

Abstract
The development of graft-versus-host disease (GVHD) can be modified by non-MHC factors. Based on our previous studies that showed an involvement of 70kD heat shock protein (hsp70) in the pathology of acute GVHD in a rat model, we determined serum levels of antibodies to hsp70, hsp90 and hsp60 in human recipients after allogeneic peripheral blood stem cell transplantation (PBSCT). Serum levels of these antibodies were correlated with GVHD status in the recipients. Twenty-nine recipients with high-risk haematological malignances, who received G-CSF mobilized allogeneic PBSCT from HLA matched family donors, were evaluated between 30 and 960 days after transplantation. Two recipients had no GVHD, 18 developed acute followed by chronic GVHD and nine developed only chronic GVHD. Patients with acute GVHD had a significant increase in IgM anti-hsp70 and/or anti-hsp90 early (30–90 days) after transplantation. In addition, an increase in IgM anti-hsp70 and/or anti-hsp90 antibodies preceded or accompanied chronic GVHD. Antibody levels returned to normal within the next 400 days in the majority of patients. Anti-hsp60 antibody levels were not different from control levels regardless of GVHD status. This study implies that the development of acute and/or chronic GVHD in humans is accompanied by an increase in anti-hsp70 and anti-hsp90 antibodies. Monitoring levels of anti-hsp70 and anti-hsp90 antibodies in stem cell transplant recipients may serve as a diagnostic tool and help to predict the onset of GVHD.
Keywords: GVHD, hsp70, hsp90, PBSC transplantation
INTRODUCTION
Peripheral blood stem cell transplantation (PBSCT) has been used in the treatment of haematological malignancies, genetic haematological disorders and aplastic anaemia [1,2]. Success of PBSCT or bone marrow transplantation (BMT) is limited greatly by the occurrence of graft-versus-host disease (GVHD) both acute and chronic. GVHD is initiated by donor T cells reactive with disparate MHC antigens of the host [3]. However, GVHD is still common in MHC-matched BMT recipients or in MHC-matched animal models of the disease [4]. Resulting GVHD varies typically in the intensity, with differences attributed to disparities in minor histocompatibility antigens [5] or other factors, among them cytokine gene polymorphism [6–9]. We propose that heat shock proteins (hsp) could also modify GVHD. Our previous studies in a rat model of acute GVHD indicate that hsp70 is involved in acute GVHD pathology [10–12].
Hsps function in cells as molecular chaperones facilitating protein folding, assembly and intracellular transport [13]. Their synthesis is greatly increased in response to a variety of stressful stimuli [14]. Despite the significant degree of evolutionary conservation, hsps are highly immunogenic and it has been postulated that they could activate antigen-presenting cells, serving as a danger signal to the immune system [15]. Immune reactivity against different members of hsp families, most frequently hsp60, hsp70 and hsp90, accompanies many infectious diseases [16], with reactivity to bacterial hsp60 and hsp71 detected in tuberculosis and leprosy [17,18] and to hsp90 in malaria [19]. Immune reactivity to hsps is also detected in autoimmune disorders, among them lupus erythematosus (hsp60, hsp70, hsp90) [20], rheumatoid arthritis (hsp60, hsp70) [21], Crohn’s disease and ulcerative colitis (hsp60, hsp70, hsp90) [22,23]. Based on these observations, it has been proposed that anti-microbial hsp antibodies may cross-react with host hsp, breaking self-tolerance, leading to autoimmunity as a result of immunological mimicry [20]. Cross-reactivity to bacterial antigens is also implicated in GVHD pathology. In animal models of the disease, the incidence of GVHD can be reduced in germ-free animals or after gastrointestinal decontamination of conventional animals [24]. In addition, in humans the lower risk of GVHD in younger bone marrow transplant recipients has been linked to reduced exposure to bacterial pathogens and lower antibacterial reactivity in younger patients [25]. GVHD is also less frequent in patients who were subjected to microbial decontamination [26].
Reactivity to hsp has been associated with rejection in solid organ transplants [27]. Increases in hsp70 expression have been observed during the rejection of lung transplants in humans [28] and rat cardiac allografts [29]. Hsps, in particular the hsp70 family, are involved in antigen processing and presentation [30,31] and can function as molecular adjuvants. Immunization with tumour-derived hsp70 or gp96 reduces metastasis and the rate of growth of murine Lewis lung carcinoma and melanoma [32]. In addition, an increased capacity for hsp70 synthesis by rat colon cancer cells corresponds with their decreased tumorigenicity [33]; also human CD4+ tumour-infiltrating lymphocytes are reactive to hsp70 expressed on B cell lines [34].
In our rat model of BMT, GVHD progression is accom-panied by increase in synthesis of hsp70 by spleen and lymph node cells, and concomitant generation of anti-hsp70 serum antibodies [10,11]. The immunosuppressant, 15-deoxyspergualin (DSG), which binds intracellularly to a constitutive form of hsp70 [35], can reduce mortality associated with GVHD in this rat model. In DSG-treated GVHD rats, hsp70 synthesis by spleen and lymph node cells was significantly reduced and serum antibodies to hsp70 were lowered to undetectable levels [12]. Based on these studies, we tested the hypothesis that humoral immune responses to hsps accompany human GVHD following PBSCT. The increased incidence of de novo chronic GVHD after PBSCT allowed us to compare the development and persistence of reactivity against hsp in human PBSCT recipients after either acute or chronic GVHD. We determined antibody levels against hsp70, hsp90, and hsp60. We found that anti-hsp70 and anti-hsp90 but not anti-hsp60 antibody levels were elevated in patients early (30–90 days) after transplantation, and correlated with the incidence of acute, or an early onset (100–120 days) of chronic GVHD. Antibodies were at the control levels during later post transplantation time periods.
METHODS
Patients
Twenty-nine patients, 21 males and eight females, 24–68 years old, with high-risk haematological malignances were included in the study. After myeloablative conditioning (total body irradiation: 175 Gy × 6 doses, VP16: 30 mg/kg × 1, cyclophosphamide: 50 mg/kg × 2) they received G-CSF mobilized PBSCT from completely HLA matched (6/6) adult siblings. HLA-A and HLA-B typing was performed by serological, and HLA-DR by high resolution molecular methods. Donor cell engraftment was confirmed by VNTR (variable number of tandem repeats) analysis at 90, 180, 360 and 720 days after transplantation. Recipients with graft rejection were deleted from the study, as were patients who suffered a relapse of the primary disease, or were overtly infected. Plasma evaluated in the study was isolated from blood samples collected between 30 and 960 days after the transplantation. Twenty-two of 29 PBSCT recipients were evaluated at two different time points. Acute GVHD, with an onset within 100 days after transplantation, was diagnosed following accepted clinical guidelines [36], as was chronic GVHD [37] with an onset after 100 days post-transplantation. Among the PBSCT recipients two of 29 did not develop GVHD, 18 developed acute GVHD (grades 1–3) first; of these, 15 patients (three patients died) developed chronic GVHD. Nine patients developed de novo chronic GVHD. Blood was collected from 11 patients during active acute GVHD, nine patients during active chronic GVHD (progressive and de novo) and 13 patients with inactive GVHD.
The immunosuppressive regimen for all PBSCT recipients included administration of cyclosporin A (CSA), 3 mg/kg body weight from day 1 post-transplantation. Levels of CSA were maintained at 250–350 ng/ml blood for 100 days. They were tapered gradually over the next 2 months or continued, based on GVHD status. Methyl prednisolone at 0·5 mg/kg was commenced on day 7, increased to 1 mg/kg on day 14 and tapered between days 28 and 56 in the absence of GVHD, continued in the presence of GVHD. Progressive acute GVHD was treated with administration of ATG (antithymocyte globulin) 15–30 mg/kg/day × 3 and increased doses of steroids. Progressive chronic GVHD was treated with CSA, steroids and azathioprine (25–150 mg/day).
Since the GVHD status of one patient was unknown he was excluded from this analysis. The study also included 17 healthy adult male and female controls and three PBSC donors before and after G-CSF mobilization.
Immunoglobulin quantification
Plasma samples were thawed once, aliquoted and stored at – 70°C until further analysis. IgG, IgM and IgA plasma levels were measured by rate nephelometry with a Beckman Coulter ARRAY® (Beckman Coulter Inc., Fullerton, CA, USA). Normal range values were derived from patient studies conducted by Beckman Coulter Inc., validated by Loyola University Medical Center Laboratories.
Elisa
Plasma antibodies to hsp70, hsp90 and hsp60 were quantified by ELISA; 96-well ELISA plates (Corning-Fisher Scientific, Itasca, IL, USA) were coated with, added to each well, 50 μl (10 μg/ml in 0·05 m bicarbonate buffer, pH 9·5) of recombinant human hsp70, hsp90, or hsp60 (StressGen Biotechnologies Corp., Canada) or mucin (Sigma-Aldrich Co., St Louis, MO, USA). After an overnight incubation at 4°C, the plates were washed three times with PBS, pH 7·4 with 0·05% Tween (PBST). PBST was used throughout assays to wash plates. Next 1% bovine serum albumin (BSA) (Sigma) in PBST was added to block non-specific binding (2 h at room temperature). After subsequent washing, 50 μl of tested plasma samples diluted 1:50–1:800 in PBST/1% BSA were added to the wells in duplicates. Each ELISA test included a set of three reference plasma samples, to allow direct comparisons between tests. The reference samples were randomly chosen healthy control samples, which were included in every experiment. Plates were incubated overnight at 4°C and washed. The plates were then incubated for 5 h at room temperature with secondary peroxidase conjugated antibodies (Sigma Biosciences, St Louis, MO, USA). To determine general levels of anti-hsp antibodies, rabbit-anti-human immunoglobulins (IgA, IgG and IgM) antibody diluted 1:30 000 was employed. For isotype characterization goat-anti-human IgG (Fab specific) diluted 1:40 000 and goat-anti-human IgM (μ−chain specific) diluted 1:50 000 were used. The plates were washed and developed with 0·4 mg/ml of o-phenylenediamine dihydrochloride (OPD) in 0·05 m phosphate citrate buffer, pH 5·0 containing 0·03% sodium perborate (Sigma Biosciences, St Louis, MO, USA). Colour development was stopped with 3 N HCl and optical density was determined at 490 nm (O.D.490) using an ELISA plate reader (Titertek Multiscan, Flow Laboratories Inc., VA, USA).
Results were calculated at 1:100 plasma dilution (for tested samples O.D.490 at 1:100 dilution was in the midpoint of the titration curve). Results were reported as the ratio of O.D.490 of tested to reference plasma samples (the average of three samples included in all experiments).
Statistical analysis
Numerical data were log transformed and evaluated by one-way and two-way analysis of variance (anova). Post hoc comparisons were made using Tukey’s test. Significance level was established at P < 0·05.
RESULTS
Levels of plasma antibodies against hsp70, hsp90 and hsp60
The presence of antibodies to hsp70, hsp90 and hsp60 was examined in the blood plasma from healthy controls. Since microbial hsps function as dominant antigens during the immune response, circulating anti-hsp antibodies were expected, a probable consequence of persistent contact with microorganisms from the environment [38]. As anticipated, low levels of anti-hsp70, anti-hsp90 and anti-hsp60 antibodies were detected in control plasma. Low levels of anti-hsp antibodies were also detected in plasma of PBSC donors. The administration of G-CSF had no effect on antihsp antibody production, since in PBSC donors they were at the control levels before and after G-CSF mobilization.
In PBSCT recipients who did not develop any GVHD after transplantation, anti-hsp antibodies remained at the control levels. In contrast, all the patients with acute GVHD, whose blood samples were collected during the active disease period (between days 30 and 90 after transplantation), had significantly elevated anti-hsp70 (P < 0·001) and/or anti-hsp90 (P < 0·001) antibodies. Their antihsp60 antibodies stayed at the control level (Fig. 1). Specifically, among 11 patients tested during the acute GVHD episode, eight had higher levels of both anti-hsp70 and anti-hsp90 antibodies (2·3–6·7 and 2·1–4·9 times above the control, respectively), two had higher only anti-hsp70 antibodies (2·0–2·4 times above the control) and one had only higher antihsp90 antibodies (twice the control). Thus, the elevated anti-hsp70 and/or anti-hsp90, but not anti-hsp60 antibody levels accompanied acute GVHD. There was no correlation between the levels of the antibodies and the severity of acute GVHD (grades 1–3) in these patients.
Fig. 1.

Antibodies to hsp70, hsp90 and hsp60 measured by ELISA (mean ± SE of the ratios of tested to reference plasma samples O.D.490) in PBSCT recipients undergoing acute GVHD (aGVHD), in GVHD-free patients (no GVHD), and in healthy controls. *Anti-hsp70 and anti-hsp90 antibodies were at significantly higher levels (P < 0·001) in acute GVHD patients than in other groups.  , Anti-hsp70; ■, anti-hsp90; □, anti-hsp60.
, Anti-hsp70; ■, anti-hsp90; □, anti-hsp60.
With the exception of GVHD-free patients, all PBSCT recipients who survived beyond 3 months post-transplantation developed progressive or de novo chronic GVHD after the procedure. Elevated levels of anti-hsp70 and anti-hsp90 antibodies were present in 13 of these patients (2·1–5·4 and 2·2–7·8 times above the control, respectively). In nine of the PBSCT recipients the increase in anti-hsp70 and/or anti-hsp90 (3·2 and 2·7 times above the control, respectively, P < 0·05) coincided with the diagnosis of chronic GVHD. Thus, the elevated anti-hsp70 and anti-hsp90 antibody levels accompanied the chronic GVHD in the majority of the PBSCT recipients. In two patients who developed chronic GVHD de novo 120 days after PBSCT, elevated anti-hsp70 and anti-hsp90 antibodies were measured twice between 30 and 90 days after the procedure. Therefore, in these patients elevated anti-hsp70 and anti-hsp90 antibody levels preceded the chronic GVHD. Anti-hsp60 antibodies stayed at the control level in all of the patients (Fig. 2) Within the studied patient population, no differences were observed in anti-hsp70 and anti-hsp90 antibody production between de novo and progressive, or between a limited and extensive form of the chronic GVHD. Therefore, any form of chronic GVHD coincided with an increase in anti-hsp70 and anti-hsp90 antibody levels.
Fig. 2.

Antibodies to hsp70, hsp90 and hsp60 measured by ELISA (mean ± SE of the ratios of tested to reference plasma samples O.D.490) in PBSCT recipients undergoing chronic GVHD (cGVHD) and in inactive GVHD patients. *Anti-hsp70 and anti-hsp90 antibodies were at significantly higher levels (P < 0·05) in chronic GVHD patients than in patients with inactive GVHD.  , Anti-hsp70; ■, anti-hsp90; □, anti-hsp60.
, Anti-hsp70; ■, anti-hsp90; □, anti-hsp60.
The number of PBSCT recipients with elevated anti-hsp70 and/or anti-hsp90 plasma antibodies diminished progressively with time after transplantation. Elevated anti-hsp70 and/or anti-hsp90 (at least twice the control level), but not anti-hsp60 antibody levels were detected in 11 of 16 patients tested between 120 and 480 days post-PBSCT, and in two of eight patients tested between 510 and 960 days post-PBSCT. Consequently, the average levels of anti-hsp70 and anti-hsp90 antibodies in patient populations tested during the later time periods (>120 days) post-transplantation were not significantly different than the control (Table 1). The reduction in the number of patients with elevated anti-hsp70 and anti-hsp90 antibodies during the later post-transplantation time periods coincided with a resolution of GVHD in response to therapeutic treatment. Thus, the return of anti-hsp70 and anti-hsp90 plasma antibodies to control levels could indicate the response to therapy.
Table 1.
Antibodies to hsp70, hsp90 and hsp60 measured by ELISA (median with range values of tested to reference plasma samples at O.D.490). Comparison of PBSCT recipients tested during different time periods post-transplantation and healthy controls
| Group | Number of patients | Anti-hsp70 | Anti-hsp90 | Anti-hsp60 |
|---|---|---|---|---|
| 30–90 days post-transplantation | 14 | 2·84 (1·37–6·74)* | 3·12 (0·23–7·8)* | 1·15 (0·23–2·01) |
| 120–480 days post-transplantation | 16 | 1·26 (0·59–5·43) | 1·03 (0·64–2·96) | 1·2 (0·28–1·36) |
| 510–960 days post-transplantation | 8 | 1·52 (0·63–4·53) | 1·33 (0·37–3·81) | 1·01 (0·5–1·66) |
| Control | 17 | 1·08 (0·75–1·36) | 1·16 (0·58–1·3) | 1·3 (0·28–1·66) |
Anti-hsp70 and antihsp90 antibodies were at significantly higher levels (P < 0·001) in patients 30–90 days post-transplantation than in other groups.
Since intestinal injury is a part of GVHD pathology, general humoral responses to proteins present in the gut could be expected in PBSCT recipients. Therefore, the presence of antibodies to mucin, a common intestinal protein which was used as a control antigen, was examined. Nevertheless, antibodies to mucin were not detected in patients’ plasma (data not shown). The absence of immune reactivity to mucin, as well as the lack of observable changes in anti-hsp60 antibody levels in the course of GVHD (Figs 1 and 2), indicate that anti-hsp antibodies present in PBSCT recipients were synthesized as a result of the specific immune response against hsp70 and hsp90.
Isotype analysis of anti-hsp70 and anti-hsp90 antibodies
The isotype of anti-hsp70 and anti-hsp90 antibodies was analysed by ELISA. Blood plasma samples were divided into two groups: plasma collected from active acute or chronic GVHD patients with elevated levels of anti-hsp70 and anti-hsp90 antibodies, and plasma collected from patients with inactive GVHD, who did not show an increase in anti-hsp antibody levels. An increase in anti-hsp70 and anti-hsp90 antibodies, observed in the active GVHD group, was associated with IgM but not with IgG or IgA antibodies (Fig. 3a,b). In this group the levels of IgM anti-hsp70 and anti-hsp90 were significantly higher than in control (2·6 times control, P < 0·001 for anti-hsp70; and 2·7 times control, P < 0·001 for anti-hsp90). Anti-hsp70 and anti-hsp90 antibodies of IgG and IgA isotypes in active GVHD group were no different than the control. As expected, anti-hsp70 and anti-hsp90 antibodies of IgM, IgG and IgA isotypes in inactive GVHD group did not differ from control.
Fig. 3.
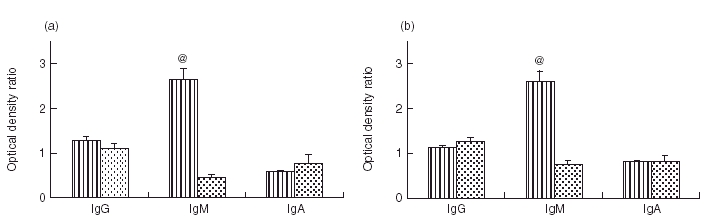
Isotype of anti-hsp70 (a) and anti-hsp90 (b) antibodies measured by ELISA (mean ± SE of the ratios of tested to reference plasma samples O.D.490). Comparison between the patients with active acute or chronic GVHD (with elevated anti-hsp70 and hsp90 antibodies), and inactive GVHD patients (with anti-hsp70 and anti-hsp90 antibodies at the control levels). @: Anti-hsp70 and anti-hsp90 antibodies of IgM isotype were at significantly higher levels (P < 0·001) in active GVHD than in inactive GVHD patients. Levels of anti-hsp70 and anti-hsp90 antibodies of IgG and IgA isotypes remained unchanged in both groups.( ), Active GVHD; (
), Active GVHD; ( ), inactive GVHD.
), inactive GVHD.
To examine the possibility that elevation in anti-hsp70 and anti-hsp90 antibody levels were not due to a general differences in IgM, IgG and IgA antibody levels, plasma concentrations of IgM, IgG and IgA immunoglobulins were measured in active and inactive GVHD patients and compared with healthy controls (Fig. 4). The total immunoglobulin levels did not show any significant differences between patient and healthy control groups. In patients with active or inactive GVHD total IgM, IgG and IgA plasma concentrations were at the low end of the normal range.
Fig. 4.

IgG, IgM and IgA immunoglobulin plasma levels measured by rate nephelometry (median with range values); comparison between active acute or chronic GVHD patients (with elevated anti-hsp70 and hsp90 antibodies), inactive GVHD patients (with anti-hsp70 and anti-hsp90 antibodies at the control levels) and healthy controls. ■, Active GVHD; ( ), inactive GVHD; □, control.
), inactive GVHD; □, control.
DISCUSSION
In this study the presence of circulating antibodies to hsp70, hsp90 and hsp60 was studied in MHC-matched PBSC transplant recipients. All the patients with active acute GVHD had significantly augmented anti-hsp70 and/or anti-hsp90 antibody levels. Similarly, elevated anti-hsp70 and/or anti-hsp90 plasma antibodies were present in the majority of the patients who developed progressive or de novo chronic GVHD. During the later post-transplantation time periods, with a resolution of GVHD due to the therapeutic treatment, anti-hsp antibodies returned to the control levels. Anti-hsp70 and anti-hsp90 antibodies were of IgM, but not IgG or IgA isotype. There was no correlation between GVHD and anti-hsp60 antibodies. These results, similar to our previous studies which indicated the involvement of hsp70 in rat GVHD (10–12), support the hypothesis that antibodies to hsps are associated with GVHD.
Our observations in the acute rat GVHD model suggested that lymphoid cells may elicit a stress response which increases hsp70 synthesis, presumably within donor as well as host cells [10]. In the present study we have examined patterns in anti-hsp antibody production but not changes in the levels of heat shock proteins (hsp70, hsp90, hsp60) in PBSCT recipients. The increase in anti-hsp70 and anti-hsp90 antibody levels indicates that hsp70 and hsp90 proteins may play a role in immune interactions of GVHD. Increase in heat shock proteins synthesis and/or their possible release due to donor–anti-host cytotoxicity resulting from GVH reaction could serve as a danger signal and improve the function of antigen presenting cells [15,31]. Extracellular hsp70 could bind to antigen-presenting cells, stimulating their inflammatory cytokine production [39]. It has been shown that host-derived antigen presenting cells are required in GVHD initiation [40]. In addition, the presence of hsps in the extracellular milieu could activate normally unresponsive antiself-hsp B cells of the host. In this regard, it has been demonstrated that previously tolerant B-cells can be activated, which results in antibody production, by the encounter of a self-mimicking antigen binding to the B-cell receptor [41]. A possibility of antihsp antibody production as a consequence of immunological mimicry could additionally strengthen the anti-hsp response.
Interestingly, our observations indicate that anti-hsp responses in GVHD are not directed against hsp60. Even though reactivity to hsp60 has been well documented in autoimmune disorders such as diabetes [42], arthritis [20] and inflammatory bowel disease (Crohn’s disease and ulcerative colitis) [22,23], anti-hsp60 antibody levels were not affected in GVHD. The apparent lack of anti-hsp60 activity in GVHD indicates that different pathways of hsp activation and synthesis are involved in the disease pathology. It is also noteworthy that different patterns of immunoglobulin production have been associated with PBSCT and inflammatory bowel disease. It has been reported that IgG and IgM levels increase in inflammatory bowel disease [43,44]. In contrast, IgG, IgM and IgA levels of PBSCT patients were low, but within a normal range (Fig. 4). In addition, there were no differences between total IgM, IgG and IgA levels in active and inactive GVHD patients, although specific anti-hsp70 and anti-hsp90 antibodies were elevated in active GVHD and decreased in inactive GVHD patients (Fig. 3a,b). It is possible that immunosuppressive agents used in patients’ treatment could target intracellular hsps specifically, similar to DSG binding to the constitutively expressed hsp70 [35], interfering with a progression of immune reactions in GVHD. It has been shown that binding of DSG to hsp70 results in the inhibition of LPS-induced nuclear translocation of the transcription factor NFκB [45]. It has been also shown that binding of exogenous hsp70 to monocytes activates NFκB [39]. Since NFκB is a crucial player in inflammatory and immune responses [46], any interference with the factor’s interactions could result in profound changes in immune processes. In our previous studies with rat GVHD, administration of DSG resulted in lower detection of hsp70 in spleen and lymph node cells, inhibition of anti-hsp70 serum antibody synthesis, IL-2 and IL-10 production, and partial inhibition of TNF-α and IFN-γ synthesis [12].
Anti-hsp70 activity in our rat acute GVHD model has been associated with IgM, as well as IgG2a and IgG2b antibodies [11]. In the present study circulating anti-hsp antibodies detected in the PBSCT recipients were of the IgM, but not the IgG isotype. This may also be a consequence of immunosuppressive therapy, administered to the patients, and its inhibitory effects on cytokine production necessary for isotype switching. Since cytokines such as IFN-γ and IL-10 regulate B cell switching to several IgG isotypes [47,48], inhibition of cytokine production can render an environment non-permissible for the immunoglobulin isotype switch. Lower IgG but not IgM or IgA total antibody levels in PBSCT recipients support this explanation. In contrast, the acute GVHD in a rat MHC-mismatched model was accompanied by IFN-γ and IL-10 synthesis [12,49], hence the presence of IgG anti-hsp70 antibodies. Alternatively, the lack of anti-hsp antibodies of IgG isotype could relate to different profiles of cytokine production and altered B cell functions in PBSCT and BMT patients. Our observations indicate that lower levels of IFNγ (are present in PBSCT when compared with BMT recipients) (unpublished observations), thus the immunoglobulin isotype switch may occur early in BMT but not PBCST recipients.
Even though, at present, the exact mechanism behind hsp70 and hsp90 involvement in GVHD is not known, the results of this study show that monitoring PBSC recipients for the presence of anti-hsp70 and anti-hsp90 plasma antibodies could be employed as an indicator of GVHD development. In addition, further investigation of hsp70 and hsp90 role in GVHD pathology may warrant new GVHD therapies with hsps as target molecules.
Acknowledgments
Supported in part by NIHKO8 Award CA72587. The authors thank Dr D. Adkins, Dr R. Brown and Dr J. Dipersio for making patients plasma samples available for analysis.
REFERENCES
- 1.Gale RP, Champlin RE. New strategies in bone marrow transplantation. New York: Wiley; 1991. [Google Scholar]
- 2.Parkman R. The application of BMT to the treatment of genetic diseases. Science. 1986;232:373–8. doi: 10.1126/science.3520819. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara JL, Cooke KR, Pan L, Krenger W. The immunopathology of acute graft-versus-host-disease. Stem Cells. 1996;14:473–89. doi: 10.1002/stem.140473. [DOI] [PubMed] [Google Scholar]
- 4.Allen RD, Dobkins JA, Harper JM, Slayback DL. Genetics of graft-versus-host disease, I. A locus on chromosome 1 influences development of acute graft-versus-host disease in major histocompatibility complex mismatched murine model. Immunology. 1999;96:254–61. doi: 10.1046/j.1365-2567.1999.00626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Els CA, D’Amaro J, Pool J, et al. Imunogenetics of human minor histocompatibility antigens: their polymorphism and immunodominance. Immunogenetics. 1992;35:161–5. doi: 10.1007/BF00185109. [DOI] [PubMed] [Google Scholar]
- 6.Bailly S, di Giovine FS, Blakemore AIF, Duff GW. Genetic polymorphism of human interleukin-1α. Eur J Immunol. 1993;23:1240–5. doi: 10.1002/eji.1830230607. [DOI] [PubMed] [Google Scholar]
- 7.Bioque G, Crusius JBA, Koutroubakis I, et al. Allelic polymorphism in IL-1β and IL-1 receptor antagonist (IL-1Ra) genes in inflammatory bowel disease. Clin Exp Immunol. 1995;102:379–83. doi: 10.1111/j.1365-2249.1995.tb03793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walley AJ, Cookson WOCM. Investigation of an interleukin-4 promoter polymorphism for associations with asthma and atopy. J Med Genet. 1996;33:689–92. doi: 10.1136/jmg.33.8.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner DM, Williams DM, Sankaran D, Lazarus M. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24:1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 10.Goral J, Mathews HL, Clancy J., Jr Expression of 70-kDa heat-shock protein during acute graft-versus-host disease. Clin Immunol Immunopathol. 1998;86:252–8. doi: 10.1006/clin.1997.4473. [DOI] [PubMed] [Google Scholar]
- 11.Goral J, Mathews HL, Clancy J., Jr Antibodies specific for the 70-kDa heat-shock protein parallel the development of acute graft-versus-host disease in (DA×LEW) F1 rats. Clin Immunol Immunopathol. 1995;75:147–53. doi: 10.1006/clin.1995.1064. [DOI] [PubMed] [Google Scholar]
- 12.Goral J, Mathews HL, Nadler SG, Clancy J., Jr Reduced levels of hsp70 result in a therapeutic effect of 15-deoxyspergualin on acute graft-versus-host disease in (DA×LEW) F1 rats. Immunobiology. 2000;202:254–66. doi: 10.1016/s0171-2985(00)80032-7. [DOI] [PubMed] [Google Scholar]
- 13.Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 14.Welch WJ, Kang HS, Beckmann RP, Mizzen LA. Response of mammalian cells to metabolic stress: changes in cell physiology and structure/function of stress proteins. Curr Top Microbiol Immunol. 1990;167:31–55. doi: 10.1007/978-3-642-75875-1_2. [DOI] [PubMed] [Google Scholar]
- 15.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 16.Zugel U, Kaufmann SHE. Immune response against heat shock proteins in infectious diseases. Immunobiology. 1999;201:22–35. doi: 10.1016/s0171-2985(99)80044-8. [DOI] [PubMed] [Google Scholar]
- 17.Young RA, Lathringa RB, Hendrix RW, Sweetser D, Young RA. Stress proteins are immune targets in leprosy and tuberculosis. Proc Natl Acad Sci USA. 1988;85:4267–70. doi: 10.1073/pnas.85.12.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams E, Basten A, Rodda S, Britton WJ. Human T-cell clones to the 70-kilodalton heat shock protein of Mycobacterium leprae define mycobacterium-specific epitopes rather than shared epitopes. Infect Immunol. 1997;65:1061–70. doi: 10.1128/iai.65.3.1061-1070.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jendoubi M, Bonnefoy S. Identification of a heat shock-like antigen in P. falciparum, related to the heat shock protein 90 family. Nucl Acids Res. 1988;16:10928–31. doi: 10.1093/nar/16.22.10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feige U, van Eden W. Infection, autoimmunity and autoimmune disease. In: Feige U, Morimoto RI, Yahara I, Polla B, editors. Stress-inducible cellular responses. Basel, Switzerland: Birkhauser-Verlag; 1996. pp. 359–73. [Google Scholar]
- 21.Hayem G, De Bandt M, Palazzo E, et al. Anti-heat shock protein 70 kDa and 90 kDa antibodies in serum of patients with rheumatoid arthritis. Ann Rheum Dis. 1999;58:292–6. doi: 10.1136/ard.58.5.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winrow VR, Mojdehi GM, Ryder SD, Rhodes JM, Blake DR, Rampton DS. Stress proteins in colorectal mucosa. Enhanced expression in ulcerative colitis. Dig Dis Sci. 1993;38:1994–2000. doi: 10.1007/BF01297075. [DOI] [PubMed] [Google Scholar]
- 23.Stevens TR, Winrow VR, Blake DR, Rampton DS. Circulating antibodies to heat-shock protein 60 in Crohn’s disease and ulcerative colitis. Clin Exp Immunol. 1992;90:271–4. doi: 10.1111/j.1365-2249.1992.tb07941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heidt PJ, Vossen JM. Experimental and clinical gnotobiotics: influence of the microflora on graft-versus-host disease after allogeneic bone marrow transplantation. J Med. 1992;23:161–73. [PubMed] [Google Scholar]
- 25.Hausmann PB, Weksler ME. Handbook of the biology of aging. New York: Von Nostrand Reinhold; 1985. pp. 414–432. [Google Scholar]
- 26.Beelen DW, Elmaagacli A, Muller KD, Hirche H, Schaefer UW. Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignances: final results of long-term follow-up of an open-label prospective randomized trial. Blood. 1999;93:3267–75. [PubMed] [Google Scholar]
- 27.Moliterno R, Valdivia L, Pan F, Duquesnoy RJ. Heat shock protein reactivity of lymphocytes isolated from heterotopic rat cardiac allografts. Transplantation. 1995;59:598–604. [PubMed] [Google Scholar]
- 28.Rizzo M, Alevy YG, Sundaresan S, et al. Increased expression of HDJ-2 (heat shock protein 40) and heat shock protein 70 in biopsy specimens of transplanted human lungs. J Heart Lung Transplant. 1998;17:241–9. [PubMed] [Google Scholar]
- 29.Davies EA, Wang BH, Stagg CA, et al. Induction of heat shock protein in cardiac allograft rejection – a cyclosporin-suppressible response. Transplantation. 1996;61:279–84. doi: 10.1097/00007890-199601270-00020. [DOI] [PubMed] [Google Scholar]
- 30.Nossner E, Goldberg JE, Naftzger C, Lyu SC, Clayberger C, Krensky AM. HLA-derived peptides which inhibit T cell function bind to members of the heat-shock protein 70 family. J Exp Med. 1996;183:339–48. doi: 10.1084/jem.183.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panjwani N, Akbari O, Garcia S, Brazil M, Stockinger B. The HSC73 molecular chaperone: involvement in MHC class II antigen presentation. J Immunol. 1999;163:1936–42. [PubMed] [Google Scholar]
- 32.Tamura Y, Peng P, Kang L, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 1997;278:117–20. doi: 10.1126/science.278.5335.117. [DOI] [PubMed] [Google Scholar]
- 33.Menoret A, Patry Y, Burg C, Le Pendu J. Co-segregation of tumor immunogenicity with expression of inducible but not constitutive hsp70 in rat colon carcinomas. J Immunol. 1995;155:740–7. [PubMed] [Google Scholar]
- 34.Yoshino I, Goedegebuure PS, Peoples GE, Lee KY, Eberlein TJ. Human tumor-infiltrating CD4+ T cells react to B cell lines expressing heat shock protein 70. J Immunol. 1994;153:4149–58. [PubMed] [Google Scholar]
- 35.Nadler SG, Tepper MA, Shacter B, Mazzucco CE. Interaction of the immunosuppressant deoxyspergualin with a member of the hsp70 family of heat shock proteins. Science. 1992;258:484–6. doi: 10.1126/science.1411548. [DOI] [PubMed] [Google Scholar]
- 36.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A matched sibling donors. Transplantation. 1974;18:295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Shulman HM, Sullivan KM, Weiden PL, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69:204–17. doi: 10.1016/0002-9343(80)90380-0. [DOI] [PubMed] [Google Scholar]
- 38.Wakui H, Itoh H, Kobayashi R, Nakamoto Y, Miura AB. Specific antibodies against the stress-inducible 72-kDa protein, a member of the heat-shock protein hsp70, in healthy human subject. Int J Biochem. 1991;23:975–8. doi: 10.1016/0020-711x(91)90132-7. [DOI] [PubMed] [Google Scholar]
- 39.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cyto-kine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nature Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 40.Schlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–5. doi: 10.1126/science.285.5426.412. 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 41.Kouskoff V, Lacaud G, Nemazee D. T cell-independent rescue of B lymphocytes from peripheral immune tolerance. Science. 2000;287:2501–3. doi: 10.1126/science.287.5462.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elias D, Markovits D, Rshef T, van der Zee R, Cohen IR. Induction and therapy of autoimmune diabetes in the non-obese diabetic (NOD/It) mouse by 65-kDa heat shock protein. Proc Natl Acad Sci USA. 1990;87:1576–80. doi: 10.1073/pnas.87.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodgson HJ, Jewell DP. The humoral immune system in inflammatory bowel disease. II. Immuoglobulin levels. Am J Dig Dis. 1978;23:123–8. doi: 10.1007/BF01073186. [DOI] [PubMed] [Google Scholar]
- 44.MacDermott RP, Nash GS, Auer IO, et al. Alterations in serum immunoglobulin G subclasses in patients with ulcerative colitis and Crohn’s disease. Gastroenterology. 1989;96:764–8. [PubMed] [Google Scholar]
- 45.Tepper MA, Nadler SG, Esselstyn JM, Sterbenz KG. Deoxyspergualin inhibits kappa light chain expression in 70Z/3 pre-B cells by blocking lipopolysaccharide-induced NF-kappa B activation. J Immunol. 1995;155:2427–36. [PubMed] [Google Scholar]
- 46.Delhase KM. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signaling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 47.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 48.Briere F, Servet-Delprat C, Bridon JM, Saint-Remy JM, Banchereau J. Human interleukin 10 induces naïve surface immunoglobulin D+ (sIgD+) B cells to secrete IgG1 and IgG3. J Exp Med. 1994;179:757–62. doi: 10.1084/jem.179.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clancy J, Jr, Goral J, Kovacs EJ. Expression of cytokine genes (TNFα, TGFβ and IFNγ) in acute lethal GVHD. In: Dinarello CA, editor. The physiological effects of cytokines. Wiley-Liss, Inc; 1990. pp. 165–170. [Google Scholar]