

Abstract
Common variable immunodeficiency (CVID) is a very frequent but heterogeneous syndrome of antibody formation. The primary defect remains unknown, but many reports describe peripheral blood T lymphocyte dysfunctions in a substantial proportion of CVID patients, which may impair T–B cell collaboration. In order to investigate whether such putative defects were intrinsic to T cells or, rather, secondary to quantitative differences in T cell subset distribution, or to other described disorders, we have used Herpesvirus saimiri (HVS) for the targeted transformation of CVID CD4+ and CD8+ T cells and subsequent functional evaluation by flow cytometry of their capacity to generate cell surface (CD154, CD69) or soluble (IL-2, TNF-α, IFN-γ) help after CD3 engagement. Unexpectedly, the results showed that 40 different CVID blood samples exposed to HVS gave rise with a significantly increased frequency to transformed CD4+ T cell lines, compared to 40 age-matched controls (27% versus 3%, P ≤ 0·00002) suggesting the existence of a CVID-specific signalling difference which affects CD4+ cell transformation efficiency. The functional analysis of 10 CD4+ and 15 CD8+ pure transformed T cell lines from CVID patients did not reveal any statistically significant difference as compared to controls. However, half of the CD4+ transformed cell lines showed CD154 (but not CD69) induction (mean value of 46·8%) under the lower limit of the normal controls (mean value of 82·4%, P ≤ 0·0001). Exactly the same five cell lines showed, in addition, a significantly low induction of IL-2 (P ≤ 0·04), but not of TNF-α or IFN-γ. None of these differences were observed in the remaining CD4+ cell lines or in any of the transformed CD8+ cell lines. We conclude that certain CVID patients show selective and intrinsic impairments for the generation of cell surface and soluble help by CD4+ T cells, which may be relevant for B lymphocyte function. The transformed T cell lines will be useful to establish the biochemical mechanisms responsible for the described impairments.
Keywords: cellular activation, cell surface, molecules, common variable immunodeficiency, immunodeficiency diseases, T cell–B cell collaboration, T lymphocytes
INTRODUCTION
The term ‘common variable immunodeficiency’ (CVID) is used to describe an incompletely defined syndrome characterized by defective antibody formation, usually accompanied by decreased serum IgG and IgA levels and generally, but not invariably, decreased serum IgM [1]. CVID is one of the most frequent of the primary immunodeficiency diseases, ranging 1: 10·000– 1: 50·000. There is no recognizable pattern of inheritance, although CVID may cluster in some families [2].
Specific B-cell abnormalities have been considered to explain the pathogenesis of CVID [3,4]. However, the fact that appropriately stimulated CVID B cells can proliferate and produce immunoglobulins [5] suggested other mechanisms involved in B cell activation as a cause of CVID. First, defects in antigen handling and presentation to T cells have been described in CVID patients, such as the association to certain MHC haplotypes [6] or the up-regulation of IL-12 in CVID monocytes [7]. Secondly, a heterogeneous range of defects have been reported in T lymphocytes, which may impair T–B cell cross-talk. Among these, quantitative T cell defects could be due to impaired development of thymus-derived (i.e. CD45RA+) T cells [8] or to their increased apoptosis [9]. Against such a common T cell lymphopenia background, it is often difficult to establish qualitative (functional) T cell defects. Nevertheless, several functional Th cell disorders have also been reported in some CVID patients, such as impaired antigen-specific proliferation or cytokine synthesis [10], impaired expression of relevant surface molecules such as CD40L [11], defective integration of early membrane signals [12], defective recruitment and activation of ZAP-70 [13], excessive PKA I activation [14], excessive cytokine (TNF-α, IFN-γ) synthesis [15,16] and more recently, lck defects [17]. Phenotypical and functional abnormalities have also been reported in the Tc lineage of some CVID patients [18]. Lastly, a few CVID patients were in fact atypical forms of otherwise well-defined congenital T lymphocyte disorders, such as XHIM [19] or XLP [20]. Such a broad spectrum of defects suggests that CVID is a heterogeneous disorder, encompassing some patients with intrinsic defects in B cells or in T cells or in antigen presentation.
Herpesvirus saimiri (HVS) strain C488, a common lymphotropic virus of squirrel monkeys, has been used to transform into extended growth human mature CD4+ and CD8+ TCRαβ+ cells. Transformed cells remain IL-2-dependent, but are antigen and mitogen-independent and acquire a Th1 functional profile [21]. We [22,23] and others [24] have shown that transformed T cells from congenital immunodeficiences preserve the original defects. We therefore reasoned that transformed T cells from CVID patients might be useful to explore, if present, any putative intrinsic T cell defect.
MATERIALS AND METHODS
Patients and controls
Forty patients (23 males and 17 females, age mean = 31·7 ± 14, age range = 11–62) with well-documented CVID according to the diagnostic criteria of the IUIS scientific group for primary immunodeficiency diseases [1] were included in the study. Patients were on regular substitution therapy with IVIG (400 mg/kg body weight at 3–4-week intervals). XHIM and XLP diagnosis were excluded by laboratory tests and/or clinical features. As normal controls, 40 age-matched healthy volunteers were used. Informed consent was obtained from all the individuals, following Spanish regulations.
Cell lines
HVS-transformed T cell lines were derived from PBLs of 40 CVID patients and 40 normal age- and sex-matched donors as described [22]. HVS transformed T cell lines were also obtained from two unrelated immunodeficiencies with antibody dysfunction, but with known primary mutations: ataxia telangiectasia [23] and X-linked agammaglobulinaemia (XLA). They were subsequently exposed once to 1 ml of infective HVS supernatant (final concentration 1 × 106 cells/ml) in 24-well plates (Costar, Cambridge, MA, USA) in the presence or absence of 1 μg/ml phytohaemagglutinin (PHA, Difco Laboratories, Detroit, MI, USA). Deprivation of PHA at day 1 is believed to increase the chance of obtaining immortalized CD4+ cells. HVS-exposed T cells had been maintained in long-term culture for more than 8 months when the experiments reported here were performed. Due to the low frequency of normal HVS CD4+ T cell lines obtained, three additional HVS CD4+ T cell lines (from non-age-matched individuals) grown during more than two years and shown to be normal for the induction of CD154 and TNF-α, were included in the study [22,23]
Phenotypical analyses
The following MoAb were used for cytofluorometric analyses: Leu4 (anti-CD3), Leu23 (anti-CD69), FastImmune anti-Hu-IL-2, FastImmune anti-Hu-TNF-α, and FastImmune anti-Hu-IFN-γ from Becton-Dickinson (Mountain View, CA, USA); MsIgG (used as negative control) from Caltag (San Francisco, CA, USA); CD154 from Pharmingen (San Diego, CA, USA). For single- and two-colour immunofluorescence, 1 × 106 cells were incubated for 30 min at 4°C with appropriate fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated MoAb in PBS/EDTA buffer containing 1% FCS. After two washes with PBS buffer, the cells were analysed in an Epics Elite Analyser cytofluorometer (Coulter, Hialeah, FL, USA). Isotype-matched irrelevant antibodies were used to define background fluorescence. Data were collected on 104 viable cells as determined by electronic gating from a tight region around the lymphocyte population on the forward scatter/side scatter parameters dot plot.
Induction of surface activation molecules
To measure CD69 and CD154 (CD40L) induction after stimulation, cells were starved in CG/RPMI medium supplemented with 2% without human rIL-2 and incubated overnight. Next morning, cells were resuspended at 2 × 105 cells/ml in 96-well plates in the absence of stimuli, in the presence of 1 μg/ml plastic-bound anti-CD3 MoAb (IOT3b) or in the presence of 10 ng/ml phorbol myristate acetate (PMA, Sigma Chemical, St Louis, MO, USA) plus 750 ng/ml Ionomycin (Sigma Chemical, St Louis, MO, USA) and incubated for 9 h at 37°C. The 9-h time point was chosen in kinetic experiments because at that time the difference between stimulated and unstimulated cells is largest (data not shown). Then, cells were washed twice in PBS, stained with anti-CD69-FITC and anti-CD154-PE MoAb for 30 min at 4°C, washed twice in PBS and analysed by flow cytometry as described above.
Induction of intracellular cytokines
To analyse intracellular cytokine induction, transformed cells were collected, washed twice in PBS and resuspended in medium supplemented just with 2% of FCS (in the absence of IL-2) and then incubated overnight. Next day, cells were resuspended at 2,5 × 106 cells/ml in 96-well plates and stimulated for 9 h (peak production time point) with or without 1 μg/ml immobilized anti-CD3 MoAb (IOT3b) or in the presence of 10 ng/ml PMA plus 750 ng/ml Ionomycin. For the last 2 h, 10 μg/ml Brefeldin A (Sigma Chemical, St Louis, MO, USA) was added to the cultures to block secretion. Cells were harvested, washed twice in PBS buffer and fixed with 500 μl of 1% formaldehyde in PBS for 10 min at room temperature. Then, the cells were stained intracellularly for cytokine content as described [22,23].
Statistical analysis
Prophet 5·0 software (a National Computing Resource for Life Science Research sponsored by the National Center for Research Resources of the National Institutes of Health) selected automatically the type of statistical analysis (Student’s t-test, Fischer’s exact test) for each comparison. Only P-values below 0·05 were considered significant. Data are presented as mean ± s.d., except where indicated.
RESULTS
Phenotypical characterization of transformed T cells from CVID patients
Samples from 40 CVID patients and 40 age-matched healthy donors were exposed to the H. saimiri (HVS) supernatant in two different culture conditions (see Materials and methods). Therefore, 80 cell lines from CVID PBLs and 80 T cell lines from normal donors were expected. However, only 59 (16 CD4+ and 43 CD8+) and 66 (2 CD4+ and 64 CD8+) pure HVS T cell lines were obtained, respectively (Table 1). This means, first, that not all samples can be transformed and secondly, that there was a stronger bias towards CD8+ cells in controls. The immunophenotype of transformed CVID T cells by immunofluorescence indicated that the surface expression of TCR/CD3 and CD4 or CD8 was similar to controls (98–100%, data not shown). The phenotypical analysis of these cell lines thus unexpectedly revealed a significantly higher proportion of CD4+ T cell lines and lower proportion of CD8+ T cell lines in CVID than in normal controls (27% versus 3% and 73% versus 97%). To test whether the effect was CVID-specific, we transformed cells from two unrelated immunodeficiencies with antibody dysfunction, but with known primary mutations: ataxia telangiectasia (AT) and X-linked agammaglobulinemia (XLA). After exposure of peripheral blood lymphocytes from 15 AT and nine XLA patients to HVS in two different culture conditions, 26 and nine pure HVS T cell lines were obtained, respectively (Table 1). No differences in the proportion of CD4+ or CD8+ T cell lines were observed in these two immunodeficiencies, compared to controls. We performed further assays in 10 (of 16) CVID HVS CD4+ T cell lines which grew better and in 15 (out of 43) CVID HVS CD8+ T cell lines selected because most belonged to patients of which we had CVID HVS CD4+ T cell lines. These lines were compared to five HVS CD4+ T cell lines (two from this study and three from previous studies, see Materials and methods) and 15 HVS CD8+ T cell lines.
Table 1.
Herpesvirus saimiri-transformed T cell line phenotype distribution in different population samples *
Percentage of transformed T cell lines with CD4 + or CD8+ phenotype resulting from the number of viable cultures indicated in brackets, obtained after exposure of peripheral blood lymphocytes from 40 CVID, 15 AT, nine XLA and 40 normal donors to HVS in two different culture conditions (see Material and methods). CVID = common variable immunodeficiency. AT = ataxia telangiectasia. XLA = X-linked agammaglobulinemia.
P ≤ 2 × 10 −5 versus controls (Fisher’s exact test). Only significant differences are highlighted.
Induction of cell surface molecules and cytokines
T lymphocytes regulate B lymphocyte function by generating both cell surface (CD154/CD40L, for instance) and soluble (IL-4 and other cytokines) signals. Thus, we next compared the intrinsic capability of a number of pure CVID and normal HVS CD4+ and CD8+ T cell lines to generate such signals in vitro in response to both membrane (anti-CD3) and CD3-independent (PMA + ionomycin) stimuli. CD154, CD69, IL-2, TNF-α and IFN-γ were evaluated. Unfortunately, IL-4 cannot be measured in transformed T cells, as they acquire a Th1 phenotype [21,25]. No statistically significant difference was found between normal and CVID HVS CD4+ T cells for any of the stimuli (Fig. 1). However, the variability of CD154 induction was clearly different in CD4+ versus CD8+ T cells. In CD4+ T cells a wide dispersion of CD154 induction was observed in CVID, but not in normal cell lines. In contrast, both normal and CVID CD8+ cell lines showed a wide range of CD154 induction (from ≤ 5% to 80% with some stimuli). This was due to the existence of two subsets in normal and CVID CD8+ T cell lines: those which expressed little CD154 before or after stimulation, and those which expressed and induced CD154 after stimulation. CD154 induction in normal primary T cells is restricted mainly to the CD4+ subset. After transformation, a similar picture emerged: all normal CD4+, but only a few CD8+, T cell lines were inducible for CD154. In CVID patients, CD8+ cells were similar to normals, but CD4+ T cell lines seemed to contain both normal responders and low responders for CD154 induction.
Fig. 1.
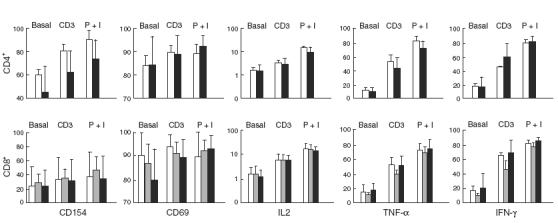
Induction of cell surface molecules and cytokines in transformed CD4+ and CD8+ T cell lines. Lines from CVID patients (10 CD4+, 15 CD8+) are compared to controls (five CD4+, 15 CD8+) and to XLA patients (five CD8+) for their response to anti-CD3 (CD3) or PMA + ionomycin (P + I). The mean ± s.e.m. is depicted. □, Controls;  , XLA; ▪, CVID. P > 0·05.
, XLA; ▪, CVID. P > 0·05.
A similar analysis of transformed CD8+ cells from XLA patients did not reveal any statically significant differences as compared to normals or to HVS CD8+ CVID patients (Fig. 1). Unfortunately, no CD4+ cells were transformed from XLA samples, precluding a similar comparison with CVID patients (Table 1).
Selective and intrinsic impairments after CD3 stimulation in certain transformed CVID T cell lines
The wide dispersion observed for the induction of CD154 in CVID HVS CD4+ led us to a more detailed analysis of these results. Thus, we observed that half of our patients (five out of 10) were under the lower limit of CD154 expression detected in normal HVS CD4+ cells after stimulation with anti-CD3 (broken line in Fig. 2a). Accordingly, we clustered our patients into two different groups: (a) those with a CD154 response to anti-CD3 stimulation above that arbitrary limit (A for above in Fig. 2a,b), mean value 78% compared to 82·4% and (b) those with a response below the indicated limit (B for below in Fig. 2a,b), mean value 46·8% compared to 82·4%. A very significant difference emerged between the CVID B subset and the two other subgroups (A CVID patients or normal controls, Fig. 2b). Of note, no significant differences were observed using CD3-independent stimuli (PMA + ION). These results suggested the existence of CVID patients within subset B with a possible intrinsic CD3-mediated defect in CD154 induction. However, since CD69 was constitutively expressed on HVS T cells and, in contrast to CD154, was only slightly up-regulated upon activation (Fig. 1), we cannot formally conclude that the observed defect was specific for CD154. The fact that the whole CVID sample showed no significant differences in the induction of any cell surface molecule, compared to controls, imposed a cautious analysis of the data.
Fig. 2.
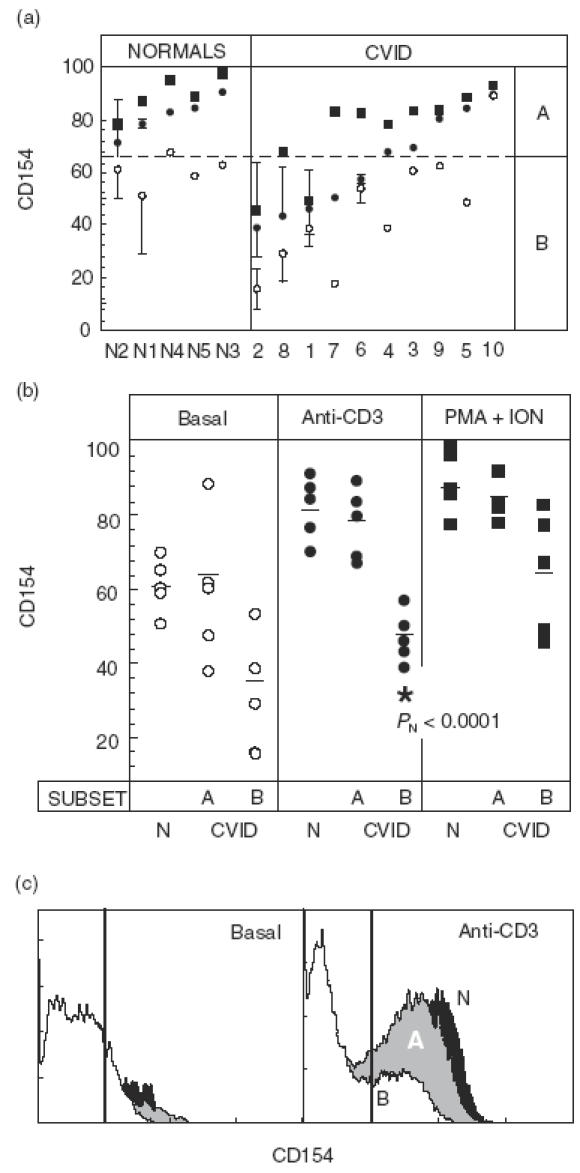
Induction of cell surface molecules in transformed CD4+ T cells. (a) Percentage of CD154 positive HVS CD4+ cells for five normal individuals and 10 CVID patients ordered by their response to anti-CD3. Numbers in the x-axis identify patients or controls. Analysed cells were unstimulated (○) or stimulated for 9 h in the presence of anti-CD3 (•) or PMA + ION (▪). When more than one experiment was performed (normally three, particularly for low responders), the mean ± s.d. is depicted. The broken line marks the lower limit of normal responses to anti-CD3. (b) Same data as in (a), grouped by the type of stimulus. Patients with a response below or above the lower limit of normals (broken line in (a)) were subgrouped as Below (B) or Above (A) responders. The short horizontal lines indicate the mean group values. The statistical significance of all comparisons among groups and subgroups within each stimulus was above 0·05 except where indicated (B versus N, PN = 0·0001). (c) Representative induction of CD154 on unstimulated (right) or anti-CD3-stimulated HVS CD4+. White, grey and black histograms represent patients 2 (CVID B subgroup), 4 (CVID A subgroup) and N1 (normal donor), respectively. The bold vertical lines indicate the upper limit of background fluorescence using isotype-matched irrelevant MoAb.
Interestingly, as observed for CD154 induction in CD4+ cells, an important proportion (4 out of 10) of CD4+ cells from CVID patients fell below the lower limit of IL-2 induction in normal controls after CD3 stimulation (broken line in Fig. 3 top, left). Although the IL-2 induction assay was of low sensitivity, those four patients belonged to the B subset defined in Fig. 2a for CD154 induction. When the CVID sample was clustered in the same A and B subsets defined in Fig. 2a, a significant difference emerged between B patients and normals (Fig. 3 top, right). We therefore conclude that the B subset contains some CVID patients with an impaired capacity to generate both cell surface (CD154) and soluble (IL-2) help by T cells. Indeed, a statistically significant correlation (r = 0·84, P = 0·0012) was observed between CD3-mediated CD154 and IL-2 induction (Fig. 4). The observed impairment in CD3-induced cytokine synthesis in the B CVID subset was IL-2-specific, since it was not observed in other tested cytokines such as TNF-α or IFN-γ (Fig. 3 middle and bottom, respectively). Unexpectedly, the A subset showed a significantly increased induction of IFN-γ via CD3 compared to normals (Fig. 3, bottom right).
Fig. 3.
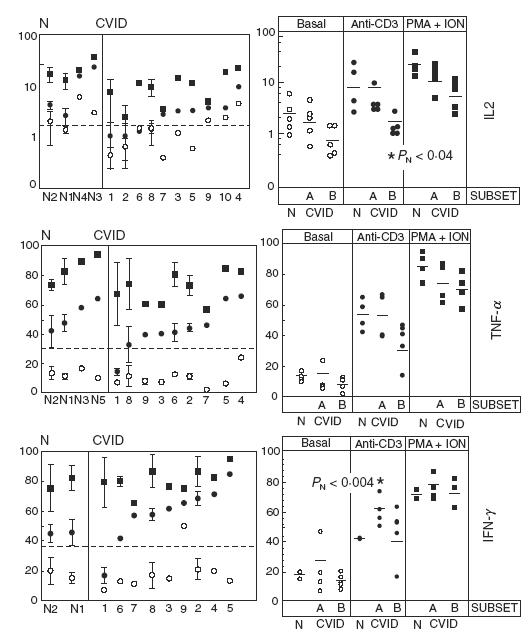
Induction of intracellular cytokines in transformed CD4+ T cells. Left: percentage of IL-2 (top), TNF-α (middle) and IFN-γ (bottom) positive HVS CD4+ cells for two normal individuals and at least nine CVID patients ordered by their response to anti-CD3. Analysed cells were stimulated as in Fig. 2. When more than one experiment was performed (normally three), the mean ± s.d. is depicted. The broken line marks the lower limit of normal responses to anti-CD3. Right: same data as in Left, grouped by the type of stimulus. Patients were subgrouped as Below (B) or Above (A) responders according to their response to CD154 induction (Fig. 2b). The short horizontal lines indicate the mean number of positive cells for each group or subgroup. The statistical significance of all comparisons among groups and subgroups within each stimulus was above 0·05 except where indicated (B versus N, PN ≤ 0·04, top right and A versus N, PN ≤ 0·004, bottom right).
Fig. 4.
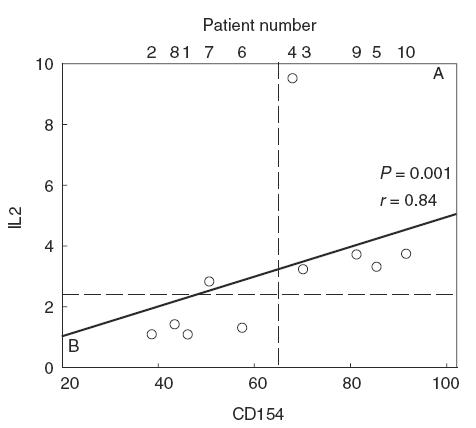
The induction of IL-2 by transformed CVID CD4+ T cells upon CD3 engagement significantly correlates with CD154 induction (P = 0·0012, r = 0,84). The broken lines mark the lower limits of normal responses to anti-CD3. B and A patients defined as in Fig. 2b clearly fall below or above the limit for IL-2, respectively, with a single exception.
Taken together the results indicated that, as a whole, transformed T cell lines from CVID patients are normal for the explored functions, but certain T cell lines showed intrinsic CD3-induced signalling disfunctions. As an example, a very interesting patient (no. 1) is shown in Fig. 5. We obtained both CD4+ and CD8+ transformed T cells from this particular patient. Her CD4+ cells showed an intrinsic defect for CD3-mediated induction of CD154, IL-2, TNF-α and IFN-γ which could be restored by PMA + ionomycin stimulation (Fig. 5 left). Thus, we conclude it was CD3-proximal. However, it was CD4+ T cell-specific, as it was not present in her transformed CD8+ T cells (Fig. 5 right).
Fig. 5.
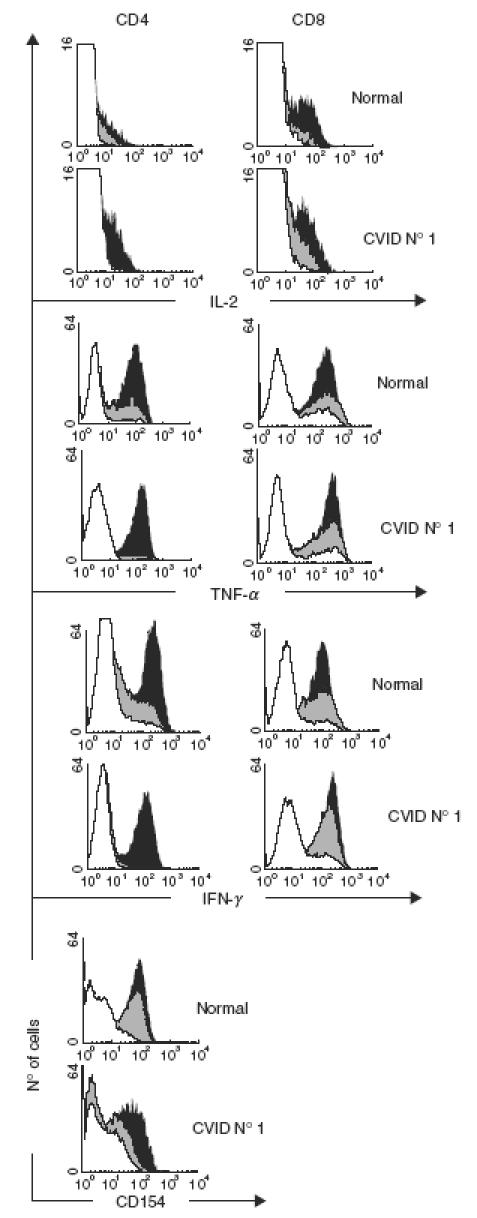
Defective CD3-mediated induction of soluble (IL-2, TNF-α, IFN-γ) and cell surface molecules (CD154) by transformed CD4+, but not CD8+, T cells from patient 1 (CVID B subgroup) compared to normal donor N1. White, grey and black histograms represent the basal expression, anti-CD3 induction and PMA + ION induction, respectively. CD154 in CVID CD8+ cells was uninducible (not shown). □, Basal; , anti-CD3; ▪, PMA + ION
DISCUSSION
Several reports have suggested that a subgroup of patients included under the CVID clinical/immunological umbrella have some degree of T cell dysfunction [8–18], which may result in depressed or inadequate contact-mediated B cell activation leading to a failure of CVID B cells to differentiate appropriately into immunoglobulin-secreting plasma cells. In a few cases, CVID patients turned out to be XHIM [19] or XLP [20], which are congenital T cell disorders. Very recently, an lck defect was shown in a CVID patient [17]. Thus, the CVID phenotype may conceal other T lymphocyte alterations. In order to investigate whether such putative defects were intrinsic to T cells or, rather, secondary to T cell subset distribution, recurrent infections or IVIG treatment, we have used HVS for the targeted transformation of human CVID T cells. Early signal transduction remains largely intact after transformation [21,25] and the cells can respond to activation with an increase of proliferation, with cytokine secretion, with cytotoxic activity and also with different forms of apoptosis. Consequently, HVS transformation of T cells from CVID patients may reflect the in vivo situation of parental T cells, when intrinsic defects are present (as in patient 1). However, there are also characteristic phenotypic and functional changes caused by HVS infection of human T cells, such as a shift towards a Th1 cytokine profile. Also, the low transformation efficiency of HVS (estimated to be 0·5–1 in 105 cells [26]) may question how representative our T cell lines are of the primary T cell subpopulations. This is also true for EBV-transformed B lymphocytes from CVID patients, although the transformation efficiency of EBV is significantly higher (around 1 in 102 [27]). However, such CVID B cell lines have been reported to preserve many phenotypic and functional features of fresh B cells [28], suggesting the existence of intrinsic and/or congenital B-cell specific defects in CVID. Therefore, the low transformation efficiency of HVS should not be a drawback if CVID-specific T cell defects were congenital or intrinsic, but may mask secondary T cell defects. In summary, abnormal transformed CVID T cells may reflect the in vivo situation, but normal transformed CVID T cells do not rule out T cell dysfunctions in vivo.
A first unexpected finding was the significantly increased proportion of CD4+ T cell lines obtained from CVID PBL samples, compared to controls or to other congenital immunodeficiencies with immunoglobulin disorders AT, XLA, Table 1(. Therefore, it is a CVID-specific feature which reveals an advantage of CD4+ cells (or a disadvantage of CD8+ cells) upon exposure to HVS. The mechanism for CD4+ versus CD8+ T cell lineage transformation by HVS is currently unknown [25]. As HVS requires a functional TCR/CD3 complex and exploits the CD2/CD58 autocrine interaction, our results suggest that a significant proportion of CVID patients have an intrinsic signalling alteration (or activation status) affecting transformation efficiency in one of the two lineages. An early signalling (qualitative) defect in CVID T cells has been reported previously by others [12–14,17,3,. Alternatively, our results may be explained by CVID-specific differences in CD4+ and/or CD8+ T cell subsets distribution, some of which may be more susceptible to HVS induced transformation. Such (quantitative) differences have also been reported by other authors [8,10].
Overall, the functional analysis of the pure transformed T cells from CVID patients for the generation of relevant cell surface (CD154) or soluble (IL-2, TNF-α, IFN-γ) help did not show any statistically significant differences compared to normals. However, CVID is a very heterogeneous disease [19] and may include several syndromes with different aetiopathogenesis, as reported previously [3–20]. By a similar approach, we have identified a significant (around 50%) subset of CVID patients (termed B), some of which show a selectively and intrinsically impaired capacity by CD4+ T cell to generate both cell surface (CD154, Fig. 2) and soluble (IL-2, Fig. 3) help after anti-CD3, but not after PMA + ION, stimulation which may be relevant for B lymphocyte function (such in patient 1). This TCR-proximal defect could be selective because, as a group, it did not affect other tested cell surfaces (CD69, Fig. 2) or soluble (TNF-α, IFN-γ, Fig. 3) molecules on transformed CD4+ cells, and it did not affect the same tested molecules (CD154, IL-2) on transformed CD8+ cells; the defect was intrinsic because it was found repeatedly in long-term cultures of pure transformed T cell lines, which may now be used for further characterization of the primary defects in specific patients’ lines such as the CD4+ line from patient 1 (see Fig. 5). However, a clear heterogeneity was observed within the CVID sample, with certain B patients showing excellent CD154 responses (patient 7) and A patients showing such high basal CD154 levels that induction cannot be measured (patient 10).
Some studies have reported low CD154 induction in stimulated PBL from a significant subset of CVID patients [11], while others have described IL-2 production defects [29]. We now show that, indeed, both disorders may be intrinsically associated in a large fraction of CVID patients (B in Fig. 4). When stimulated with anti-CD3, the reciprocal subset of CVID patients (i.e. B in Fig. 3, bottom) produced significantly elevated amounts of IFN-γ. Other authors have reported high IFN-γ synthesis by primary CVID T cells [30] Thus, it is possible that an intrinsic abnormality in some CVID patients (subset B) impairs Th-cell dependent help delivery to B cells, whereas in other CVID patients (subset A) such help is available, but in the wrong (i.e. Th1) direction, skewing the system away from the Th2 cytokine pattern needed for B cell antibody production. In contrast to some reports describing functional defects in CD8+ cells from CVID patients [16,18,30], our data rule out an intrinsic defect in the CD8+ lineage for the generation of cell-surface (CD154, CD69) and soluble (IL-2, TNF-α, IFN-γ) help factors for B cells (Fig. 1). However, we cannot exclude secondary or quantitative effects in primary T cells as explained above.
Collectively, our negative data support that most CVID T lymphocytes do not carry congenital or intrinsic defects in the explored functions and, thus, those CVID cases would not result from a T-cell-specific immunodeficiency. However, certain patients did show functional defects which appear intrinsic despite the low transformation efficiency. Further studies will be required to explore the mechanisms involved in the intrinsic dysfunctions reported here, and in their relevance for B cell function in CVID patients.
Acknowledgments
This work was supported by Fondo de Investigación Sanitaria (FIS) grants 96/0860 and 00/0936. We thank Manuela Beltrán Vicente (supported in part by FINNOVA) and Centro de Técnicas Inmunológicas (Universidad Complutense de Madrid) for technical support. Hoffmann-La Roche and Dr Craig W. Reynolds (Frederick Cancer Research and Development Center, NCI, Frederick, MD, USA) are gratefully acknowledged for the continuous supply of recombinant human IL-2.
REFERENCES
- 1.Report of IUIS Scientific Group. Primary immunodeficiency diseases. Clin Exp Immunol. 1999;118:1–28. doi: 10.1046/j.1365-2249.1999.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammarstroöm L, Vorechovsky I, Webster ADB. Selective IgA deficiency (SigAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120:255–31. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30:2516–20. doi: 10.1002/1521-4141(200009)30:9<2516::AID-IMMU2516>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Denz A, Eibel H, Illges H, Kienzle G, Schlesier M, Peter HH. Impaired up-regulation of CD86 in B cells of ‘type A’ common variable immunodeficiency patients. Eur J Immunol. 2000;30:1069–77. doi: 10.1002/(SICI)1521-4141(200004)30:4<1069::AID-IMMU1069>3.0.CO;2-M. 10.1002/(sici)1521-4141(200004)30:04<1069::aid-immu1069=3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 5.Nonoyama S, Farrington M, Ishida H, Howard M, Ochs HD. Activated B cells from patients with common variable immunodeficiency proliferate and synthesize immunoglobulin. J Clin Invest. 1993;92:1282–7. doi: 10.1172/JCI116701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vorechovsky I, Cullen M, Carrington M, Hammarstrom L, Webster AD. Fine mapping of IGAD1 in IgA deficiency and common variable immunodeficiency: identification and characterization of haplotypes shared by affected members of 101 multiple-case families. J Immunol. 2000;164:4408–16. doi: 10.4049/jimmunol.164.8.4408. [DOI] [PubMed] [Google Scholar]
- 7.Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol. 2000;164:488–94. doi: 10.4049/jimmunol.164.1.488. [DOI] [PubMed] [Google Scholar]
- 8.Pandolfi F, Trentin L, San Martin JE, Wong JT, Kunick JT, Moscicki RA. T cell heterogeneity in patients with common variable immunodeficiency as assessed by abnormalities of T cell subpopulations and T cell receptor gene analysis. Clin Exp Immuno. 1992;89:198–203. doi: 10.1111/j.1365-2249.1992.tb06932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Renzo M, Zhou Z, Becker K, Cunningham-Rundles C. Enhanced apoptosis of T cells in common variable immunodeficiency (CVID): role of defective CD28 co-stimulation. Clin Exp Immunol. 2000;120:503–11. doi: 10.1046/j.1365-2249.2000.01239.x. 10.1046/j.1365-2249.2000.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funauchi M, Farrant J, Moreno C, Webster AD. Defects in antigen-driven lymphocyte responses in common variable immunodeficiency (CVID) are due to a reduction in the number of antigen-specific CD4+ T cells. Clin Exp Immunol. 1995;101:82–8. doi: 10.1111/j.1365-2249.1995.tb02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrington M, Grosmaire LS, Nonoyama S, et al. CD40 ligand expression is defective in a subset of patients with common variable immunodeficiency. Proc Natl Acad Sci USA. 1994;91:1099–103. doi: 10.1073/pnas.91.3.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majolini MB, D’Elios MM, Boncristiano M, et al. Uncoupling of T-cell antigen receptor and downstream protein tyrosine kinases in common variable immunodeficiency. Clin Immunol Immunopathol. 1997;84:98–102. doi: 10.1006/clin.1997.4372. 10.1006/clin.1997.4372. [DOI] [PubMed] [Google Scholar]
- 13.Boncristiano M, Majolini MB, D’Elios MM, et al. Defective recruitment and activation of ZAP-70 in common variable immunodeficiency patients with T cell defects. Eur J Immunol. 2000;30:2632–8. doi: 10.1002/1521-4141(200009)30:9<2632::AID-IMMU2632>3.0.CO;2-C. 10.1002/1521-4141(200009)30:9<2632::aid-immu2632=3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 14.Aukrust P, Aandahl EM, Skålhegg BS, et al. Increased activation of Protein Kinase A Type I contributes to the T cell deficiency in common variable immunodeficiency. J Immunol. 1999;162:1178–85. [PubMed] [Google Scholar]
- 15.Aukrust P, Lien E, Kristoffersen AK, et al. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency. Possible immunologic and clinical consequences. Blood. 1996;87:674–81. [PubMed] [Google Scholar]
- 16.North ME, Ivory K, Funauchi M, Webster AD, Lane AC, Farrant J. Intracellular cytokine production by human CD4+ and CD8+ T cells from normal and immunodeficient donors using directly conjugated anti-cytokine antibodies and three-colour flow cytometry. Clin Exp Immunol. 1996;105:517–22. doi: 10.1046/j.1365-2249.1996.d01-795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawabe T, Horiuchi T, Nakamura M, et al. Defect of lck in a patient with common variable immunodeficiency. Int J Mol Med. 2001;7:609–14. doi: 10.3892/ijmm.7.6.609. [DOI] [PubMed] [Google Scholar]
- 18.Jaffe JS, Strober W, Sneller MC. Functional abnormalities of CD8+ T cells define a unique subset of patients with common variable immunodeficiency. Blood. 1993;82:192–20. [PubMed] [Google Scholar]
- 19.Spickett GP, Farrant J, North ME, Zhang J, Morgan L, Webster ADB. Common variable immunodeficiency: how many diseases? Immunol Today. 1997;18:325–8. doi: 10.1016/s0167-5699(97)01086-4. [DOI] [PubMed] [Google Scholar]
- 20.Gilmour K, Cranston T, Jones A, et al. Diagnosis of X-linked lymphoproliferative disease by analysis of SLAM-associated protein expression. Eur J Immunol. 2000;30:1691–7. doi: 10.1002/1521-4141(200006)30:6<1691::AID-IMMU1691>3.0.CO;2-K. 10.1002/1521-4141(200006)30:6<1691::aid-immu1691=3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 21.Meinl E, Hohlfeld R. T cell transformation with Herpesvirus saimiri: a tool for neuroimmunological research. J Neuroimmunol. 2000;103:1–7. doi: 10.1016/s0165-5728(99)00217-9. [DOI] [PubMed] [Google Scholar]
- 22.Pacheco Castro A, Alvarez-Zapata D, Serrano Torres P, Regueiro JR. Signalling through a CD3 gamma deficient T CR/CD3 complex in immortalized mature CD4+ and CD8+ T lymphocytes. J Immunol. 1998;161:3152–60. [PubMed] [Google Scholar]
- 23.Rivero ME, Porras O, Pelaez B, Pacheco-Castro A, Gatti R, Regueiro JR. Membrane and transmembrane signaling in Herpesvirus saimiri-transformed human CD4+ and CD8+ T lymphocytes is ATM-independent. Int Immunol. 2000;12:927–35. doi: 10.1093/intimm/12.6.927. [DOI] [PubMed] [Google Scholar]
- 24.Gallego MD, Santamaría M, Peña J, Molina IJ. Defective actin reorganization and polymerization of Wiskott-Aldrich T cells in response to CD3-mediated stimulation. Blood. 1997;90:3089–97. [PubMed] [Google Scholar]
- 25.Bröker BM, Fickenscher H. Herpesvirus saimiri strategies for T cell stimulation and transformation. Med Microbiol Immunol. 1999;187:127–36. doi: 10.1007/s004300050084. 10.1007/s004300050084. [DOI] [PubMed] [Google Scholar]
- 26.Fickenscher H, Bökel C, Knappe A, et al. Functional phenotype of transformed human alphabeta and gammadelta T cells determined by different subgroup C strains of Herpesvirus saimiri. J Virol. 1997;71:2252–63. doi: 10.1128/jvi.71.3.2252-2263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugden B, Mark W. Clonal transformation of adult human leukocytes by Epstein–Barr virus. J Virol. 1977;23:503–8. doi: 10.1128/jvi.23.3.503-508.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saxon A, Keld B, Díaz-Sánchez D, Guo BC, Sidell N. B cells from a distinct subset of patients with common variable immunodeficiency (CVID) have increased CD95 (Apo1/fas), diminished CD38 expression, and undergo enhanced apoptosis. Clin Exp Immunol. 1995;102:17–25. doi: 10.1111/j.1365-2249.1995.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hauber I, Fischer MB, Maris M, Eibl MM. Reduced IL-2 expression upon antigen stimulation is accompanied by deficient IL-9 gene expression in T cells of patients with CVID. Scand J Immunol. 1995;41:215–9. doi: 10.1111/j.1365-3083.1995.tb03556.x. [DOI] [PubMed] [Google Scholar]
- 30.North ME, Webster AD, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency). abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28-(‘suppressor’) CD8+ subsets. Clin Exp Immunol. 1998;111:170–5. doi: 10.1046/j.1365-2249.1998.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]