of surface molecules mentioned above. FACS showed that, unlike cells cultured without stimulus, p33 induced an enhancement in the percentage of B7.2+ as well as MHC class II+ cells (Fig. 4a(i) and (ii), respectively). Thus, when B cells were stimulated by p33, we detected 36% B7.2+ B cells versus 20% B7.2+ B cells in the control culture (Fig. 4a(i)). Furthermore, we observed that p33 triggered higher expression of B7.2 and MHC class II molecules. Statistical analysis revealed that the mean of B7.2 expression in the presence of p33 was 1737 unlike 1565 in B cells cultured with medium alone and that the mean of MHC class II expression was 2141 versus 1857 in control B cells. Moreover, the percentage of B cells with high MHC class II expression in cultures stimulated with p33 was 61% as compared with 33% in cultures without stimulus (indicated as M2 in Fig. 4a(ii)). Surprisingly, p33 did not increase the expression of B7.1 in B cells but p33-stimulated B cells showed an enhancement of the B-cell population with low expression of B7.1 (Fig. 4a(iii)). These results clearly show that p33 is able to activate normal murine B cells.
Fig. 4.
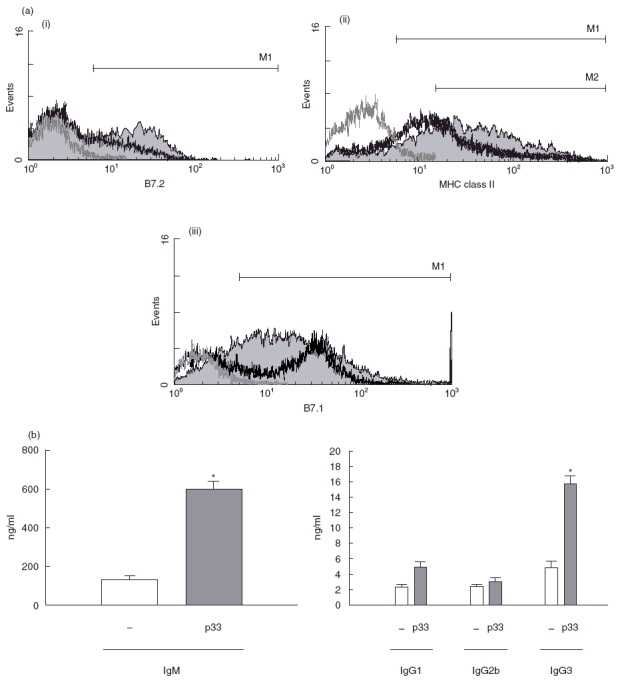
(a) p33 up-regulates expression of B-cell activation markers. B cells were cultured in the presence of macrophages with p33 (20 μg/ml) or medium alone for 3 days. The cells were then stained with PE-labelled antimouse CD19+ and FITC-labelled antimouse B7.1, B7.2 or FITC-labelled antimouse CD19 and PE-labelled antimouse MHC class II molecules. CD19+ cells were gated and analysed for the expression of surface molecules mentioned above. We show on p33 stimulated B cells (grey fill histogram): (i) expression of B7.2 + B cells; (ii) expression of MHC class II + B cells; (iii) expression of B7.1 + B cells. In all figures thick black line histograms represent B cells cultured without stimulus, thin grey line histograms represent unstained cells. This set of results is representative of three individual experiments. (b) p33 effect on immunoglobulin secretion. B cells in the presence of macrophages (3 × 106/well) were incubated with medium alone (□) or with 20 μg/ml p33for 6 days. Cell free supernatants were harvested and analysed for Ig isotype production by ELISA. Data are presented as represent the mean ± s.d. The asterisks indicate significant differences (*P < 0·05,**P < 0·01) compared with unstimulated B-cells.
Next, we evaluated whether p33-stimulated B cells undergo terminal differentiation. B cells were stimulated by p33 for 6 days and the production of immunoglobulin isotypes was measured in the culture supernatant by ELISA. As shown in Fig. 4b we observed that p33 stimulated strong immunoglobulin production with a predominance of IgM and IgG3 isotypes, reaching values of 600 and 15·6 ng/ml, respectively.
We did not observe modification in IgG2b levels. Moreover, we found that the IgM secreted by p33-stimulated B cells did not specifically recognize the T. cruzi mitogenic antigens (FI and p33) (data not shown).
p33 shows homology with an E. coli hypothetical oxidoreductase
In order to characterize T. cruzi p33 we performed some biochemical assays.
The nature of the 33 kDa protein was determined by mass spectrometry after tryptic digestion. The peptides obtained from the crude extract were then analysed by MALDI-TOF mass spectrometry (Fig. 5a). We firstly tested whether the obtained set of peptide masses could be used to directly identify the protein. A database search using the set of peptide masses did not result in any significant matches, suggesting that this protein was not recorded in databases and that the peptide masses were insufficient to identify the protein by homology.
Fig. 5.

Sequencing of p33 by mass spectrometry. The p33 band was separated by SDS-PAGE, excised from the gel and digested with trypsin and the resulting peptides analysed as described in Materials and Methods. (a) Analysis of peptides from p33 by MALDI-TOF mass spectrometry. Only the masses of peptides which were further detectable by nanoESI-IT mass spectrometry are indicated. (b) Sequencing of peptide at m/z 1439·6 by nanoESI-IT tandem mass spectrometry. The doubly charged ion corresponding to this peptide was isolated within the ion trap and subjected to fragmentation; shown is the fragment or MS/MS spectrum from this ion. The assigned peptide sequence is indicated, detailing the observed backbone fragment ions according to standard nomenclature. The precursor ion (M + H+) and related ion resulting from neutral loss of water are indicated. The vertical axis in both (a) and (b) represent the relative peak intensity in arbitrary units.
We then subjected the crude peptide extract to microdesalting, and the cleaned peptide pool was analysed by nanoESI-IT mass spectrometry. Some, but not all of the peptides detected by MALDI-TOF were also detected by nanoESI-IT; this result is typically encountered when analysing peptides in the low picomole to subpicomole range (see, for example, Ogueta et al. [24]), and may be due to losses during the desalting step, intrinsic differences in the ionization methods or supression effects produced by the simultaneous occurrence of several peptides. These peptides were then sequentially subjected to fragmentation in order to identify their sequence; from the nine peptides detected, three of them (at m/z 965·4, 1572·6 and 1808·8 in Fig. 5a) failed to yield interpretable MS/MS spectra. From the other six peptide ions (as exemplified in Fig. 5b with peptide at m/z 1439·6), we could obtain high quality fragments. A database search using the fragment spectra failed to give a significant match, indicating that none of these peptide sequences was listed in databases.
The fragment spectra were then interpreted manually (see Fig. 5b), yielding unambiguous sequences in all cases, except for one, in which only a partial sequence could be deduced (Table 1). A thorough database search with the obtained sequences failed to identify peptides with 100% sequence identity, as expected from previous results. However, four of the peptides were found to have a significant homology, ranging from 75 to 100%, with E. coli hypothetical oxidoreductase Q46857, a member of the aldo/ketoreductase family. Consistently, all the peptides from this protein which showed homology with those from p33 yielded a basic residue previous to their N-terminal end, indicating that the tryptic site was also conserved (Table 1); in addition, the theoretical mass of this protein (31 kDa) was in good agreement with that of p33. Peptide at m/z 1506·6 was also found to have homology with another E. coli hypothetical oxido reductase (P77256), although the homology was not so significant, and the N-terminal tryptic site was not conserved. In conclusion, the results summarized in Table 1 support the evidence that p33 is a hypothetical oxidoreductase from T. cruzi, and thereby a member of an aldo/ketoreductase family.
Table 1.
Sequence of peptides from p33 and homology with other proteins
| Average mass (M + H+)* | 1514·6‡ | 869·4 | 1406·9 | 1439·6 |
| Peptide sequence † | HIDTAEF | SPAQVVIR | WDLQHGLVTIPK | IQENFNVWDFK |
| | | | | | :| | |: | | | | | | | | | | | | | | | | | | | | |: | |
| Hypothetical oxido reductase (Q46857) | ..RSIDTAAAY… | KTPAQIVIRW… | RWHLDSGLVVIPKSVTPSRIAENFDVWDFRL.. | |
| Average mass (M + H+)* | 1506·6 | |||
| Peptide sequence † | FEGGPFFNPDHDK | |||
| | | | | | |: | ||||
| Hypothetical oxido reductase (P77256) | ..AIGGGPAWNGDLDR.. | |||
Determined by MALDI-TOF mass spectrometry.
Determined by nanoESI-IT mass spectrometry. Notice that by this method amino acids I and L, and K and Q are indistinguishable. For simplicity, only one of each are indicated; in this table we have chosen the amino acid which gives sequence identity with the indicated proteins. Since peptides are generated by trypsin in conditions where almost complete digestion is expected, we have assumed that K must be present instead of Q at C-terminal position, and that Q is more probable than K in internal positions.
Only a partial sequence of this peptide could be determined; therefore, the observed mass does not agree with the depicted sequence.
p33 is recognized by polyclonal rabbit sera anti T. cruzi mMDH
At this point we have observed that the mitogenic protein obtained from T. cruzi epimastigotes has a molecular weight of 33 kDa, an alkaline pI and moreover displays homology with oxidoreductases. In search of T. cruzi proteins with these characteristics, we found that T. cruzi mMDH presents biochemical similarities [25]. By Western blotting we observed that an antimMDH polyclonal antiserum strongly reacted with purified p33 (Fig. 6a, lane 2). As positive control we ran a total parasite homogenate, named F105, which contains mMDH in parallel (Fig. 6a, lane 1). Pre-immune rabbit antiserum did not react with purified p33 (Fig. 6a, lane 3).
Fig. 6.

(a) Purified p33 is recognized by polyclonal rabbit anti-T cruzi mMDH antisera. F105 (lane 1) and p33 (lane 2) were subjected to SDS-PAGE, applied onto nitrocellulose membrane and incubated with polyclonal rabbit antiserum raised against purified mMDH. In lane 3, p33 was incubated with rabbit preimmune serum. (b) mMDH is present in the trypomastigote form of T. cruzi. Total homogenate of trypomastigotes (T) was obtained and Western blot analysis was performed as described above. Lane 1: T was incubated with polyclonal rabbit antiserum against mMDH; lane 2: T probed with rabbit preimmune serum. The arrow shown on the right side of both figures indicates the molecular weight of 33 kDa marker
We then searched for the presence of p33 in the infective form of the parasite. Western blot analysis of parasite cell extracts demonstrated that mMDH is present in different T. cruzi development stages. In fact, we detected that antimMDH polyclonal antiserum recognized a 33 kDa protein in trypomastigote lysate (Fig. 6b, lane 1). As a negative control preimmune rabbit antiserum was processed in parallel (Fig. 6b, lane 2).
Finally, we decided to confirm the identity of p33 as MDH by determining its MDH enzyme activity. We observed that p33 showed 0·7 U/mg of specific MDH activity.
Taken together these results demonstrate that p33, which is present in the epimastigote as well as in the infective trypomastigote form of T. cruzi, is mMDH and that it exerts B cell polyclonal activation in a T-independent way.
DISCUSSION
In this study we have identified and characterized a 33 kDa protein obtained from T. cruzi capable of inducing B-cell activation, proliferation and differentiation into antibody secreting cells in the complete absence of T cells (T-independent manner).
We demonstrated that p33 induced B cell activation since it was able to promote the up-regulation of B7.2 as well as that of MHC class II molecules on the B-cell surface. Earlier studies have shown that polyclonal activators such as bacterial DNA, micobacterial membranes and LPS may induce the expression of costimulatory molecules such as B7.1 and B7.2 [27,30,31]. This ability is thought to account for the potency of several adjuvants used in vaccines. Hence, the up-regulation of these molecules by p33 is likely to enhance the antigen presenting capacity of B cells. It is important to point out that when B cells were stimulated by p33, we observed an increase in the percentage of B cells with low B7.1 expression. Selective expression of B7.1 versus B7.2 on antigen presenting cell surfaces has been shown in many models to preferentially influence Th1 and Th2 type responses, respectively [32]. However, this issue remains controversial since conflicting results have arisen from different experimental models both in vivo and in vitro [33,34]. The biological consequence of this preferential expression of B7.2 induced by p33 is under current investigation in our laboratory.
Our data show that p33-stimulated B cells undergo terminal differentiation and release mainly IgM and IgG3 which do not specifically recognize T. cruzi mitogenic antigen, indicating that p33 is a T. cruzi B cell polyclonal activator. This behaviour is in agreement with that found for LPS, a typical T-independent polyclonal activator.
In order to identify the 33 kDa protein with mitogenic property, we first analysed the amino acid sequence of the peptides obtained from the trypsinated-p33. These sequences were screened for homologies with known sequences in the data bank and we found a significant homology ranging from 75% to 100% with E. coli hypothetical oxidoreductase, a member of the aldo/keto reductase superfamily. Finally, p33 was identified as a T. cruzi mMDH by enzyme activity and Western blotting using specific polyclonal antimMDH sera.
Trypanosoma cruzi presents two isoforms of MDH which differ both in cellular localization and biological properties. One of the isoenzymes is located in the mitochondrion (mMDH) and the other in the glycosome (glcMDH). On the basis of isoelectrofocusing of cell free extracts, the MDH isoenzyme with a strongly basic isoelectric point is mitochondrial whereas the acidic isoform is glycosomal [35]. The isoforms show neither amino acid sequence similarities nor immunological cross reaction with each other. Reynoso Hunter et al. [25] have reported that the amino acid sequence previously predicted for the putative MDH from T. cruzi (Gen Bank, accession number AF 051893) matched perfectly with glcMDH but did not correspond to the mitochondrial isoform. Although the p33 mitogenic protein was recognized by specific polyclonal antimMDH sera, the p33-peptide fragments sequenced in this study did not show homology with the partial mMDH amino acidic sequence reported previously [25]. This lack of homology could be explained by considering that at present only one third of the complete molecule of mMDH has been sequenced and it is likely that the peptide sequenced by us corresponds to the remaining two thirds.
Mitochondrial MDH was described previously in T. cruzi epimastigotes; in this work we report that this enzyme is also present in the trypomastigote form of the parasite. Considering that the trypomastigote is the infective form of T. cruzi this data enhances the relevance of our findings in the context of Chagas' disease pathogenesis.
Eukaryotic mMDH is one of the enzymes in the tricarboxylic acid cycle. Trypanosoma cruzi mMDH is supposed to play its usual role in this cycle, but the results presented herein show for the first time, as far as we know, that mMDH may promote polyclonal B cell activation. These findings are novel and it would be interesting to find out how an intracellular protein that is usually present in the parasite mitochondrion might induce B cell proliferation. It is possible that after infection, immune system cells destroy the microorganisms releasing the pathogen products. In keeping with this, it has previously been reported that products which are not normally present on microbial cell surface membranes, such as unmethylated CpG motifs in bacterial DNA, trigger B-cell mitogenicity [27].
In the mouse model of T. cruzi infection the abrogation or reduction of polyclonal B and T cell responses leads to an increased resistance to infection and to the control of chronic tissue pathology [36,37]. Therefore, the polyclonal activation induced by membrane-associated as well as intracellular antigens could be a strategy used by the parasite in order to survive and lodge chronically in the host even if the immune system started to control its replication.
It is probable that the enzyme activity is not involved in the induction of polyclonal activation since mMDH retains its mitogenic activity after heat treatment. We suspect that the B cell activation induced by mMDH is related more to its structure than to the enzymatic activity itself.
In conclusion, our results show that mMDH, present in infective as well as noninfective stages of T. cruzi, is involved in the induction of normal murine B-cell proliferation. Essentially, we believe that understanding polyclonal lymphocyte activation, through its impact on immunopathology and parasite escape, could facilitate the development of novel therapies for preventing or reversing T. cruzi infection.
Acknowledgments
This work was supported by grants from ‘Consejo de Investigaciones Científicas y Técnicas’ (CONICET), Fundación Antorchas and ‘Agencia Córdoba Ciencia’ and ‘Agencia Nacional de Promoción Científica y Técnica, FONCYT’. CM, EZ thank CONICET for the fellowships granted. AG. and CA are members of the Research Career from CONICET. We thank Dr C. Nowicki for providing T. cruzi mMDH specific antiserum and Dr N. Gerez de Burgos and Dr M. S. Remedi for the enzyme activity determination.
REFERENCES
- 1.Brener Z, Gazinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol. 1997;114:103–10. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]
- 2.Leite de Moraes MC, Hontebeyrie-Joskowicz M, Leboulenger F, Savino W, Dardenne M, Lepault F. Studies on the thymus in Chagas' disease. II. Thymocyte subset fluctuations in Trypanosoma cruzi-infected mice: relationship to stress. Scand J Immunol. 1991;33:267–75. doi: 10.1111/j.1365-3083.1991.tb01772.x. [DOI] [PubMed] [Google Scholar]
- 3.Minoprio P, Eisen H, Forni L, D'Imperio Lima MR, Joskowicz M, Coutinho A. Polyclonal lymphocyte response to murine Trypanosoma cruzi infection. I. Quantitation of both T- and B- cell responses. Scand J Immunol. 1986;24:661–8. doi: 10.1111/j.1365-3083.1986.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 4.Russo M, Starobinas N, Ribeiro-Dos Santos R, Minoprio P, Eisen H, Hontebeyrie-Joskowicz M. Susceptible mice present higher macrophage activation than resistant mice during infections with myotropic strains of Trypanosoma cruzi. Parasite Immunol. 1989;11:385–95. doi: 10.1111/j.1365-3024.1989.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 5.Spinella S, Liegeard P, Guilbert B, Hontebeyrie-Joskowicz M. Anti-Ia treatment modulates specific and polyclonal antibody responses in Trypanosoma cruzi-infected mice. J Autoimmunity. 1989;2:791–802. doi: 10.1016/0896-8411(89)90005-x. [DOI] [PubMed] [Google Scholar]
- 6.Minoprio P, Burlen O, Pereira P, Guilbert B, Andrade L, Hontebeyrie-Joskowicz M, Coutinho A. Most B cells in acute Trypanosoma cruzi infection lack parasite specificity. Scand J Immunol. 1988;28:553–61. doi: 10.1111/j.1365-3083.1988.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 7.Minoprio P, Itohara S, Heusser C, Tonegawa S, Coutinho A. Immunobiology of murine T. cruzi infection: The predominance of parasite-nonspecific responses and the activation of TCRI T cells. Immunol Rev. 1989;112:183–207. doi: 10.1111/j.1600-065x.1989.tb00558.x. [DOI] [PubMed] [Google Scholar]
- 8.Fleming B, Rook GAW. T-cell dependent polyclonal activation by soluble mycobacterial extracts of B cells in peripheral blood mononuclear cell populations from leprosy patients and normal donors. Immunology. 1982;47:589–95. [PMC free article] [PubMed] [Google Scholar]
- 9.Rott O, Cash E. Influenza virus hemagglutinin induces differentiation of mature resting B cells and growth arrest of immature WEHI-231 lymphoma cells. J Immunol. 1994;152:5381–91. [PubMed] [Google Scholar]
- 10.Yamashita T, Watanabe T, Saito S, Araki Y, Sendo F. Shistosoma japonicum soluble egg antigens activate naive B cells to produce antibodies: definition of parasite mechanisms of immune deviation. Immunology. 1993;79:189–95. [PMC free article] [PubMed] [Google Scholar]
- 11.Goldenberg SS, Carneiro MN, Silva-Pereira AA, Mares-Guia L. Release of lipopolysaccharide (LPS) from cell surface of Trypanosoma cruzi by EDTA. Int J Parasitol. 1983;13:11–18. doi: 10.1016/s0020-7519(83)80062-9. [DOI] [PubMed] [Google Scholar]
- 12.Cordeiro Da Silva A, Espinoza AG, Taibi A, Ouaissi A, Minoprio P. 24000 MW Trypanosoma cruzi antigen is a B-cell activator. Immunology. 1998;94:189–96. doi: 10.1046/j.1365-2567.1998.00498.x. 10.1046/j.1365-2567.1998.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montes CL, Vottero-Cima E, Gruppi A. Trypanosoma cruzi cytosolic alkaline antigens (FI) induce polyclonal activation in murine normal B cells. Scand J Immunol. 1996;44:93–100. doi: 10.1046/j.1365-3083.1996.d01-285.x. [DOI] [PubMed] [Google Scholar]
- 14.Reina San Martin B, Degrave W, et al. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nature Med. 2000;6:890–7. doi: 10.1038/78651. [DOI] [PubMed] [Google Scholar]
- 15.Montes CL, Zuñiga E, Minoprio P, Vottero-Cima E, Gruppi A. Trypanosoma cruzi alkaline antigen induces polyclonal B-cell activation of normal murine spleen cells by T-cell-independent, BCR-directed stimulation. Scand J Immunol. 1999;50:159–66. doi: 10.1046/j.1365-3083.1999.00577.x. 10.1046/j.1365-3083.1999.00577.x. [DOI] [PubMed] [Google Scholar]
- 16.Reina San Martín B, Cosson A, Minoprio P. Lymphocyte polyclonal activation: a pitfall for vaccine design against infectious agents. Parasitol Today. 2000;16:62–7. doi: 10.1016/s0169-4758(99)01591-4. [DOI] [PubMed] [Google Scholar]
- 17.Camargo EP. Growth and differentiation in T. cruzi origen of metacyclic trypanosomes in liquid media. Rev Med Trop S Paulo. 1964;6:93–7. [PubMed] [Google Scholar]
- 18.Piuvezam MR, Russo DM, Burns JM, Jr, Skeiky YAM, Grabstein KH, Reed SG. Characterization of responses of normal human T cells to Trypanosoma cruzi antigens. J Immunol. 1993;150:916–24. [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Hunkapiller MW, Lujan E, Ostrander F, Hood LE. Isolation of microgram quantities of protein from polyacrylamide gels for amino acid sequence analysis. Meth Enzymol. 1983;91:227–36. doi: 10.1016/s0076-6879(83)91019-4. [DOI] [PubMed] [Google Scholar]
- 21.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometry sequencing of silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–8. doi: 10.1021/ac950914h. 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 22.Konecny P, Redinbaugh MG. Amplification of differentially displayed PCR products isolated from untreated denaturing polyacrylamide gels. Biotechniques. 1997;22:246–50. doi: 10.2144/97222bm09. [DOI] [PubMed] [Google Scholar]
- 23.Marina A, García MA, Albar JP, Yagüe J, López de Castro JA, Vázquez J. High-sensitivity analysis and sequencing of peptides and proteins by quadrupole ion trap mass spectrometry. J Mass Spectrom. 1999;34:17–27. doi: 10.1002/(SICI)1096-9888(199901)34:1<17::AID-JMS746>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 24.Ogueta S, Rogado R, Moreno F, Redondo JM, Vázquez J. Identification of phosphorylation sites in proteins by nanospray quadrupole ion trap mass spectrometry. J Mass Spectrom. 2000;35:556–65. doi: 10.1002/(SICI)1096-9888(200004)35:4<556::AID-JMS969>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Reynoso Hunter G, Hellman U, Cazzulo JJ, Nowicki C. Tetrameric and dimeric malate dehydrogenase isoenzymes in Trypanosoma cruzi epimastigotes. Mol Biochem Parasitol. 2000;105:203–14. doi: 10.1016/s0166-6851(99)00176-0. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida A. L-Malate dehydrogenase from Bacillus subtilis. Meth Enzymol. 1969;13:141–8. [Google Scholar]
- 27.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–9. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 28.Liu MA, Friedman A, Oliff AL, et al. A vaccine carrier derived from Neisseria meningitidis with mitogenic activity for lymphocytes. Proc Natl Acad Sci USA. 1992;89:4633–7. doi: 10.1073/pnas.89.10.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y. Janeway CA Jr Microbial induction of costimulatory activity for CD4 T-cell growth. Int Immunol. 1991;3:323–32. doi: 10.1093/intimm/3.4.323. [DOI] [PubMed] [Google Scholar]
- 30.Hauschildt S, Kleine B. Bacterial stimulators of macrophages. Int Rev Cytol. 1995;161:263–331. doi: 10.1016/s0074-7696(08)62499-7. [DOI] [PubMed] [Google Scholar]
- 31.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–57. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 32.Kuchroo VK, Das MP, Brown JA, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–18. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 33.Frosch S, Kuntzlin D, Fleischer B. Infection with Trypanosoma cruzi selectively upregulates B7-2 molecules on macrophages and enhances their costimulatory activity. Infect Immun. 1997;65:971–7. doi: 10.1128/iai.65.3.971-977.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenschow DJ, Herold KC, Rhee L, et al. CD28/B7 Regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–93. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- 35.Cannata JJB, Cazzullo JJ. Glycosomal and mitochondrial malate dehydrogenases in epimastigotes of Trypanosoma cruzi. Mol Biochem Parasitol. 1984;11:37–49. doi: 10.1016/0166-6851(84)90053-7. [DOI] [PubMed] [Google Scholar]
- 36.Minoprio P, Eisen H, Joskowicz M, Pereira P, Coutinho A. Supression of polyclonal antibody production in Trypanosoma cruzi infected mice by treatment with anti-L3T4 antibodies. J Immunol. 1987;139:545–50. [PubMed] [Google Scholar]
- 37.Minoprio P, Coutinho A, Spinella S, Hontebeyrie-Joskowicz M. Xid immunodeficiency imparts increased parasite clearance and resistance to pathology in experimental Chagas' disease. Int Immunol. 1991;3:427–33. doi: 10.1093/intimm/3.5.427. [DOI] [PubMed] [Google Scholar]