

Abstract
In the present study, we investigated the effect of RWJ-67657, a p38 MAP kinase inhibitor, upon in vivo LPS-induced monocyte cytokine production and upon monocyte LPS-hyporesponsiveness. Thirty minutes before a single injection of LPS (4 ng/kg BW), healthy male volunteers received a single oral dose of RWJ-67657 at increasing dosages (0–1400 mg). Blood samples (pre-medication, 3, 6 and 24 h after LPS) were immediately incubated with LPS (reflecting LPS-hyporesponsiveness) or without LPS (reflecting in vivo monocyte stimulation) for 4 h at 37°C. Following red blood cells lysis and white blood cell permeabilization, cells were labelled with α-CD14-FITC and α-IL-1β, α-IL-12 or α-TNFα (PE-labelled), fixed, and analysed using flow cytometry. In vivo LPS injection resulted in an increased percentage of circulating monocytes producing IL-1β, TNFα and IL-12 only at 3 h after the LPS injection. This was dose-dependently inhibited by RWJ-67657 treatment. LPS-hyporesponsiveness to in vitro LPS treatment was most prominent at 3 and 6 h after the in vivo LPS injection; compared with pre-medication monocytes, at these intervals a reduced percentage of monocytes produced IL-1β, TNFα or IL-12 after the in vitro LPS stimulus. At t = 6 h, this LPS-hyporesponsiveness could dose-dependently be inhibited by RWJ-67657 treatment of the volunteers. We therefore conclude that p38 MAP kinase inhibition with RWJ-67657 inhibited monocyte production of cytokines following in vivo LPS injection. Treatment with RWJ-67657 also reversed the LPS-hyporesponsiveness. Whether this result can be extended to the clinical situation remains to be elucidated. Patients with sepsis or an otherwise high risk for multi-organ failure are potential study groups.
Keywords: p38 MAP kinase, LPS-hyporesponsiveness, human endotoxaemia, monocytes, cytokines
INTRODUCTION
Septic shock is a leading cause of acute hospital admissions and, in addition, often complicates the clinical course of patients hospitalized for other reasons. It is an inflammatory response of the immune system to a bacterial infection [1]; the inflammatory response is initiated by bacterial toxins, such as LPS, which activates monocytes to release large amounts of pro-inflammatory cytokines [1]. Recently, however, it has become evident that the pro-inflammatory response is followed by a counter-regulatory anti-inflammatory response, which may be intended to prevent unnecessary tissue destruction from uncontrolled inflammation [1]. However, this anti-inflammatory response induces a state of immunological anergy, with an increased risk of secondary infections [1]. Intravenous injection of LPS into healthy human subjects not only induces the cascade of inflammatory pathways [1], but also initiates a temporary refractory state, referred to as LPS-hyporesponsiveness [2,3]. Intravenous LPS injection therefore can be used as a model for the study into the pro- and anti-inflammatory responses in sepsis.
The inflammatory cascade after intravenous LPS injection is initiated by LPS-induced production of pro-inflammatory cytokines by monocytes, such as the cytokine TNFα. TNFα is considered the most important mediator initiating the septic response. Next to TNFα, monocytes also produce other pro-inflammatory mediators, such as IL-1β, IL-12, IL-6 and reactive oxygen species, thereby amplifying the pro-inflammatory response [4]. This LPS-induced monocyte activation is mediated through the cell-surface receptors CD14 and Toll-like receptor 4 (TRL4) [5,6].The mechanism by which CD14 and TRL4 transmits a stimulatory response is still not exactly known. It has, however, been shown that the p38 MAP kinase is activated upon LPS binding to CD14 [7]. The MAP kinases are an important group of serine/threonine signalling kinases that, by modulating phosphorylation and hence, the activation status of transcription factors, link transmembrane signalling with gene induction events in the nucleus. P38 MAP kinase is involved in the LPS-induced production of TNFα, IL-1β and IL-12 [8–10].
LPS-hyporesponsiveness is a state of immunological hyporesponsiveness to LPS, a phenomenon in which monocytes/ macrophages play a central role [11]. This LPS-hyporesponsiveness is characterized by decreased monocyte production of cytokines, such as TNFα and IL-1β [2] upon a second LPS challenge. Although this phenomenon has been extensively studied both in vitro and in vivo in various animal species and cell types [12–14], the cellular and molecular changes that contribute to it are not fully understood. Since the p38 MAP kinase is involved in cytokine production following LPS activation, the present study was designed to investigate (i) whether a p38 MAP kinase inhibitor (RWJ-67657) suppressed monocyte cytokine production after an in vivo LPS injection, and (ii) whether this p38 MAP kinase inhibitor affected LPS-hyporesponsiveness.
MATERIALS AND METHODS
Subjects
This study was approved by the local Medical Ethics Committee. After signing informed consent and providing a medical history, a physical, haematological and biochemical examination was carried out on a random selection of 10 healthy male volunteers who were then admitted to the research unit of our Intensive Care Unit for pharmacokinetic and pharmacodynamic studies of RWJ-67657. Subjects were admitted the evening before medication and LPS infusion. A radial arterial catheter was placed for blood sampling and continuous monitoring of heart rate and blood pressure. Thirty minutes before the infusion of LPS, the volunteers received a single oral dose of RWJ-67657 (4-[4-(Fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyrindinyl)-1H-imidazol-2-yl]-3-butyn-1-ol), supplied in an oral pharmaceutical formulation by R.W. Johnson Pharmaceutical Research Institute, Bassersdorf, Switzerland). Three dose levels were tested, placebo-controlled: placebo (n = 2), 1400 mg (n = 2), 700 mg (n = 3) and 350 mg (n = 3). At time point t = 0, LPS (E-Coli, batch EC-6, US Pharmacopeia, Twinbrook Parkway, Rockville, MD, USA) was administered as a 1 min infusion at a dose of 4 ng/kg body weight (10 000 LPS units/mg). Four blood samples were obtained in vacutainers containing sodium heparin: pre-medication (t = 0), and 3, 6 and 24 h after administration of LPS. Blood samples were immediately processed as described before [15] and below.
Sample processing
Antibodies
The following monoclonal antibodies were used: fluorescein isothiocyanate (FITC)-labelled mouse anti-human CD14 (clone UCHM1; IQ Products, Groningen, The Netherlands); phycoerythrin (PE)-labelled mouse anti-human TNFα (clone Mab11; Pharmingen, San Diego, CA); PE-labelled mouse anti-human IL-1β (clone AS10; Becton Dickinson, San Jose, CA) and PE-labelled mouse anti-human IL-12 (clone C11·5; Pharmingen); PE-labelled mouse isotype control IgG1 (clone MCG1; IQ Products).
Reagents
Monensin (Sigma-Aldrich Chemie, Bv, Zwyndrecht, The Netherlands); FACS™ lysing solution (Becton Dickinson); FACS™ permeabilizing solution (Becton Dick-inson); complete RPMI 1640 (Gibco-Invitrage Co., Breda, The Netherlands) supplemented with 60 μg/ml gentamycin; washing buffer (phosphate-buffered saline with 0·5% bovine serum albumin and 0·1% NaN3; fixation buffer (0·5% paraformaldehyde in phosphate-buffered saline); freezing buffer.
Sample incubation
Immediately after sampling, 1 ml heparinized whole blood was mixed with 1 ml RPMI and stimulated with 2 μg/ml LPS (E-coli, 0·55:B5, Whittaker MA Bioproducts, Walkersville, MD, USA) (stimulated sample). Heparinized blood (1 ml) was used as unstimulated control and only mixed with 1 ml RPMI. In both the stimulated and unstimulated sample, monensin [16] was added to enable accumulation of the cytokines in the Golgic omplex by interrupting intracellular transport processes. Stimulated and unstimulated samples were incubated in sterile glass tubes for 4 h at 37°C and 5% CO2.
Sample labelling
After incubation, both stimulated and unstimulated samples were aliquoted (0·2 ml per tube) and 5 μl anti-CD14 were added to each tube. Tubes were incubated at RT in the dark for 30 min. Following incubation with 1 ml lysing buffer for 5 min in the dark, tubes were centrifuged and aspirated. Cells were then washed with 2 ml washing buffer, after which 0·2 ml freezing buffer was added to each tube. Tubes were frozen at –80°C.
All tubes (from t = 0, t = 3, t = 6 and t = 24 h) from one subject were thawed on the same day. After thawing, and two washes, the remaining pellets were resuspended in 0·5 ml permeabilizing buffer and incubated in the dark for 10 min. Then, cells were washed with ice-cold washing buffer. After aspiration, stimulated and unstimulated aliquots were incubated for 30 min in the dark at RT with either anti-TNFα, anti-IL-1β, anti-IL-12 or isotype control at saturating dilutions. After washing with washing buffer, cells were fixed with fixation buffer and kept at 4°C in the dark until measured (within 24 h).
Flow cytometry
Cells were analysed using the Coulter Epics flow cytometer (Argon-ion 488 nm laser, Beckmann Coulter Inc., Miami FL, USA). Two thousand monocytes were acquired while life-gating on monocytes using the CD14 positive cell signal and saved for later analysis. Data analysis was performed using Winlist32 (Verity softwarhouse Inc., Topsham, ME, USA).
Analysis
During analysis, a gate was set on CD14-positive monocytes. A single parameter fluorescence histogram for the monocytes was defined for evaluation of intracellular cytokine production.
Experiment 1: Intracellular cytokine production in human experimental endotoxaemia
Since our unstimulated samples were incubated without stimulation, these samples reflect monocyte cytokine production, which is due to in vivo stimulation. These samples can thus be used for evaluation of intracellular cytokine production following in vivo LPS injection. To evaluate intracellular cytokine production following in vivo LPS stimulation, and to evaluate the effect of the p38 MAP kinase inhibitor, for each individual the unstimulated sample at t = 0 was used as the unstimulated control. Thus, for each cytokine, the unstimulated sample at t = 0 was used to set a gate in the histogram, so that at least 99% of the monocytes in this sample were negative for cytokine production. This gate-setting was then used in the histograms of the samples taken at 3, 6 and 24 h after in vivo LPS injection, and resulted in the percentage of cytokine-producing monocytes at 3, 6 and 24 h after LPS injection.
Experiment 2: Intracellular cytokine production after in vitro LPS stimulation of monocytes during human endotoxaemia
For evaluation of cytokine production in volunteers injected in vivo with LPS followed by in vitro LPS stimulation, and for evaluation of the effect of the p38 MAP kinase inhibitor on this stimulation, samples that were not stimulated in vitro were compared with the samples taken at the same time but stimulated in vitro with LPS. Thus, using the unstimulated sample, a gate was set in the histogram so that at least 99% of the cells in this sample were negative for cytokine production. This gate-setting was then used in the corresponding stimulated sample, which resulted in a percentage of cytokine-producing monocytes in the stimulated samples.
Statistics
Results are expressed as mean ± s.e.m. In experiment 2, results are expressed relative to the pre-infusion value. The effect of RWJ-67657 in both experiment 1 and experiment 2 was evaluated using linear regression (least sum of squares method). R2 and the slope were calculated, and whether the slope was significantly different from 0 was tested. The slope was considered to be different from 0 if P < 0·05.
RESULTS
Experiment 1: The percentage of intracellular cytokine-producing monocytes in human experimental endotoxaemia; effect of p38 MAP kinase inhibitor
In this experiment, we first evaluated the intracellular cytokine production of monocytes from the two healthy human volunteers, who were not treated with the p38 MAP kinase inhibitor, after an in vivo LPS injection (n = 2). Thus, intracellular cytokine production of monocytes was measured at t = 0 (before LPS) and at 3, 6 and 24 h after LPS injection. An in vivo injection of LPS resulted in an increased percentage of circulating monocytes producing IL-1β, IL-12 and TNFα at 3 h after the LPS injection compared with numbers before the injection (Fig. 1). No effect of the LPS injection on percentage of monocytes producing cytokines was observed 6 or 24 h after the LPS injection.
Fig. 1.
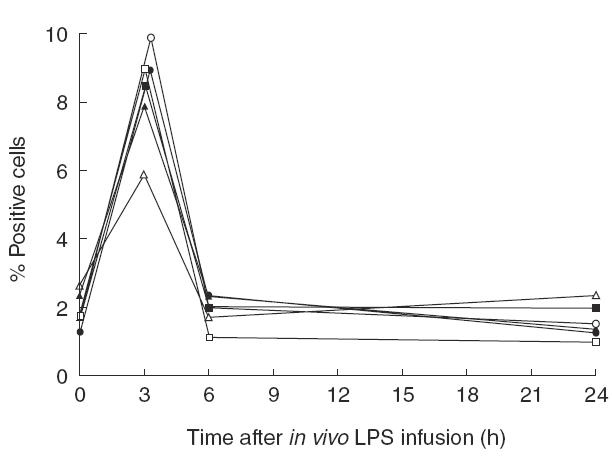
The percentage of circulating monocytes producing IL-1β, TNFα or IL-12 before, and at 3, 6 and 24 h after the injection of LPS (4 ng/kg body weight) of the two control individuals not treated with RWJ-67657. Each line represents an individual, open symbols for one individual, closed symbols for the other individual. (□,▪) IL-1β; (○,•) TNFα; (▵,▴) IL-12.
In the second part of this experiment, the effect of the p38 MAP kinase inhibitor on this intracellular cytokine production of monocytes was evaluated. Since an effect of LPS on intracellular cytokine production of monocytes was only observed at 3 h after in vivo LPS injection, an effect of the p38 MAP kinase inhibitor could also only be observed at this interval. Fig 2 shows the percentage of monocytes producing intracellular IL-1β, IL-12 and TNFα 3 h after LPS injection for untreated subjects or subjects treated with 350, 700 or 1400 mg p38 MAP kinase inhibitor. It can be seen from this figure that treatment with p38 MAP kinase inhibitor decreased the percentage of monocytes producing IL-1β, TNFα or IL-12 after an intravenous LPS injection in a dose-dependent way.
Fig. 2.
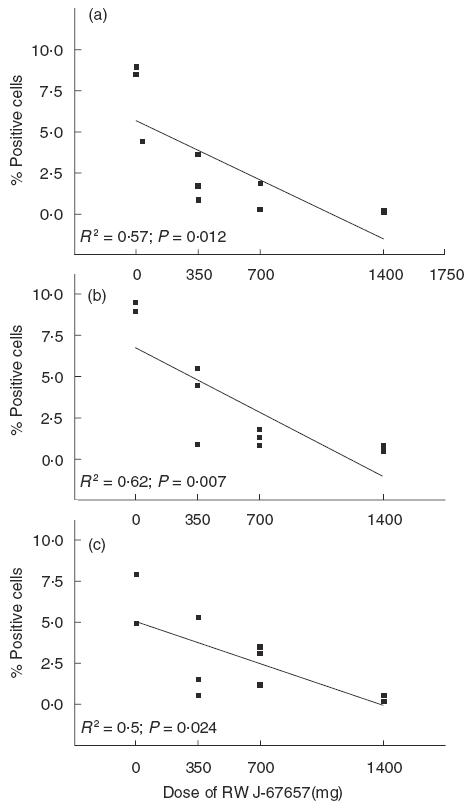
Percentage of IL-1β (a), TNFα (b) and IL-12 (c) producing monocytes of individuals with (350, 700 or 1400 mg) or without treatment with RWJ-67657. The line represents the regression line. Each square represents an individual. The P-value is given when the slope of the regression line significantly differs from 0.
Experiment 2: Intracellular cytokine production after in vitro LPS stimulation of monocytes during human endotoxaemia: effect of p38 MAP kinase inhibitor
This experiment was designed to evaluate whether monocytes from subjects injected in vivo with LPS showed LPS-hyporesponsiveness on an in vitro LPS challenge, as well as to evaluate the effect of the p38 MAP kinase inhibitor on LPS-hyporesponsiveness.
As can be seen from Fig 3, LPS-hyporesponsiveness was most prominent 3 and 6 h after in vivo LPS injection; at these intervals, a second in vitro LPS challenge resulted in a reduced percentage of monocytes producing intracellular IL-1β, IL-12 or TNFα compared with the percentage of monocytes producing these intracellular cytokines after in vitro LPS challenge at t = 0 (i.e. before the subjects received in vivo LPS injection). Twenty-four hours after in vivo LPS, the percentage of monocytes producing IL-12 after the in vitro LPS challenge appeared to have returned to normal. However, the percentage of monocytes producing IL-1β or TNFα is still decreased compared with the production before the in vivo LPS challenge.
Fig. 3.
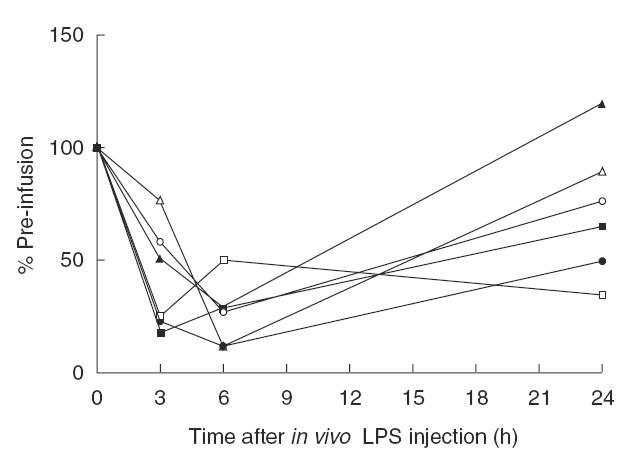
The effect of a second in vitro LPS challenge on percentage of monocytes producing IL-1β, TNFα or IL-12, before, and 3, 6 and 24 h following LPS injection in the two control individuals not treated with RWJ-67657. Results are expressed relative to the value before injection of LPS (t = 0). Each line represents an individual, open symbols for one individual, closed symbols for the other individual. (□,▪) IL-1β; (○,•) TNFα; (▵,▴) IL-12.
The effect of the p38 MAP kinase inhibitor on LPS-hyporesponsiveness was only apparent at 6 and 24 h after in vivo LPS injection; at t = 3 h, no significant effect of the p38 MAP kinase inhibitor was observed on the percentage of monocytes producing cytokines after in vitro LPS challenge (results not shown).
Fig 4 shows the effect of RWJ-67657 on cytokine production following a second in vitro LPS stimulus 6 and 24 h after in vivo LPS injection. For IL-1β (Fig 4a,d), at 6 and 24 h after in vivo LPS injection, percentages of monocytes producing IL-1β were significantly and dose-dependently increased after the in vitro LPS challenge in volunteers that were treated with the p38 MAP kinase inhibitor; the percentage of IL-1β-producing monocytes returned to normal values, i.e. pre-infusion values with increasing doses of p38 MAP kinase inhibitor.
Fig. 4.
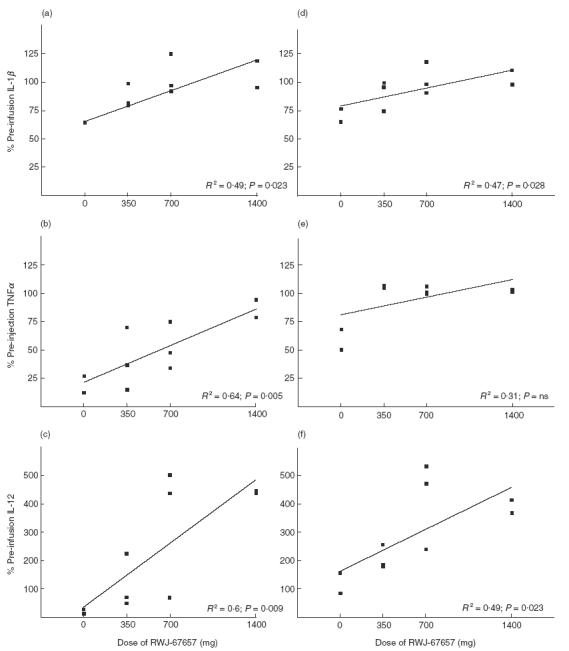
The effect of RWJ-67657 (0, 350, 700 or 1400 mg) on the percentage of monocytes producing IL-1β (a,d), TNFα (b,e) or IL-12 (c,f) after an in vitro LPS challenge of blood taken 6 (a-c) and 24 h (d-f) after an in vivo LPS injection. The line represents the regression line. Each square represents an individual. The P-value is given when the slope of the regression line significantly differs from 0.
The results for TNFα are shown in Fig. 4b,c. At 6 h after the in vivo LPS injection, the percentage of TNFα-producing monocytes after in vitro LPS challenge was also dose-dependently affected by treatment of the volunteers with the p38 MAP kinase inhibitor; the percentage of TNFα-producing monocytes returned to normal values (pre-infusion values) after treatment with 1400 mg of the p38 MAP kinase inhibitor. Also at 24 h after in vivo LPS injection, the percentage of TNFα-producing monocytes after the in vitro LPS challenge was affected by the p38 MAP kinase inhibitor. However, the effect was not dose dependent at the doses tested; even after treatment with 350 mg p38 MAP kinase inhibitor, the effect was maximal, i.e. the percentage of TNFα-producing monocytes after the in vitro LPS challenge had returned to pre-infusion values.
Interesting results were found for IL-12. Although the percentage of monocytes producing IL-12 after the in vitro LPS challenge 6 or 24 h after in vivo LPS injection was also dose-dependently affected by the p38 MAP kinase inhibitor (Fig 4c,f), after treatment with 700 mg, and especially after treatment with 1400 mg of the inhibitor, the percentage of IL-12-producing monocytes was increased to values exceeding the control value, i.e. the value before injection of LPS or p38 MAP kinase inhibitor, by about 300–400%.
DISCUSSION
In the present study, we investigated the effect of a p38 MAP kinase inhibitor on intracellular cytokine production of monocytes from healthy male volunteers injected with a low dose LPS, as well as the effect of this inhibitor on LPS-hyporesponsiveness. Whole blood rather than isolated cells of the healthy volunteers was stimulated, since the whole blood system is considered to mimic in vivo conditions best, with cytokines and other soluble factors being present that are able to influence cytokine production [17]. We measured intracellular cytokine production of TNFα, IL-1β and IL-12 using flow cytometry, which enabled us to detect cytokine production at the single cell level. TNFα and IL-1β were chosen for their central role in the induction of the LPS effect [4]; IL-12 was chosen for its role in providing a link between innate immunity and adaptive immunity: IL-12 produced by monocytes in response to LPS is a powerful factor for generation of a Th1-type immune response [18].
Using this method, 3 h after an in vivo injection of low dose LPS into healthy male volunteers, we were able to detect circulating monocytes that were producing IL-1β, TNFα and IL-12. As far as TNFα is concerned, this is in line with the previous observation from a similar study in our laboratory showing that plasma TNFα is increased after an in vivo LPS challenge [19]. Previous observations from our laboratory [20] and from others [21–23] had shown that plasma IL-1β after an in vivo LPS injection was undetectable and therefore, we did not measure plasma IL-1β in this study. Neither, unfortunately, did we measure plasma IL-12. However, it has been shown that the present dose of LPS does not increase plasma IL-12 levels [24].
Pre-infusion treatment with the p38 MAP kinase inhibitor significantly and dose-dependently decreased the percentage of monocytes that were positive for intracellular TNFα, IL-1β or IL-12, indicating that the p38 MAP kinase inhibitor does indeed inhibit monocyte cytokine production following LPS injection. This is consistent with the report from our laboratory, described above, in which we evaluated the effect of the p38 MAP kinase inhibitor on plasma cytokines and clinical responses to LPS [20]. It was shown that the plasma concentration of TNFα following in vivo LPS injection was significantly, dose-dependently decreased following treatment with the p38 MAP kinase inhibitor. The present results are also consistent with in vitro experiments showing inhibition of cytokine production by LPS-stimulated monocytes/macrophages following treatment with a p38 MAP kinase inhibitor [7,9].
The present study also shows an effect of RWJ-67657 on LPS-hyporesponsiveness. It was shown that 3, 6 and 24 h after in vivo LPS injection in healthy males, the percentages of cytokine-producing monocytes after a second, in vitro LPS challenge were decreased compared with the percentages of cytokine-producing monocytes upon an in vitro LPS in the blood sample taken before the in vivo LPS. Despite the number of studies evaluating LPS-hyporesponsiveness of monocytes [2,25,26], to our knowledge this is the first study to evaluate LPS-hyporesponsive monocytes using intracellular cytokine production measured by flow cytometry. The present data indicate that LPS-induced LPS-hyporesponsiveness is due to a decrease in percentage of monocytes capable of responding to the second LPS challenge. This decreased percentage of monocytes responding to LPS may then lead to decreased overall cytokine production following a second LPS challenge.
Although intensive research has been carried out into the mechanism of LPS-induced LPS-hyporesponsiveness [26–29], the mechanism still remains largely unknown. It has been suggested, however, that p38 MAPK is involved in the induction of an immunosuppressive macrophage phenotype in septic animals [30]. Kraatz et al. [31] suggested that p38 MAP kinase is unable to become activated in LPS-hyporesponsive monocytes [31], i.e. if p38 MAP kinase is activated after the first LPS stimulus, it cannot be activated following a second stimulus. This may explain why, after a second LPS stimulus, cytokine production is decreased.
This may be in line with the present results. We showed that inhibition of p38 MAP kinase 6 and 24 h after the in vivo LPS challenge dose-dependently reversed the LPS-induced LPS-hyporesponsiveness as far as production of IL-1β and TNFα was concerned. We suppressed p38 MAP kinase during the first LPS challenge. At the time of the second challenge, this p38 MAP kinase had not been activated before and could respond to LPS normally (depending on the dose of RWJ-67657 used), resulting in normal cytokine production. Our results may thus agree with the suggestion of Kraatz et al. [31] that, once p38 MAP kinase has been activated by LPS activation, it cannot be activated by a second LPS challenge. This suggestion implies that at 6 and 24 h after oral admission of the inhibitor, the inhibitor must have been released from the p38 MAP kinase; indeed, binding of pyrindinyl imidazoles to MAP kinases is reversible [32]. The fact that no effect of the p38 MAP kinase inhibitor was observed 3 h after the injection of LPS may indicate that at that time, the inhibitor given in vivo was still bound to p38 MAP kinase during the in vitro LPS challenge and thus, the p38 MAP kinase was still inhibited, leading to decreased cytokine production.
Interestingly, the p38 MAP kinase inhibitor not only reversed the effect of the second LPS stimulus on IL-12 production by monocytes, it even increased the percentage of IL-12-producing monocytes after the (second) in vitro LPS challenge compared with the effect of the in vitro LPS challenge on monocytes from volunteers before the in vivo LPS injection. This increase in percentage IL-12-producing monocytes would suggest that inhibition of p38 MAP kinase during human endotoxaemia would shift the specific immune response towards a Th1-type response and may thus restore the reduced Th1-type response seen in human endotoxaemia [33]. The mechanism of increased percentage of IL-12-producing monocytes following a second LPS challenge in volunteers treated with RWJ-67657 remains unclear from the present study.
The LPS-induced state of transient hyporesponsiveness to subsequent stimulation with LPS may, on the one hand, be beneficial by protecting the host from developing a shock syndrome caused by hyperactivation of monocytes and macrophages when bacteria and LPS persist. On the other hand, it has also been suggested that suppressed production of cytokines associated with this LPS-hyporesponsiveness may render a survivor of sepsis susceptible to secondary infections. The fact, therefore, that the p38 MAP kinase inhibitor, RWJ-67657, reversed the LPS-induced LPS-hyporesponsiveness may or may not make it a potential drug in the treatment of sepsis. There may be, however, other applications for this drug. Cardiac operations with cardiopulmonary bypass cause a systemic inflammatory response which can lead to organ failure and post-operative morbidity. To overcome this problem, patients are treated pre-operatively with corticosteroids. This, however, results in susceptibility to infections post-operatively [34]. RWJ-67657 may be useful in these situations; given pre-operatively, it inhibits the inflammatory reaction peri-operatively. Moreover, since it also inhibits LPS-hyporesponsiveness, it may render patients less sensitive to post-operative infections. This remains to be investigated.
REFERENCES
- 1.Adrie C, Pinsky MR. The inflammatory balance in human sepsis. Intensive Care Med. 2000;26:364–75. doi: 10.1007/s001340051169. [DOI] [PubMed] [Google Scholar]
- 2.Granowitz EV, Porat R, Mier JW, et al. Intravenous endotoxin suppresses the cytokine response of peripheral blood mononuclear cell in healthy humans. J Immunol. 1993;151:1637–45. [PubMed] [Google Scholar]
- 3.van der Poll T, Coyle SM, Moldawer LL, Lowry SF. Changes in endotoxin-induced cytokine production by whole blood after in vivo exposure of normal humans to endotoxin. J Infect Dis. 1996;174:1356–60. doi: 10.1093/infdis/174.6.1356. [DOI] [PubMed] [Google Scholar]
- 4.van der Poll T, van Deventer SJH, Hack CE, et al. Effects of leukocytes after injection of tumor necrosis factor into healthy humans. Blood. 1992;79:693–8. [PubMed] [Google Scholar]
- 5.Ulevitch RJ. Recognition of bacterial endotoxins by receptor-dependent mechanisms. Adv Immunol. 1993;53:267. doi: 10.1016/s0065-2776(08)60502-7. [DOI] [PubMed] [Google Scholar]
- 6.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chemistry. 1999;274:10689–92. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 7.Manthey CL, Wang SW, Kinney SD, Yao Z. SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS induced mRNAs in monocytes. J Leukoc Biol. 1998;64:409–17. doi: 10.1002/jlb.64.3.409. [DOI] [PubMed] [Google Scholar]
- 8.Baldassare JJ, Bi Y, Bellone CJ. The role of p38 Mitogen-Activated Protein Kinase in IL-1β transcription. J Immunol. 1999;162:5367–73. [PubMed] [Google Scholar]
- 9.Scherle PA, Jones EA, Favata MF, et al. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J Immunol. 1998;161:5681–6. [PubMed] [Google Scholar]
- 10.Feng GJ, Goodridge HS, Harnett MM, et al. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania Phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J Immunol. 1999;163:6403–12. [PubMed] [Google Scholar]
- 11.Freudenberg MA, Galanos C. Induction of tolerance to lipopolysaccharide (LPS)-d-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun. 1988;56:1352–7. doi: 10.1128/iai.56.5.1352-1357.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufmann A, Gemsa D, Sprenger H. Differential desensitization of lipopolysaccharide-inducible chemokine gene expression in human monocytes and macrophages. Eur J Immunol. 2000;30:1562–7. doi: 10.1002/1521-4141(200006)30:6<1562::AID-IMMU1562>3.0.CO;2-Q. 10.1002/1521-4141(200006)30:6<1562::aid-immu1562>62;3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 13.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and Toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–74. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 14.Lush CW, Cepinskas G, Kvietys PR. LPS tolerance in human endothelial cells: reduced PMN adhesion, E-selectin expression, and NF-kB mobilization. Am J Physiol. 2000;278:H853–H861. doi: 10.1152/ajpheart.2000.278.3.H853. [DOI] [PubMed] [Google Scholar]
- 15.Bouman A, Moes H, de Heineman MJ, Leij FMLH, Faas MM. The luteal phase of the ovarian cycle: increasing sensitivity of monocytes to endotoxin. Fertil Steril. 2001;76:555–9. doi: 10.1016/s0015-0282(01)01971-9. [DOI] [PubMed] [Google Scholar]
- 16.Ibbotson GC, Wallace JL. Beneficial effects of prostaglandin E2 in endotoxic shock are unrelated to effects on PAF-acether synthesis. Prostaglandins. 1989;37:237–50. doi: 10.1016/0090-6980(89)90060-9. [DOI] [PubMed] [Google Scholar]
- 17.Petrovsky N, Harrison LC. Diurnal rhythmicity of human cytokine production. J Immunol. 1997;158:5163–8. [PubMed] [Google Scholar]
- 18.Trinchieri G. Interleukin-12 and its role in the generation of Th1 cells. Immunol Today. 1993;14:335–8. doi: 10.1016/0167-5699(93)90230-I. [DOI] [PubMed] [Google Scholar]
- 19.Fijen JW, Zijlstra JG, de Boer P, et al. Suppression of the clinical and cytokine response to endotoxin by RWJ-67657, a p38 Mitogen-Activated Protein-kinase inhibitor, in healthy human volunteers. Clin Exp Immunol. 2001;124:16–20. doi: 10.1046/j.1365-2249.2001.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fijen JW, Muller Kobold AC, de Boer P, et al. Leukocyte activation and cytokine production during experimental human endotoxemia. European J Intern Med. 2000;11:89–95. doi: 10.1016/s0953-6205(00)00068-6. [DOI] [PubMed] [Google Scholar]
- 21.Deventer SJH, Buller HR, Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolitic, and complement pathways. Blood. 1990;76:2520–6. [PubMed] [Google Scholar]
- 22.Spinas GA, Bloesch D, Kaufmann MT, Keller U, Dayer JM. Induction of plasma inhibitors of interleukin 1 and TNF-α activity by endotoxin administration to normal humans. Am J Physiol. 1990;259:R993–R997. doi: 10.1152/ajpregu.1990.259.5.R993. [DOI] [PubMed] [Google Scholar]
- 23.Michie HR, Manoque KR, Spriggs DR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481–6. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- 24.Zimmer S, Pollard V, Marshall GD, et al. The Moyer Award. Effects of endotoxin on the Th1/Th1 response in humans. J Burn Care Rehab. 1996;17:491–6. [PubMed] [Google Scholar]
- 25.Rodrick ML, Moss NM, Grbic JT, et al. Effects of in vivo endotoxin infusions on in vitro cellular immune responses in humans. J Clin Immunology. 1992;12:440–50. doi: 10.1007/BF00918856. [DOI] [PubMed] [Google Scholar]
- 26.Wittmann M, Larsson VA, Schmidt P, Begemann G, Kapp A, Werfel T. Suppression of interleukin-12 production by human monocytes after preincubation with lipopolysaccharide. Blood. 1999;94:1717–26. [PubMed] [Google Scholar]
- 27.Randow F, Döcke WD, Bundschuh DS, Hartung T, Wendel A, Volk HD. In vitro prevention and reversal of lipopolysaccharide desensitization by IFN-γ, IL-12, and Granulocyte-Macrophage Colony-Stimulating Factor. J Immunol. 1997;158:2911–8. [PubMed] [Google Scholar]
- 28.Haas JG, Baeuerle PA, Rietmüller G, Ziegler-Heitbrock HWL. Molecular mechanisms in down-regulation of tumor necrosis factor expression. Proc Natl Acad Sci USA. 1990;87:9563–7. doi: 10.1073/pnas.87.24.9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziegler-Heitbrock HWL, Blumenstein M, Käfferlein E, et al. In vitro desensitization to lipopolysaccharide suppresses tumour necrosis factor, interleukin-1 and interleukin-6 gene expression in a similar fashion. Immunol. 1992;75:264–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Song GY, Chung CS, Jarrar D, Chaudry IH, Ayala A. Evolution of an immune suppressive macrophage phenotype as a product of p38 MAPK activation in polymicrobial sepsis. Shock. 2001;15:42–8. doi: 10.1097/00024382-200115010-00007. [DOI] [PubMed] [Google Scholar]
- 31.Kraatz J, Clair L, Rodriquez JL, West MA. Macrophage TNF secretion in endotoxin tolerance: Role of SARK, p38 and MAPK. J Surg Res. 1999;83:158–64. doi: 10.1006/jsre.1999.5587. 10.1006/jsre.1999.5587. [DOI] [PubMed] [Google Scholar]
- 32.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–46. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 33.Lauw FN, Ten hove T, Dekkers PEP, de Jonge E, van Deventer SJH, van der Poll T. Reduced Th1, but not Th2, cytokine production by lymphocytes after in vivo exposure of healthy subjects to endotoxin. Infect Immun. 2000;68:1014–8. doi: 10.1128/iai.68.3.1014-1018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler J, Rocker GM, Westaby S. Inflammatory response to cardiopulmonary bypass. Ann Thorac Surg. 1993;55:552–9. doi: 10.1016/0003-4975(93)91048-r. [DOI] [PubMed] [Google Scholar]