

Abstract
Although infection of single-stranded RNA viruses can enhance expression of major histocompatibility complex (MHC) class I genes, the mechanism underlying this process remains unclear. Recent studies have indicated that exposure of non-immune cells to double-stranded deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) of viral origin can directly increase the expression of MHC class I and related molecules without immune cell interaction. In this report, we show that transfection of single-stranded hepatitis A virus RNA into cultured hepatocytes results in the induction of genes for MHC class I, LMP2 and transporter for antigen processing (TAP1), in addition to the generation of viral proteins. We suggest that this stimulatory effect is due to the double-stranded RNA formed during replication of single-stranded viral RNA, and involves both double-stranded, RNA-dependent protein kinase PKR and the secretion of IFNβ.
Keywords: infectious immunity-virus, MHC, antigen presentation, protein kinases/phosphatases
INTRODUCTION
Viral infection can modulate the cellular expression of major histocompatibility complex (MHC) genes through the action of both viral proteins and nucleic acids. For example, the immediate early protein of HSV effectively blocks the MHC class I antigen presentation pathway by inhibiting the function of the transporter for antigen processing (TAP) genes [1–3]. In contrast, infection by other viruses, such as flavivirus [4], West Nile virus [5], parainfluenza virus [6] and hepatitis viruses [7,8], increases MHC expression. It is thought that interferons (IFNs) induced following viral infection are responsible for the abnormal or aberrant expression of MHC and related molecules, and contribute to the presentation of viral antigens to immune cells [9,10]. The source of the IFNs can be immune cells, in the case of IFNα and IFNγ, or virally-infected epithelial cells in the case of IFNβ.
Viral double-stranded RNA (dsRNA) acts as a ligand for PKR, a double-stranded, RNA-dependent protein kinase involved in the activation of a variety of cellular responses [11]. After binding to dsRNA, PKR dimerizes and autophosphorylates, then catalyses the phosphorylation of serine/threonine residues on target substrates, including the α subunit of the protein synthesis initiation factor eIF-2 and the NFκB inhibitor, IκB [11]. Because PKR induces type I IFN, and IFN can induce MHC expression [11], it is likely that secreted IFN is responsible for the MHC induction by double-stranded nucleic acids (dsNA) after infection by dsRNA viruses. As PKR does not bind to dsDNA or RNA/DNA duplexes [12], but requires A-form helical structures characteristic of stable dsRNA [13], the underlying mechanism by which non-dsRNA viruses induce MHC appears not to involve PKR activation. Recent studies have suggested that other pathways may be responsible for the induction of MHC expression and antigen presentation induced by dsDNA [14].
Thus, when introduced into the cytoplasm of normal cells, dsNAs induce abnormal or aberrant expression of MHC and related genes [14]. However, this is not a direct effect of IFN and is associated with the structure of double-stranded, but not single-stranded, DNA or RNA rather than particular sequence motifs [14]. dsNA fragments not only induce aberrant MHC class II and increased MHC class I expression, but also increase expression of TAP, LMP2, invariant chain (Ii) and co-stimulatory molecule (B7) genes in a variety of cells with normal phenotypes. This effect involves the activation of Stat 1, Stat 3, MAPK and NFκB [14]. These data are consistent with studies describing increased MHC class I expression following transfection of plasmid DNA into cultured cells [15,16].
Although known mechanisms can explain the mechanism of MHC induction following infection by dsDNA or dsRNA viruses, the molecular events related to single-stranded (ss) viral infection, such as with hepatitis A virus (HAV), remain unclear. Activation of the MHC class I pathway is essential for the clearance of viruses within infected hepatocytes. Thus, the hepatitis virus is not directly cytopathic on infected hepatocytes; rather, cytolysis is caused by antigen-specific CD8+ cytotoxic T lymphocytes (CTLs) [7,8]. Activation of specific CTLs requires the presentation of viral antigen epitopes by MHC class I molecules, such that viral proteins are processed in proteasomes and actively transported to the rough endoplasmic reticulum (rER) by TAP where it binds to MHC class I molecules [17,18].
MATERIALS AND METHODS
HAV ssRNA preparation
An infectious HAV cDNA clone, pHAV38-2, was generated as previously described [19,20], and provided by Dr Robert Purcell and co-workers (Hepatitis Virus Section, NIAID, NIH, Bethesda, MD). Single-stranded viral RNA was prepared in vitro by transcription from plasmids containing full-length cDNA. Plasmids were linearized with HaeII, XhoI or EcoRI to generate intact or truncated constructs, purified by phenol/chloroform extraction and ethanol precipitation, then used as template for in vitro transcription, as previously described [21] (Fig. 1). Briefly, 10 μg of template DNA were mixed with 1 mm each of ATP, CTP, GTP and UTP, 10 mm DTT, 4% RNasin, 2 μl SP6 RNA polymerase and 1 × transcription buffer (Promega) in a total volume of 100 μl, and incubated at 37°C for 2 h. After purification using TRIzol (Gibco-BRL, Gaithersburg, MD, USA) according to the manufacturer’s protocol. RNA quality and quantity were determined by agarose gel electrophoresis and absorbance measurement.
Fig. 1.
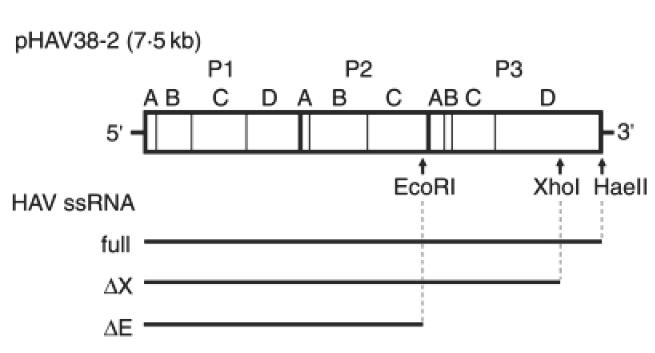
Schematic representation of the HAV genome and ssRNA constructs used for transfection. Restriction enzyme sites within the full-length cDNA construct pHAV38-2 [19,20] are shown. In vitro transcription following digestion with HaeII yields full-length RNA (full), whereas XhoI or EcoRI yields truncated RNAs (ΔX and ΔE, respectively) that do not contain the region essential for RNA-dependent RNA synthesis [19,20].
Transfection
Transfection with HAV RNA was performed using the DMRIE-C Reagent (Gibco-BRL). Briefly, the human hepatoblastoma cell line Hu-H7 was grown in DMEM supplemented with 10% FCS to 60% confluency and washed with 5 ml OPTI-MEM I (Gibco-BRL). RNA–lipid complexes consisting of 25 μg HAV RNA, 75 μl DMRIE-C and 3 ml OPTI-MEM I were added to the cells and incubated for 4 h at 37°C, after which the medium was replaced with normal culture medium. Transfection with other nucleic acids was performed using Lipofectamine Plus (Gibco-BRL) as previously described [14,22].
RNA isolation and Northern analysis
Total RNA isolation and Northern blot analysis were performed as previously described [14,22]. RNA was prepared using the RNeasy Mini Kit (Qiagen Inc., Chatsworth, CA, USA). Cells were washed with PBS, recovered with 600 μl lysis solution, passed through a QIAshredder (Qiagen), and 600 μl 70% ethanol added. Mixtures were passed through a spin column, then washed with 600 μl RW1 solution and twice with 500 μl RPE solution. RNA was eluted in 30 μl diethylpyrocarbonate (DEPC)-treated water, and RNA samples were electrophoresed on denaturing agarose gels and capillary blotted onto nylon membranes (Schleicher & Schuell, Keen, NH, USA).
After ultraviolet cross-linking, hybridization was performed. Probes were labelled with 32P-dCTP using a Ladderman Labelling Kit (PanVera, Madison, WI, USA) in the presence of a random primer, dNTPs and Bca DNA polymerase. After membranes were pre-hybridized with 10 ml QuickHyb Hybridization Solution (Stratagene, La Jolle, CA, USA) for 1 h at 68°C, 1 × 107 cpm radiolabelled probe pre-denatured with 100 μl sonicated salmon sperm DNA were added. After hybridization for 3 h, the membranes were washed, exposed to an Imaging Plate, and analysed using the BAS 1500 System (Fuji Medical Systems, Stanford, CT, USA).
Immunofluorescence
Cells grown in 2-well chamber slides (Nunc, Rochester, NY, USA) were fixed with absolute ethanol for 10 min at room temperature and washed with Dulbecco’s phosphate-buffered saline (DPBS). Cells were incubated with DPBS containing 1% BSA and 5% normal goat serum for 30 min at room temperature, then with anti-MHC class I antibody (Pharmingen) or anti-HAV envelope protein (provided by Dr Robert Purcell and co-workers, Hepatitis Virus Section, NIAID, NIH, Bethesda, MD) for 2 h at room temperature. Cells were subsequently washed three times with DPBS before the addition of fluorescein isothiocyanate (FITC)-labelled second antibody and further incubated for 30 min. Slides were washed, mounted with Antifade (Oncor, Gaithersburg, MD, USA) and examined by fluorescence microscopy.
FACS analysis
Cells were washed and pelleted in 96-well plates, suspended in DPBS and incubated with FITC-labelled monoclonal anti-human MHC class I antibody (Serotec, Raleigh, NC, USA) for 30 min. Cells were then washed three times, resuspended in DPBS and kept in the dark at 4°C until FACS analysis. Optimal antibody dilution was predetermined at a level where non-specific cell surface binding was not observed. Subclass-matched immunoglobulin was used as a negative control. Analysis was performed using FACSort (Becton Dickinson, Franklin Lakes, NV, USA).
SDS-polyacrylamide gel electrophoresis and Western blotting
The protocol was slightly modified as previously described [14,23]. Cells were placed on ice before harvesting, washed with ice-cold DPBS, released by gentle scraping, and collected by low-speed centrifugation at 833 g for 10 min in a table-top centrifuge. After a second wash in DPBS, cells were resuspended in cold lysis buffer (50 mm HEPES pH 7·0, 2 mm MgCl2; 250 mm NaCl; 0·1 mm EDTA; 0·1 mm EGTA; 1 mm DTT; 2 mm Na3VO4; 10 mm Na4P2O7; 10 mm NaF; 0·1% NP-40; 0·5 mm p-amidinophenyl methanesulphonyl fluoride hydrochloride (p-APMSF)) plus a protease inhibitor cocktail (2·5 mg/ml pepstatin A; 2·5 mg/ml chymostatin; 0·25 mg/ml leupeptin; 0·25 mg/ml antipain). Cells were lysed on ice for 60 min, vortexed vigorously and centrifuged in a microcentrifuge at 12 000 r.p.m. (10000 × g) for 10 min at 4°C; supernatant fluids were collected and frozen in aliquots at – 70°C. Before electrophoresis in sodium dodecyl sulphate (SDS)-containing gels, cell lysates (50 μg protein) were incubated with 62·5 mm Tris-HCl buffer pH 6·8 containing 2% SDS, 5% 2-mercaptoethanol, 7% glycerol and 0·01% bromophenol blue for 30 min at room temperature. SDS-gel electrophoresis was performed using 10–20% SDS Tris-glycine gels with molecular weight markers (NOVEX, Madison, WI, USA). After gel electrophoresis, samples were transferred to nitrocellulose membranes by electroblotting at 30 V for 2 h. After anti-body binding, proteins were identified using the ECL method (Amersham Life Science, Cleveland, OH, USA). Briefly, following blocking with a solution of 0·6% Tween 20, 10% skim milk and 1% crystalline bovine serum albumin (BSA) overnight at room temperature, the buffer was replaced with a 1:500 dilution of primary antibody (Santa Cruz, Santa Cruz, CA, USA) in blocking buffer diluted 1:10 in PBS-Tween and incubated for 1 h. Membranes were washed and peroxidase-conjugated second antibody (Santa Cruz) added for 1 h. Membranes were washed again and protein detected by incubation with ECL detection reagent (Amersham, Arlington Heights, IL, USA) for 1–10 min, followed by exposure to X-ray film.
Preparation of nuclear protein and electrophoretic gel mobility shift analysis (EMSA)
Preparation of nuclear protein and EMSA was performed as previously described [22,24,25]. Cells were washed, scraped into 1 ml DPBS, pelleted in a microfuge, and resuspended in five volumes of Buffer A (10 mm HEPES-KOH, pH 7·9, 10 mm KCl, 1·5 mm MgCl2, 0·1 mm EDTA) containing 0·3 m sucrose and 2% Tween 40. To release nuclei, cells were frozen and thawed once, then repetitively pipetted more than 100 times using a micropipette with a yellow tip (200 μl capacity). Samples were overlayed on 1 ml 1·5 m sucrose in Buffer A and microfuged for 10 min at 4°C. Pelleted nuclei were washed with 1 ml Buffer A, centrifuged for 30 s, then resuspended in 50 μl Buffer B (20 mm HEPES-KOH, pH 7·9, 420 mm NaCl, 1·5 mm MgCl2, 0·2 mm EDTA, 25% glycerol). Samples were incubated on ice for 20 min with occasional vortexing and centrifuged for 20 min at 4°C. Supernatant fractions containing nuclear protein were aliquoted and stored at – 70°C. Buffers A and B also contained 0·5 mm dithiothreitol (DTT), 0·5 mm phenylmethylsulphonylfluoride (PMSF), 2 ng/ml Pepstatin A and 2 ng/ml Leupeptin. All procedures were performed on ice or at 4°C. Oligonucleotides were labelled with 32P-γATP using T4 polynucleotide kinase and purified on 8% native polyacrylamide gels. Electrophoretic mobility shift analysis was performed using 3 μg nuclear extracts. Radiolabelled double-stranded oligonucleotide probes (50 000 cpm) were added and incubated for 20 min at 4°C. Mixtures were analysed on 5% native polyacrylamide gels and autoradiographed.
Enzyme-linked immunosorbent assay (ELISA)
The concentration of human IFNβ in culture supernatant fluids was measured using a Human Interferon-β ELISA Kit (FUJIREBIO, Wilmington, DE, USA) according to the manufacturer’s protocol. Briefly, 100 μl pre-spun culture supernatant fluid were mixed with 50 μl enzyme-labelled anti-IFNβ antibody in an antibody-coated 96-well microplate. Samples were incubated at room temperature for 2 h with shaking, washed three times with washing solution, then further incubated with 100 μl substrate for 30 min. Absorbance at 450 nm was measured after adding 100 μl reaction stopper solution and concentration determined using a human IFNβ standard.
Other reagents
Sheep anti-human IFNβ and rabbit anti-human TNFα were from Biosource International (Rockville, MD, USA). 2-Aminopurine and methimazole were from Sigma (St Louis, MO, USA).
RESULTS
Transfection of single-stranded hepatitis A virus RNA induces expression of MHC class I and related genes essential for antigen processing and presentation
To study the effect of HAV replication on MHC-related gene expression, we used in vitro transcription to create intact and truncated HAV ssRNA molecules from a full-length HAV cDNA clone (pHAV38-2). Single-stranded RNA produced from this clone has been shown to be virulent and induced hepatitis when directly injected into liver tissue of susceptible primates, such as tamarins and chimpanzees [19,20,26]. We created full-length ssRNA copies of the HAV genome (full), and two truncated HAV ssRNAs (ΔX and ΔE) that lacked the 3′-end of the HAV genome responsible for RNA-dependent RNA synthesis (Fig. 1). We transfected the Hu-H7 human hepatoblastoma cell line, grown in 10 cm cell culture dishes at 60% confluency, with 10 μg intact or truncated HAV ssRNA and examined the expression of viral and cell surface MHC class I proteins.
Using indirect immunofluorescence staining, HAV protein was detected in approximately 20% of Hu-H7 cells transfected with intact HAV ssRNA (Fig. 2a), while none was observed in cells transfected with truncated HAV ssRNA (Fig. 2b). Increased cell surface MHC class I expression was also detected only in cells transfected with intact HAV ssRNA (Fig. 2c) rather than in cells transfected with truncated HAV ssRNA (Fig. 2d). Interestingly, increased MHC class I expression was observed in more than 70% of cells transfected with intact HAV ssRNA (Fig. 2c), which was much higher than the proportion of cells that expressed HAV protein (Fig. 2a). This discrepancy between detectable HAV protein and MHC class I induction suggests that HAV protein was not responsible for the increased MHC class I expression.
Fig. 2.
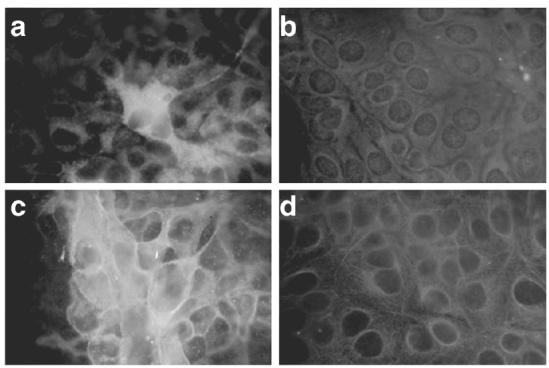
Immunofluorescence staining of HAV antigen (a and b) and MHC class I (c and d) expressed in Hu-H7 cells. Hu-H7 cells were transfected with full-length (a and c) or truncated, ΔX (b and d) HAV ssRNA created by in vitro transcription. Immunofluorescence staining was performed 48 h after transfection.
To study changes in mRNA levels of MHC class I genes and related genes necessary for antigen processing, intact or truncated HAV ssRNA was transfected into Hu-H7 cells and Northern blot analysis performed 12–48 h after transfection. Mock transfection, using distilled water instead of ssRNA, served as a control. Only constitutive MHC class I mRNA expression was observed in control Hu-H7 cells, with minimal expression of LMP2 and TAP1 mRNA (Fig. 3, lane 1). Although transfection of intact HAV ssRNA did not significantly enhance mRNA levels of constitutively expressed MHC class I, LMP2 and TAP1 mRNA were strongly induced (Fig. 3). Transfection of truncated HAV ssRNA failed to alter gene expression levels (Fig. 3a, lanes 8–13). These results suggest that HAV infection activates antigen processing and MHC class I presentation pathways in hepatocytes without help from other cell types, including IFNγ produced by T lymphocytes.
Fig. 3.
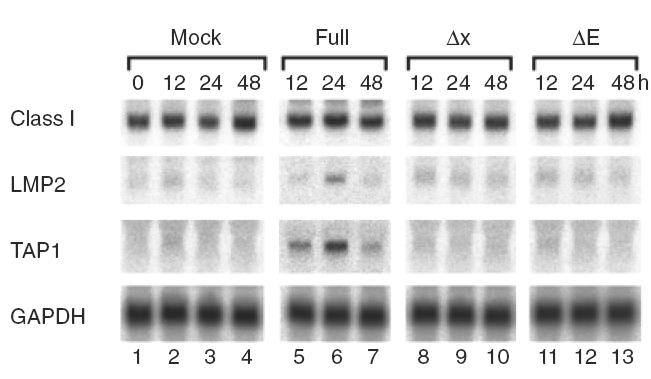
Northern blot analysis of MHC class I, LMP2 and TAP1 following transfection with HAV ssRNA constructs. Full-length or truncated HAV ssRNA was generated by in vitro transcription from template as shown in Fig. 1 and transfected into Hu-H7 cells. Total cellular RNA was prepared at the indicated time points, and Northern blotting performed as described in Materials and Methods. Mock transfection was performed using water instead of HAV ssRNA.
Double-stranded, but not single-stranded, RNA induces MHC class I expression in liver cells
To determine if dsRNA generated during replication of HAV ssRNA was responsible for the activation of MHC class I and related genes in hepatocytes, we transfected Hu-H7 cells with synthetic ssRNA or dsRNA polymers. Northern blot analysis indicated that transfection of dsRNA (poly IC-poly IC), but not ssRNA (poly A), significantly increased MHC class I mRNA levels and induced LMP2 and TAP1 mRNA levels 24 h after transfection (Fig. 4a). Transfection with 5 μg dsRNA was equivalent to treatment with 100 U/ml recombinant human IFNγ (Fig. 4a, lanes 4 and 5). Enhancement of cell surface MHC class I expression by dsRNA was confirmed by FACS analysis, so that, like mRNA levels, transfection with dsRNA, but not ssRNA, increased cell surface MHC class I expression to similar levels observed using IFNγ (Fig. 4b). These results indicate that the intact HAV ssRNA genome would not be able to induce MHC class I and related genes unless it formed double-stranded structures during replication. The results also suggest that constitutively expressed MHC class I mRNA is further enhanced by significant amounts of dsRNA formed intracellularly, and that the increased levels of cell surface class I protein (Fig. 2c) may involve post-transcriptional regulation.
Fig. 4.
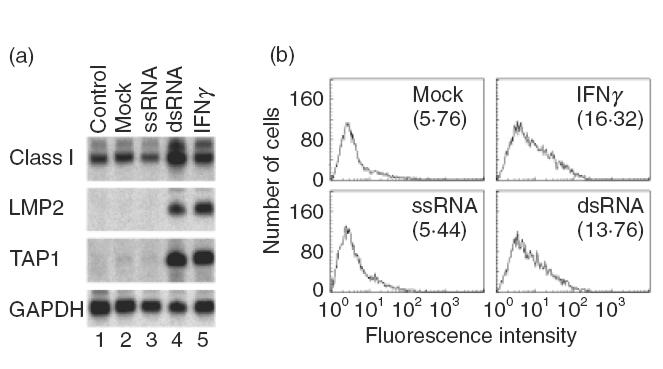
Induction of MHC class I mRNA (a) and cell surface protein (b) by dsRNA in Hu-H7 cells. Hu-H7 cells were transfected with ssRNA (poly A) or dsRNA (poly IC-poly IC). Northern blot analysis (a) and FACS analysis (b) were performed 24 and 48 h after transfection, respectively. Mock transfection with RNase-free water served as a negative control and human recombinant IFNγ (100 U/ml) as a positive control. Numbers in parentheses in (b) are mean fluorescence intensity.
PKR mediates MHC induction by dsRNAs
If the factor responsible for MHC class I pathway up-regulation were dsRNA, the effect would most likely be mediated by the double-stranded RNA-dependent protein kinase PKR [11]. To investigate this possibility, we treated cells with 2-aminopurine (2AP), a specific PKR inhibitor [27–30]. Based on preliminary studies to optimize inhibition, cells were treated 12 h before the transfection of dsRNA and continued until 48 h after transfection, whereupon cellular RNA was purified. No difference in the transfection efficiency using the pGL3-control vector, as measured by a luciferase activity, was observed between control and 2AP-treated cells (relative light units of 3·22 × 106 ± 0·96 × 106 and 3·52 × 106 ± 0·78 × 106, respectively). This suggests that any effect observed using 2AP would not be the result of altered dsRNA delivery into cells through altered transfection efficiency.
Our results indicated that 2AP inhibited the ability of dsRNA to induce MHC class I, LMP2 and TAP1 genes in Hu-H7 cells in a dose-dependent manner (Fig. 5, lanes 1–6), suggesting that PKR activation was essential for the induction of these genes. Interestingly, PKR mRNA strongly induced by dsRNA in Hu-H7 cells was also inhibited by 2AP (Fig. 5, lanes 1–6). These data additionally suggest that the induction of PKR is itself positively regulated by PKR activation.
Fig. 5.
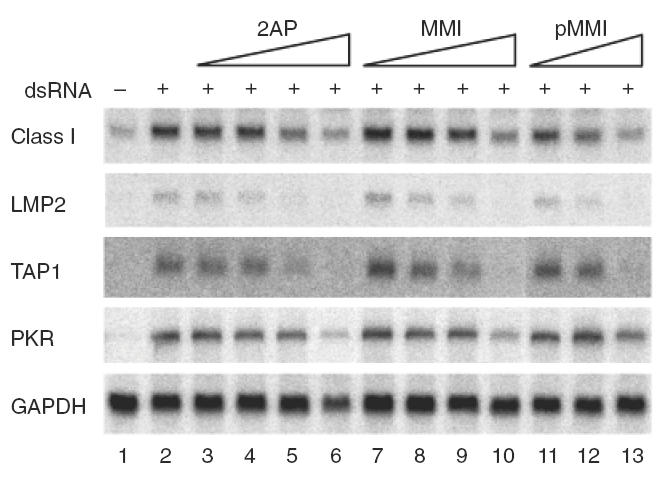
The PKR inhibitor 2AP, as well as MMI and pMMI, abolishes dsRNA-initiated MHC induction in Hu-H7 cells. After transfection of Hu-H7 cells with dsRNA (poly IC-poly IC), 2AP at final concentrations of 0·01, 0·1, 1·0 or 10 mm (lanes 3–6), 0·01, 0·1, 1·0 or 10 mm of MMI (lanes 7–10) or pMMI at concentrations of 0·01, 0·1 or 0·5 mm, were added to the culture medium. Total cellular RNA was prepared 48 h after transfection. The effect of 2AP is not due to altered transfection efficiency (see text).
Although the mechanism by which 2AP inhibits PKR activity is not known, tautomerization of 2AP may be involved [31]. Therefore, we tested the effect of methimazole (MMI) and one of its derivatives, 1-methyl-5-phenyl-imidazoline-2(3) thione (pMMI), on the ability of dsRNA to induce MHC class I pathway genes. MMI is known to suppress immune function in vivo [32,33], while pMMI undergoes tautomerization similar to that of 2AP; both compounds can suppress aberrant MHC class I expression in vivo and in vitro [34,35]. Our results showed that both MMI and pMMI inhibited the stimulatory effect of dsRNA on MHC class I pathway genes in a dose-dependent manner (Fig. 5). However, the inhibitory effect of 2AP and pMMI was stronger than that of MMI when compared at the same dose (Fig. 5). These results additionally suggest that immunosuppressive compounds with similar structures to 2AP can also inhibit the effects of dsRNA.
Activation of NFκB is involved in the action of dsRNA in Hu-H7 cells
It is known that PKR activation by dsRNA results in the activation of various signalling molecules, including IκB phosphorylation and NFκB activation [34,36]. NFκB is an important transcription factor for MHC class I and related genes [37,38], as well as for IFNβ induction [39]. Phosphorylation of IκB results in the release of NFκB dimers, allowing nuclear localization and unmasking of DNA binding domains, while phosphorylated IκB undergoes ubiquitination and proteosomal degradation [40].
Therefore, we studied IκB degradation and NFκB activation following dsRNA transfection of Hu-H7 cells. In addition, Stat 1 and Star 3 phosphorylation, important for MHC induction [14], was also studied. Cytoplasmic protein was purified and Western blot analysis performed using specific antibodies as described in the Materials and Methods section. Results showed that dsRNA transfection into Hu-H7 cells caused rapid degradation of IκBβ from 6 to 12 h (Fig. 6a). Although degradation of IκBα was not evident, phosphorylation of Stat 1 and Stat 3 was also noted at 12–24 h after transfection (Fig. 6a).
Fig. 6.
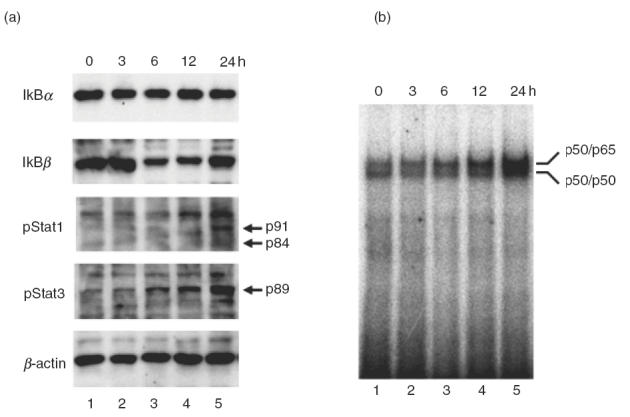
Activation of NFκB by dsRNA. Hu-H7 cells were transfected with dsRNA and cytoplasmic and nuclear proteins prepared independently and subjected to Western blot analysis (a) and EMSA (b), respectively. Detailed protocols are described in Materials and Methods section and elsewhere [14,22].
In order to test the nuclear localization and specific DNA binding of NFκB, an electrophoretic mobility shift assay (EMSA) was performed using nuclear extracts and oligonucleotide probes containing NFκB binding elements as previously described [14]. Increased specific binding of dimerized p50 and p65 NFκB subunits to the DNA probe was detected in nuclear extracts 12–24 h after dsRNA transfection (Fig. 6b). These results indicate that activation of downstream signalling cascades and transcription factors that positively regulate MHC class I expression was induced following PKR activation.
Effect of soluble factor(s) for activation of MHC expression
We then attempted to determine whether the mRNA-mediated induction of MHC class I, LMP2 and TAP1 genes was a direct effect of dsRNA through activation of intracellular signalling molecules, or an autocrine or paracrine effect due to cytokines induced by dsRNA. If the latter were the case, the most likely candidates would be IFNβ and TNFα, which are produced by liver cells and induce MHC expression [41,42]. To elucidate the roles of IFNβ and TNFα, we added specific neutralizing IFNβ and TNFα antibodies in the culture media of Hu-H7 cells after dsRNA transfection and studied MHC class I, LMP2 and TAP1 mRNA levels. Final concentrations of the anti-IFNβ and anti-TNFα antibodies were 1000 U/ml and 20 μg/ml, respectively, and were sufficient to neutralize cytokine activity [43,44]. Northern blot analysis indicated that anti-IFNβ antibody almost completely inhibited the ability of dsRNA to induce MHC class I, LMP2, TAP1 and PKR mRNA levels, whereas anti-TNFα had no effect (Fig. 7a). Flow cytometry analysis of cell surface MHC class I expression confirmed the effect of the anti-IFNβ antibody (data not shown).
Fig. 7.
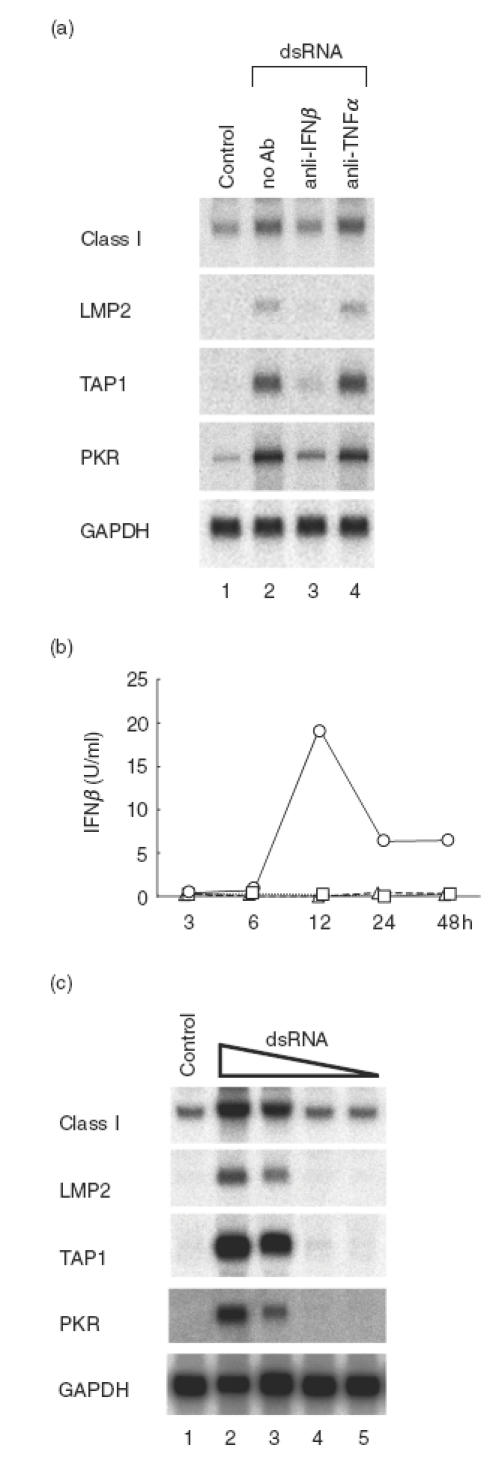
Secreted IFNβ is responsible for the action of dsRNA in the induction of MHC class I and related genes. Hu-H7 cells were transfected with dsRNA and treated with neutralizing concentrations of anti-hIFNβ and anti-hTNFα antibodies (1000 U/ml and 20 μg/ml, respectively) [43,44], and Northern blot analysis performed (a). Concentrations of secreted hIFNβ in culture supernatant fluids were measured by ELISA at the indicated time points following transfection with RNase-free dsRNA (○), ssRNA (□) or mock transfection with RNase-free water (▵). Hu-H7 cells were transfected with various concentrations of dsRNA. Northern blot analysis performed and showed similar results. Lanes 2–5: 5 μg, 1·0 μg, 0·1 μg and 0·01 μg of poly IC-poly IC was transfected, respectively, into Hu-H7 cells grown in a 10 cm dish to 60% confluency (c).
To examine whether IFNβ was actually secreted by Hu-H7 cells, we performed an ELISA to measure IFNβ in culture media 3–48 h after dsRNA transfection. IFNβ secretion was detected after 12 h only in media from dsRNA-transfected cells (Fig. 7b). Although IFNβ levels decreased at 24–48 h, this may have been due to the degradation of the IFNβ protein in the culture medium. These results indicate that secreted IFNβ induced by dsRNA appears to be the major factor responsible for the induction of MHC class I pathway genes. Since the concentration of IFNβ detected in dsRNA-transfected cells was very low (<20 U/ml), it likely that high local concentrations of IFNβ around secreting cells are responsible for the autocrine/paracrine effect. Consistent with these results, IFNβ mRNA was induced by dsRNA in Hu-H7 cells that do not constitutively express IFNβ (Fig. 7c). This IFNβ induction was dsRNA dose-dependent, and correlated with increased MHC class I mRNA and induction of LMP2 and TAP1 mRNA (Fig. 7c). These results further support the hypothesis that secreted IFNβ is the major factor responsible for the induction of MHC and related genes.
DISCUSSION
Using full-length and truncated HAV ssRNA constructs, we have provided evidence that replication of single-stranded genomic RNA results in the induction of MHC class I genes within transfected cells. Because this effect involved PKR, it strongly suggests that dsRNA formed during replication of the ssRNA genome was the responsible factor. We additionally showed that the autocrine/paracrine action of secreted IFNβ appeared to mediate the effect of dsRNA in the induction of MHC class I, LMP2 and TAP1 genes.
Although aberrant MHC expression after infection by ssRNA viruses such as parainfluenza virus has been described [6], the underlying molecular mechanisms remain unclear. Similarly, although the relationship between viral infection and activation of PKR or IFN pathways has been discussed, there has been no direct evidence showing that replication of viral ssRNA creates stable double-stranded forms in sufficient amounts to bind and activate PKR. Our study suggests that the dsRNA formed during ssRNA replication are stable and last long enough, even if transient, to activate PKR. PKR activation results in phosphorylation of secondary signalling molecules such as eIF-2 and the NFκB inhibitor, IκB [36,45]. We showed specific degradation of IκBβ and activation of NFκB in Hu-H7 cells following dsRNA transfection, as well as phosphorylation of Stat 1 and Stat 3. In most cases, IκBα degradation is the major pathway in NFκB induction, with only a few stimuli leading to IκBβ degradation [46,47]. Although the effect of different IκB kinases and the differential phosphorylation of serine residues on IκB molecules may be important for differential degradation, the precise mechanism responsible for such effects remain unclear [46].
The results from these in vitro studies strongly suggest that upon HAV infection of hepatocytes, the dsRNA formed during viral replication activated PKR and induced IFNβ. The secreted IFNβ in turn acted on hepatocytes in an autocrine/paracrine manner to induce expression of MHC class I, LMP2 and TAP1 genes. Expression of these genes would then contribute to the processing and presentation of viral antigens to the immune system to initiate the clearance of viruses within infected cells. Even though IFNβ is synthesized by Hu-H7 cells, the cytokine cannot act directly within the cell, but must be secreted outside the cell and bind its specific cell surface receptor [48]. We have previously shown, by creating intercrossed mice carrying a thyroid IFNγ transgene and IFNγ receptor knock-out, that overexpressed IFNγ in the thyroid cannot act on thyroid cells without a cell surface receptor [49].
Our data also suggested that high local concentrations of secreted IFNβ may be required for the stimulatory effect of dsRNA. Thus, the paracrine action of IFNβ may not only be important for the presentation of viral antigens by infected cells, but also allow surrounding uninfected cells to acquire a resistance against viral spreading by activating 2′-5′ oligoadenylate synthetases (2–5 AS) [50]. Spotty necrosis seen in liver tissues with viral hepatitis may be the result of focal viral replication, which directly induces MHC expression and the presentation of viral antigens. Interestingly, increased PKR expression has been reported in liver tissues from patients with chronic hepatitis [51]. In order to test this hypothesis, a direct cytotoxicity assay will be required, as we have recently shown using mouse dendritic cells, MHC-matched effector cells and ovalbumin peptide [52]. However, at present there is no model system available to study CTL activity against HAV-infected hepatocytes using MHC-matched effector cells. In addition, we found that the effects of soluble factors secreted into culture supernatant fluid were variable in other cells tested. Therefore, despite the fact that secreted IFNβ seems to be the major factor responsible for the MHC induction following dsRNA transfection in Hu-H7 cells, direct activation by other intracellular signalling molecules and/or transcription factors may also be important in other situations.
The present data are very similar to results obtained using dsDNA with regard to induction of MHC and related genes [14]. However, unlike dsRNA, dsDNA does not induce IFNβ [14] and therefore, the MHC induction pathways appear to be different. The initial cytoplasmic factor that responds to dsDNA, equivalent to PKR for dsRNA, has not yet been identified. Nevertheless, it has been shown that NFκB, Stat 1, Stat 3 and MAPK pathways are involved when thyroid cells respond to dsDNA [14]. One of the factors that might mediate the effect of dsDNA is the DNA-dependent protein kinase DNA-PK [53,54], a nuclear serine/ threonine kinase that recognizes dsDNA ends and plays a key role in repairing DNA breakage, such as during V(D)J recombination, with the help of Ku [55–57]. Although recent studies have indicated that DNA-PK phosphorylates IκB to activate NFκB [58], the relationship between DNA-PK and MHC expression is not yet known. Because PKR is activated in the cytoplasm by dsRNA, but not by dsDNA or DNA/RNA hybrids [12], and because DNA-PK is activated in the nucleus by single-strand breaks within dsDNA ends, but not by dsRNA [53], it would be expected that the underlying mechanism between the two pathways is quite different. Nevertheless, it is interesting to note that the activation of MHC and related genes seems to be a common outcome for cells exposed to dsDNA or dsRNA, but not to ssDNA or ssRNA.
The adenine analogue 2AP undergoes tautomerization [31] and is known to inhibit the action of PKR [27–30]. Cyclic tautomeric thiones, as well as MMI and its derivatives, demonstrate immunoregulatory functions, especially the ability to suppress MHC class I expression induced by IFNγ and dsNAs [34,35]. We showed that low concentrations of such compounds inhibited the effect of dsRNA in the activation of MHC class I and related genes. Thus, it may be possible to control immune responses against infected cells by modulating intracellular MHC expression. Reducing aberrantly-induced MHC expression on target cells by suppressing PKR action would result in the suppression of the specific immune response without affecting generalized immune cell function. Although 2AP is a possible mutagen [31], MMI is a widely used drug with a long history of treatment in patients with thyroid disorders, and its derivatives have been tested in experimental animals [34]. Therefore, selective suppression of dsRNA-dependent MHC gene activation by such compounds might be applicable as a therapeutic approach to severe inflammation due to viral infection, such as fluminant hepatitis. Conventional immunosuppressive drugs, such as corticosteroids and cyclosporin, cause immune deficiency and can result in opportunistic infection. Specific suppression of aberrantly-induced MHC expression might be a critical means of targeting treatment to abnormal antigen presentation, not only in cases of viral infection but also in autoimmune diseases, transplantation and in some cases of miscarriage.
Our results strongly suggest that other ssRNA viruses may also activate PKR and induce MHC expression as long as dsRNA structures are formed during the viral replication cycle. The study also suggests that it may be possible to measure the expression levels of MHC or related genes following viral infection or transfection of viral genomes as a universal tool to detect viral replication. For example, an assay could be developed to study the ability of viruses to replicate in different cell types under a variety of conditions, even in cases of unknown ssRNA viruses where specific antiserum or nucleic acid probes are not available. This strategy may also be useful in the analysis of viral genomes, as it would allow the identification of regions essential for replication.
Acknowledgments
The authors thank Drs Suzanne U. Emerson, Jens Bukh and Robert H. Purcell, Hepatitis Virus Section, NIAID, NIH for providing cDNA clones.
References
- 1.York IA, Roop C, Andrews DW, et al. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525–35. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 2.Fruh K, Ahn K, Djaballah H, et al. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–8. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 3.Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–5. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, King NJ, Dos Remedios CG. Flavivirus induces MHC antigen on human myoblasts: a model of autoimmune myositis? Muscle Nerve. 1992;15:1271–7. doi: 10.1002/mus.880151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argall KG, Armati PJ, King NJ, et al. The effects of West Nile virus on major histocompatibility complex class I and II molecule expression by Lewis rat Schwann cells in vitro. J Neuroimmunol. 1991;35:273–84. doi: 10.1016/0165-5728(91)90181-6. [DOI] [PubMed] [Google Scholar]
- 6.Gao JBP, Banerjee AK. Human parainfluenza virus type 3 up-regulates major histocompatibility complex class I and II expression on respiratory epithelial cells: involvement of a STAT1- and CIITA-independent pathway. J Virol. 1999;73:1411–8. doi: 10.1128/jvi.73.2.1411-1418.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehermann B, Chisari FV. Cell mediated immune response to the hepatitis C virus. Curr Top Microbiol Immunol. 2000;242:299–325. doi: 10.1007/978-3-642-59605-6_14. [DOI] [PubMed] [Google Scholar]
- 8.Cerny A, Chisari FV. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology. 1999;30:595–601. doi: 10.1002/hep.510300312. [DOI] [PubMed] [Google Scholar]
- 9.Biron CA. Cytokines in the generation of immune responses to, and resolution of, virus infection. Curr Opin Immunol. 1994;6:530–8. doi: 10.1016/0952-7915(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 10.Hisamatsu H, Shimbara N, Saito Y, et al. Newly identified pair of proteasomal subunits regulated reciprocally by interferon gamma. J Exp Med. 1996;183:1807–16. doi: 10.1084/jem.183.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–24. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- 12.Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–94. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- 13.Bevilacqua PC, George CX, Samuel CE, et al. Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry. 1998;37:6303–16. doi: 10.1021/bi980113j. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Mori A, Ishii KJ, et al. Activation of target tissue immune recognition molecules by double strand nucleic acids. Proc Natl Acad Sci USA. 1999;96:2285–90. doi: 10.1073/pnas.96.5.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox BA, Drury M, Hu HM, et al. Lipofection indirectly increases expression of endogenous major histocompatibility complex class I molecules on tumor cells. Cancer Gene Ther. 1998;5:307–12. [PubMed] [Google Scholar]
- 16.Park JH, Chang SH, Kim MC, et al. Up-regulation of the expression of major histocompatibility complex class I antigens by plasmid DNA transfection in non-hematopoietic cells. FEBS Lett. 1998;436:55–60. doi: 10.1016/s0014-5793(98)01097-7. [DOI] [PubMed] [Google Scholar]
- 17.York IA, Rock KL. Antigen processing and presentation by the class I major histocompatibility complex. Annu Rev Immunol. 1996;14:369–96. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]
- 18.Shepherd JC, Schumacher TN, Ashton-Rickardt PG, et al. TAP1-dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell. 1993;74:577–84. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 19.Funkhouser AW, Raychaudhuri G, Purcell RH, et al. Progress toward the development of a genetically engineered attenuated hepatitis A virus vaccine. J Virol. 1996;70:7948–57. doi: 10.1128/jvi.70.11.7948-7957.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emerson SU, Lewis M, Govindarajan S, et al. cDNA clone of hepatitis A virus encoding a virulent virus: induction of viral hepatitis by direct nucleic acid transfection of marmosets. J Virol. 1992;66:6649–54. doi: 10.1128/jvi.66.11.6649-6654.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagi M, Purcell RH, Emerson SU, et al. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–43. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki K, Lavaroni S, Mori A, et al. Thyroid transcription factor 1 is calcium modulated and coordinately regulates genes involved in calcium homeostasis in C cells. Mol Cell Biol. 1998;18:7410–22. doi: 10.1128/mcb.18.12.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki K, Mori A, Saito J, et al. Follicular thyroglobulin suppresses iodide uptake by suppressing expression of the sodium/iodide symporter gene. Endocrinology. 1999;140:5422–30. doi: 10.1210/endo.140.11.7124. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki K, Kobayashi Y, Katoh R, et al. Identification of thyroid transcription factor-1 in C cells and parathyroid cells. Endocrinology. 1998;139:3014–7. doi: 10.1210/endo.139.6.6126. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki K, Lavaroni S, Mori A, et al. Autoregulation of thyroid-specific gene transcription by thyroglobulin. Proc Natl Acad Sci USA. 1998;95:8251–6. doi: 10.1073/pnas.95.14.8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raychaudhuri G, Govindarajan S, Shapiro M, et al. Utilization of chimeras between human (HM-175) and simian (AGM-27) strains of hepatitis A virus to study the molecular basis of virulence. J Virol. 1998;72:7467–75. doi: 10.1128/jvi.72.9.7467-7475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zinn K, Keller A, Whittemore LA, et al. 2-Aminopurine selectively inhibits the induction of beta-interferon, c-fos, and c-myc gene expression. Science. 1988;240:210–3. doi: 10.1126/science.3281258. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y, Conway TW. 2-Aminopurine inhibits the double-stranded RNA-dependent protein kinase both in vitro and in vivo. J Interferon Res. 1993;13:323–8. doi: 10.1089/jir.1993.13.323. [DOI] [PubMed] [Google Scholar]
- 29.Cheshire JL, Williams BR, Baldwin AS., Jr Involvement of double-stranded RNA-activated protein kinase in the synergistic activation of nuclear factor-kappaB by tumor necrosis factor-alpha and gamma-interferon in preneuronal cells. J Biol Chem. 1999;274:4801–6. doi: 10.1074/jbc.274.8.4801. [DOI] [PubMed] [Google Scholar]
- 30.Tiwari RK, Kusari J, Kumar R, et al. Gene induction by interferons and double-stranded RNA: selective inhibition by 2-aminopurine. Mol Cell Biol. 1988;8:4289–94. doi: 10.1128/mcb.8.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siden RR. Academic Press Inc. California: San Diego; 1994. DNA structure and function. [Google Scholar]
- 32.Singer DS, Kohn LD, Zinger H, et al. Methimazole prevents induction of experimental systemic lupus erythematosus in mice. J Immunol. 1994;153:873–80. [PubMed] [Google Scholar]
- 33.Mozes E, Zinger H, Kohn LD, et al. Spontaneous autoimmune disease in (NZB × NZW) F1 mice is ameliorated by treatment with methimazole. J Clin Immunol. 1998;18:106–13. doi: 10.1023/a:1023242732212. [DOI] [PubMed] [Google Scholar]
- 34.Kohn LD, Napolitano G, Singer DS, et al. Graves’ disease: a host defense mechanism gone awry. Int Rev Immunol. 2000;19:633–64. doi: 10.3109/08830180009088516. [DOI] [PubMed] [Google Scholar]
- 35.Kohn LD, Shimojo N, Kohno Y, et al. Kluwer Academic Publishers. Boston: Reviews in endocrine and metabolic disorders; 2000. An animal model of Graves’ disease: understanding the cause of autoimmune hyperthyroidism; pp. 59–67. [DOI] [PubMed] [Google Scholar]
- 36.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, et al. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–90. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giuliani C, Saji M, Napolitano G, et al. Hormonal modulation of major histocompatibility complex class I gene expression involves an enhancer A-binding complex consisting of Fra-2 and the p50 subunit of NF-kappa B. J Biol Chem. 1995;270:11453–62. doi: 10.1074/jbc.270.19.11453. [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi SI, Shong M, Giuliani C, et al. Iodide suppression of major histocompatibility class I gene expression in thyroid cells involves enhancer A and the transcription factor NF-kappa B. Mol Endocrinol. 1998;12:19–33. doi: 10.1210/mend.12.1.0052. [DOI] [PubMed] [Google Scholar]
- 39.Kirchhoff S, Wilhelm D, Angel P, et al. NFkappaB activation is required for interferon regulatory factor-1-mediated interferon beta induction. Eur J Biochem. 1999;261:546–54. doi: 10.1046/j.1432-1327.1999.00308.x. 10.1046/j.1432-1327.1999.00308.x. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 41.Lampson GP, Tytell AA, Field AK, et al. Inducers of interferon and host resistance. I. Double-stranded RNA from extracts of Penicillium funiculosum. Proc Natl Acad Sci USA. 1967;58:782–9. doi: 10.1073/pnas.58.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wordemann M, Fandrey J, Jelkmann W. Tumor necrosis factor-alpha production by human hepatoma cell lines is resistant to drugs that are inhibitory to macrophages. J Interferon Cytokine Res. 1998;18:1069–75. doi: 10.1089/jir.1998.18.1069. [DOI] [PubMed] [Google Scholar]
- 43.Dansky-Ullmann C, Salgaller M, Adams S, et al. Synergistic effects of IL-6 and IFN-gamma on carcinoembryonic antigen (CEA) and HLA expression by human colorectal carcinoma cells: role for endogenous IFN-beta. Cytokine. 1995;7:118–29. doi: 10.1006/cyto.1995.1016. 10.1006/cyto.1995.1016. [DOI] [PubMed] [Google Scholar]
- 44.Kinter AL, Poli G, Fox L, et al. HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J Immunol. 1995;154:2448–59. [PubMed] [Google Scholar]
- 45.Kohn LD, Suzuki K, Mori A, et al. Immune activation of double-stranded polynucleotides. US Patent Application Pending.
- 46.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 47.Takeshita F, Ishii KJ, Ueda A, et al. Positive and negative regulatory elements contribute to CpG oligonucleotide-mediated regulation of human IL-6 gene expression. Eur J Immunol. 2000;30:108–16. doi: 10.1002/1521-4141(200001)30:1<108::AID-IMMU108>3.0.CO;2-4. 10.1002/(sici)1521-4141(200001)30:01<108::aid-immu108>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 48.Sen GC, Ransohoff RM. Interferon-induced antiviral actions and their regulation. Adv Virus Res. 1993;42:57–102. doi: 10.1016/s0065-3527(08)60083-4. [DOI] [PubMed] [Google Scholar]
- 49.Caturegli P, Hejazi M, Suzuki K, et al. Hypothyroidism in transgenic mice expressing IFN-gamma in the thyroid. Proc Natl Acad Sci USA. 2000;97:1719–24. doi: 10.1073/pnas.020522597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharp TV, Raine DA, Gewert DR, et al. Activation of the interferon-inducible (2′-5′) oligoadenylate synthetase by the Epstein-Barr virus RNA, EBER-1. Virology. 1999;257:303–13. doi: 10.1006/viro.1999.9689. [DOI] [PubMed] [Google Scholar]
- 51.Shimada A, Shiota G, Miyata H, et al. Aberrant expression of double-stranded RNA-dependent protein kinase in hepatocytes of chronic hepatitis and differentiated hepatocellular carcinoma. Cancer Res. 1998;58:4434–8. [PubMed] [Google Scholar]
- 52.Ishii KJ, Suzuki K, Coban C, et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602–7. doi: 10.4049/jimmunol.167.5.2602. [DOI] [PubMed] [Google Scholar]
- 53.Ohtsuki K, Yamada E, Nakamura M, et al. Mouse spleen cell nuclear protein kinases and the stimulating effect of dsDNA on NHP phosphorylation by cyclic AMP-independent protein kinase in vitro. J Biochem (Tokyo) 1980;87:35–45. doi: 10.1093/oxfordjournals.jbchem.a132745. [DOI] [PubMed] [Google Scholar]
- 54.Walker AI, Hunt T, Jackson RJ, et al. Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 1985;4:139–45. doi: 10.1002/j.1460-2075.1985.tb02328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smider V, Rathmell WK, Lieber MR, et al. Restoration of X-ray resistance and V (D) J recombination in mutant cells by Ku cDNA. Science. 1994;266:288–91. doi: 10.1126/science.7939667. [DOI] [PubMed] [Google Scholar]
- 56.Taccioli GE, Gottlieb TM, Blunt T, et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V (D) J recombination. Science. 1994;265:1442–5. doi: 10.1126/science.8073286. [DOI] [PubMed] [Google Scholar]
- 57.Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J. 1997;16:5098–112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu L, Kwak YT, Bex F, et al. DNA-dependent protein kinase phosphorylation of IkappaB alpha and IkappaB beta regulates NF-kappaB DNA binding properties. Mol Cell Biol. 1998;18:4221–34. doi: 10.1128/mcb.18.7.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]