

Abstract
In an infectious process complement activation is necessary for a proper immune and inflammatory response, but when exacerbated may cause tissue injuries. In infective endocarditis (IE) patients tend to develop high titres of circulating immune complexes (CIC) that activate complement. The aim of this study was to evaluate for the first time complement activation in IE for possible correlation with extracardiac manifestations and clinical prognosis. Twenty patients with IE, 14 healthy controls and 15 patients presenting mitral and aortic valve lesions (with no signs of either infection or other associated diseases), were studied. Plasma levels of C3adesArg, SC5b-9, C1rs-C1Inh and C3b(Bb)P were determined by ELISA and C3d by double decker immunoelectrophoresis. C3 and C4 levels were assayed by turbidimetry and CIC by ELISA. Elevation of plasma levels of all complement activation products, with the exception of C3b(Bb)P, indicated a significant classical pathway activation in IE patients when compared to controls (C3d: P < 0·00004; C3adesArg: P < 0·03, SC5b-9: P < 0·01, C1rs-C1Inh: P < 0·00007). CIC levels were significantly increased (P < 0·005) and C3 reduced in IE patients (P < 0·05). Elevated C3d (P < 0·02) and C3adesArg (P < 0·03) levels were associated with pulmonary manifestations. In addition, C3d was significantly elevated in the patients who died when compared to those who had a good recovery (P < 0·02). Our data demonstrate the activation of the complement classical pathway, most probably mediated by CIC, in IE and suggests C3d and C3adesArg as possible markers for extracardiac lesion and severity of the disease.
Keywords: complement, immune complex, infective endocarditis
INTRODUCTION
Infective endocarditis (IE) is thought to initiate with the adherence of circulating microorganisms to preinjured cardiac tissue [1]. Proliferation of these microorganisms leads to the formation of the typical thrombotic lesion of the disease, with an accumulation of bacteria, platelets and fibrin [2]. Despite the sensibility of the infective pathogens to antibiotic therapy, IE remains a severe clinical condition. This is due mainly to the high concentration of microorganisms trapped in the vegetation and their apparent lack of access to phagocytosis [3].
In the course of IE, patients tend to develop high titres of specific antibodies against the causative microorganism. Despite the strong antibody response, the infection is not terminated. The continuous formation of circulating immune complexes (CIC) is followed by deposition in tissue, leading consequently to tissue injury [4]. Despite controversial discussion on the exact nature of the immune-mediated injury in IE, there is increasing evidence that CIC play a major pathogenic role in this disease [5,6]. In several studies, a high prevalence of CIC has been observed in IE patients [7–9]. Extravalvular manifestations of IE present many clinical features of an immune complex disease, and studies on tissue lesions are compatible with those known from immune complex deposition [10]. Although it is well known that CIC activate the complement system, its demonstration in IE patients has not yet been documented. In fact, the involvement of complement in the disease has been based mainly on studies of complement protein levels [4,6,11], haemolytic titres and complement deposition in tissue lesions as demonstrated by immunohistological analysis [12,13].
The complement system represents one of the major mediators of inflammation and immune response. Once activated through the classical, alternative and the newly described mannan binding lectin (MBL) pathways, split products especially of C3 and C5 provoke multiple biological effects. The anaphylatoxins C3a and C5a are potent inflammatory mediators, causing smooth muscle contraction, increased vascular permeability and degranulation by mast cells and basophils of histamine and other vasoactive substances. In addition, C5a is strongly chemotactic for phagocytic cells, inducing their activation and release of lysosomal enzymes and oxygen radicals [14]. C3b and C4b prevent immune complex aggregation and promote their transportation, mediated predominantly through their binding to the complement receptor CR1 on red blood cells, to the mononuclear phagocytic system [15]. C3b and C4b also act as opsonins, supporting phagocytosis and elimination of pathogens. Finally, the terminal membrane attack complex C5b-9 is potentially injurious to all cell membranes, causing their direct cell lysis. In sublytic amounts, C5b-9 has the ability to release inflammatory mediators from macrophages, neutrophils and platelets, such as arachidonic acid, prostaglandin E2, thromboxane, leukotriene B4, IL-1 and oxygen radicals [16].
Thus, complement activation may exert a considerable proinflammatory effect in the course of IE and in the development of the extravalvular manifestations. In the present study we therefore investigated the activation of complement in patients with IE and present evidence that complement split products may be suitable markers of extracardiac manifestations and severity of the disease.
PATIENTS AND METHODS
Patients
Twenty patients (14 men and six female) with a mean age of 37·9 years (range 15–76 years), who fulfilled the diagnosis criteria for IE, based on clinical, histopathological, blood culture and echocardiographic features according to Durack et al. 1994 [17], were included in this study after informed consent. Eighteen patients (90%) presented extravalvular manifestations. The most frequent extracardiac manifestations observed were splenomegaly (30%), renal (50%), neurological (25%) and pulmonary (35%) complications (Table 1). Eight patients presented a disease onset period of less than 4 weeks (acute evolution) and 12 patients had a disease onset of more than 4 weeks (chronic evolution). Regarding clinical outcome, 14 patients were healed by clinical and/or surgical treatment and six died. Eleven patients (55%) showed the presence of microorganisms in their blood cultures [Staphylococcus aureus (n = 4), Streptococcus viridans (n = 2), Haemophilus influenza, Enterobacter sp., Enterococcus faecalis, Pseudomonas aeruginosa and Staphylococcus (one positive each)].
Table 1.
Diagnostic criteria and clinical findings in patients with infective endocarditis
| Patient | Diagnostic criteria | Evolution | Blood culture | Extracardiac manifestations |
|---|---|---|---|---|
| 1 | 2 major | Cure | Staphylococcus aureus | Pulmonar, renal |
| 2 | Pathologically definitive | Surgery | Negative | Renal |
| 3 | 1 major + 3 minor | Death | Negative | Central nervous system (CNS), renal |
| 4 | 2 major | Death | Staphylococcus aureus | Splenomegaly, pulmonar, dermatological, renal |
| 5 | 1 major + 3 minor | Death | Pseudomonas aeroginosa | Renal, pulmonar |
| 6 | Pathologically definitive | Surgery | Negative | Renal |
| 7 | 2 major | Death | Staphylococcus aureus | Osteoarticular manifestations. |
| 8 | Pathologically definitive | Death | Haemophilus influenza | CNS |
| 9 | Pathologically definitive | Surgery | Negative | CNS |
| 10 | 2 major | Surgery | Streptococcus viridans | CNS, splenomegaly, renal |
| 11 | 2 major | Surgery | Staphylococcus aureus | Renal, pulmonar |
| 12 | 1 major + 3 minor | Surgery | Enterobacter sp. | Splenomegaly |
| 13 | 2 major | Surgery | Enterococcus faecalis | CNS, splenomegaly |
| 14 | 1 major + 3 minor | Death | Negative | Pulmonar |
| 15 | 2 major | Surgery | S. aureus (coagulase neg) | Renal |
| 16 | 2 major | Surgery | Negative | Splenomegaly, pulmonar, renal |
| 17 | 5 minor | Cure | Streptopcoccus viridans | None |
| 18 | 2 major | Surgery | Negative | Splenomegaly, osteomuscular |
| 19 | 1 major + 3 minor | Surgery | Negative | None |
| 20 | 1 major + 3 minor | Surgery | Negative | Pulmonar |
Controls
Clinical controls. Clinical controls comprised 15 patients with valvular heart disease (nine male and six female), with a mean age of 42·0 years (range 25–79), 11 of them presenting mitral and four of them aortic valve commitments. All patients were clinically stable, without history of intravenous drug abuse, signs of infection, acute rheumatic fever or any other associated disease.
Healthy controls. Fourteen volunteers (seven male and seven female, 20–53 years, mean 33·0 years) served as normal controls. None of them presented any history of previous drug use, infection or other associated disease.
Plasma and serum samples
Venous blood was drawn from each subject to obtain serum and ethylenediaminetetraacetic acid (EDTA) plasma. Blood samples were centrifuged at 4°C and plasma and serum were divided into aliquots and stored at –70°C until assayed. Samples from the IE patients were taken at the beginning of the antibiotic therapy (between the 2nd and 4th day).
Complement assays
SC5b-9. In brief, SC5b-9 was measured by enzyme-linked immunosorbent assay (ELISA) using a monoclonal antibody against a neoantigen according to Mollnes et al. [18], with minor modifications. Fifty μl of anti SC5b-9 (MoAb aE11 1 μg/ml) in carbonate buffer (pH 9·6) were used to coat microtitre plates (Nunc, Wiesbaden, Germany) overnight at 4°C. The following steps of blocking and washing were performed with PBS-gelatine 0·25% (Sigma, St Louis, MO, USA) and with PBS containing 0·1% Tween 20 (Serva, Heidelberg, Germany), respectively. The plasma samples were diluted 1:3 in PBS-EDTA and applied in duplicates. After incubation for 60 min at room temperature, rabbit antihuman C5 IgG (Dakopatts, Glostrup, Denmark) followed by peroxidase-conjugated goat antirabbit IgG was added and the reaction was visualized by addition of o-phenylamine-diamine (OPD) (Dakopatts, Glostrup, Denmark) and H2O2. Optical density (O.D.) was read on an ELISA reader (Sorin, Biomedica Vercelli, Italy) at 492 and 630 nm. Zymosan-activated serum calibrated with purified SC5b-9 was used as standard.
C3d. The concentration of C3d was evaluated by double-decker rocket immunoelectrophoresis assay according to Brandslund [19].
C1rs-C1Inh and C3b(Bb)P. Specific activation of the classical and alternative pathways was analysed by measuring the protein–protein complexes C1rs-C1inhibitor (C1rs-C1Inh) and C3b(Bb)P, respectively, by ELISA, basically as described in Cat et al. 1993 [20]. Briefly, microtitre plates (Nunc) were coated with rabbit antiC1Inh IgG (Dakopatts) or goat antiproperdin IgG (Baxter, Unterschleisheim, Germany). After blocking unspecific binding sites, appropriate dilutions of the EDTA plasma samples were incubated. The activation-specific protein complexes were detected by goat antiC1s IgG (Baxter) or rabbit antiC3c IgG (Dakopatts), respectively, followed by the appropriate peroxidase-labelled third antibody (Dianova, Hamburg, Germany). The reaction was visualized by the addition of o-phenylenediamine/H2O2 as substrate. Purified C1rs-CInh complex or inulin-activated normal serum C3b(Bb)P] were used as standards normal ranges (x ± 2 s.d.) as measured in EDTA-plasma of 55 healthy blood donors were 110–260 U/ml C1rs-C1Inh and 5–20 U/ml C3b(Bb)P, respectively].
C3a/C3adesArg. C3a/C3adesArg was determined by ELISA (Progen, Heidelberg, Germany) according to the manufacturer’s instruction. C3 and C4 levels were determined by turbidimetry, using polyclonal specific antisera following manufacturer’s instruction (Boeringer,Mannhein, Germany).
Other assays
Rheumatic factor. Analysis of RF was performed by agglutination of IgG-coated latex particles according to standard techniques.
CIC. The determinations of CIC levels were realized by ELISA using goat IgG anti-C1q or human anti-C3 (Sigma, St Louis, MO, USA) according to Pereira et al. [21].
Statistical analysis
The Mann–Whitney unpaired nonparametric and Student’s t-tests were used to compare data between patients and controls as indicated. Correlation coefficients were determined by the method of linear correlation of Pearson. Differences were considered to be significant at P < 0·05.
RESULTS
All markers for complement activation with the exception of C3b(Bb)P showed statistically significant results (P < 0·05) when comparing the IE patients with both the control groups (Figs 1 and 2 and 2). The most significant results were observed in the levels of C3d IE × healthy controls (HC): P = 0·000015 and IE × clinical controls (CC): P = 0·000035], C3adesArg (IE × HC: P = 0·00049 and IE × CC: P = 0·029), C1rs-C1Inh (IE × HC: P = 0·000025 and IE × CC: P = 0·000069) and SC5b-9 (IE × HC: P = 0·00014 and IE × CC: P = 0·013). These results demonstrated a preferential activation of the classical pathway in IE patients. The plasma concentrations of C3adesArg and of SC5b-9 were also significantly higher in clinical controls when compared to healthy controls (P = 0·000493 and P = 0·0008, respectively) (Fig. 2). Regarding activation of the alternative pathway, only five (25%) of the 20 IE patients had elevated levels of C3b(Bb)P when compared with the controls (P = n.s.). The mean levels of C3 and C4 remained within the normal range in IE patients as well as in controls (Fig. 3). The levels of C3 were lower in IE patients in comparison with the CC (P = 0·048). C3 and C4 levels did not correlate with any of the activation products, indicating that the levels of C3 and C4 are not reliable in demonstrating complement activation.
Fig. 1.
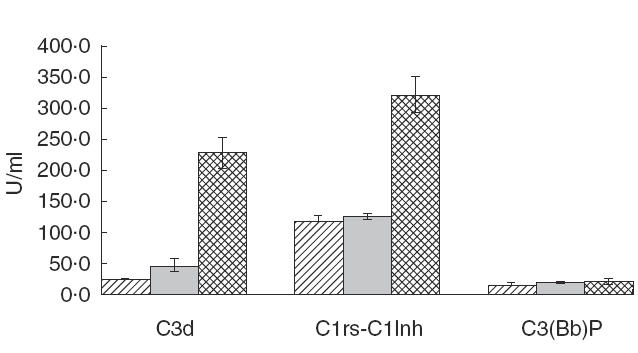
Levels of complement activation products C3d, C1rsC1Inh and C3(Bb)P in the patients with infective endocarditis and controls. C3d: P = 0·00001 (HC versus IE) and P = 0·0003 (CC versus IE). C1rsC1Inh: P = 0·00002 (HC versus IE) and P = 0·00007 (CC versus IE). C3(Bb)P: P = n.s.  , Healthy controls;
, Healthy controls;  , clinical controls;
, clinical controls;  , patients’ IE.
, patients’ IE.
Fig. 2.
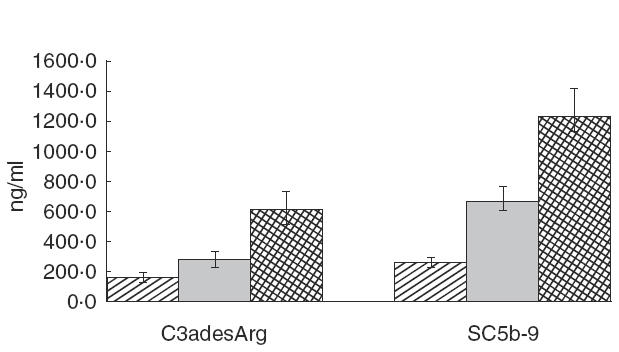
Levels of complement activation products C3adesArg and SC5b-9 in the patients with infective endocarditis and controls. C3adesArg: P = 0·0005 (HC versus IE) and P = 0·03 (CC versus IE). SC5b-9: P = 0·0001 (HC versus IE) and P = 0·013 (CC versus IE). , Healthy controls; , clinical controls; , patients’ IE.
Fig. 3.
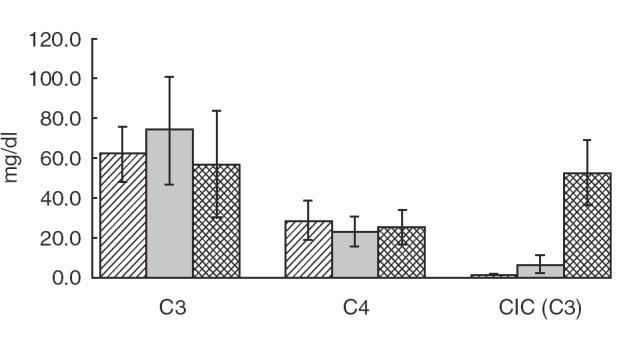
Levels of complement C3, C4 and CIC(C3) in the patients with infective endocarditis and controls. C3: P = n.s. (HC versus IE) and P = 0·048 (CC versus IE). C4: P = n.s. (HC versus IE) and P = 0·0007 (CC versus IE). CIC (C3): P = 0·0055 (HC versus IE) and P = 0·004 (CC versus IE). , Healthy controls;, clinical controls; , patients’ IE.
Increased CIC levels were found in 7/20 (35%) of the IE patients (CIC-C3) but in only 1/20 (5%) (CIC-C1q) (this patient was positive by both methods). None of the healthy controls had detectable levels of CIC. Among the clinical controls, however, two individuals (13%) showed low but detectable levels of CIC (C3). There was a statistically significant difference for CIC values when comparing IE patients with both the clinical (P = 0·004) and healthy controls (P = 0·0055) (Fig. 3). All IE patients with positive CIC showed increased levels of C1rs-C1Inh when compared to the controls but normal levels of C3b(Bb)P, indicating a preferential activation of the classical pathway. An inverse correlation between CIC and C4 levels (P = 0·013) supported the notion of complement consumption by the classical pathway.
Rheumatoid factor was positive in six (30%) IE patients and three of them were positive for CIC. No significant correlation with the presence of rheumatoid factor and complement activation was observed.
Mean plasma concentrations of C3d, C3adesArg, SC5b-9, C1rs-C1Inh and C3b(Bb)P were higher in patients with pulmonary manifestations when compared with those without such manifestations; however, the only significant results were for C3d (P = 0·003) and C3adesArg (P = 0·03). No significant correlations of complement activation and/or CIC levels with splenomegaly, renal and neurological manifestations were observed.
There was no significant difference in the levels of complement activation products between the patients with and without positive blood culture. Although all the patients with positive blood cultures showed elevated levels of C1rs-C1Inh, indicating activation of the classical pathway, none of them had evidence of complement activation by the alternative pathway. Patients infected with Staph. aureus showed higher levels of C3d, C3adesArg, SC5b-9 and C1rs-C1Inh than patients infected with other microorganisms. Patients with positive blood cultures showed more CIC frequently than those with negative blood culture (54% versus 12·5%), suggesting an association of CIC production with the presence of bacteria.
Regarding the clinical outcome, we observed that the levels of C3d and C1rs-C1Inh were higher in terminal patients when compared to those who had a good recovery, with differences in levels of C3d being statistically significant (P = 0·02).
DISCUSSION
Numerous studies have been performed to elucidate the pathophysiological mechanisms involved in the course of IE [2,3,5]. The binding of circulating bacteria, in most cases of low virulence, to an injured valve cannot account alone for inducing the primary thrombotic lesion and subsequent embolization nor for the broad and severe clinical manifestations of the disease. It is now clear that many clinical and histological features of IE, thought initially to be induced by bacterial tissue invasion, are mediated immunologically [4,1,2,3,4,5,6,7,
The presence of CIC in IE was suggested first upon findings of complement consumption [22] and cryoglobulinaemia [23]. Today, it is well accepted that high levels of CIC are a common feature of bacterial endocarditis [24], correlating with disease activity and with renal and vascular lesions [24,25]. The involvement of complement in IE has been drawn from different studies. Complement consumption indicated by a reduction in serum complement levels that returned to normal after antibiotic therapy has been demonstrated elsewhere [7]. Decreased levels of haemolytic titres of the whole complement classical pathway, indicating complement activation, have also been described [4,8,11]. In addition, deposition of complement components on basement membranes was reported from immunofluorescence studies on renal biopsies and on cardiac valves [26,27]. Elevated complement split products in IE patients have been reported by Pocidalo et al. [28] and Enriquez and Reyes [29], the latter suggesting C3d as a useful differential diagnostic marker between IE and acute rheumatic fever. In our study, we present evidence of complement activation which is based on a comprehensive analysis of the system. All complement activation products, with the exception of C3b(Bb)P, showed significantly elevated levels in IE patients when compared to the controls, supporting the notion that complement activation products are useful diagnostic markers. We also observed that levels of both C3 and C4 in IE remained in the normal range, indicating a lack of sensitivity of these traditional complement parameters to reflect complement activation [30]. As several complement proteins, including C3 and C4, behave as acute phase proteins, assessment of complement protein levels is often inadequate to detect complement activation, since an increased synthesis may mask accelerated catabolism. In addition, the range of normal complement protein concentration is wide, low levels may be related to deficiency states and differences may be significant among distinct racial groups. Therefore, measurement of activation products such as split products or protein–protein complexes has proved to be more accurate in assessing complement activation [30]. By the measurement of pathway-specific protein–protein complexes C1rs-C1Inh and C3b(Bb)P] we were able to identify the participation of the classical pathway in the disease process and its implication in the extracardiac lesions observed in IE. It is well established that the classical pathway is mainly antibody-driven and that antibody–antigen complexes play a major role in the development of extracardiac manifestations of both clinical and experimental IE [4–10]. In fact, the extracardiac manifestations in the kidney, skin and joints are believed to be the result of immune-complex deposition and subsequent complement activation in target tissues. Experimental as well as clinical studies have documented thoroughly the involvement of CIC and complement in tissue injury of IE-related glomerulonephritis [5]. It has been proposed that those lesions are caused by CIC formed intravascularly in the presence of antibody excess and not a consequence of in situ renal antigen–antibody complex formation [31]. Although observed as the most consistent association, there is evidence that CIC-mediated tissue injury in IE is related not only to the kidney, but also to tissue injury of the skin, synovium, choroid plexus and spleen [32,33].
In our study, pulmonary manifestations were associated with the activation of the classical pathway. Lung injury has already been related to intravascular complement activation by different authors [34,35]. Moreover, deposition of IgG immune-complexes were shown to cause lung injury dependent on complement activation in experimental models [36]. Our data, therefore, suggest that complement activation is related to pulmonary damage in the course of IE.
The mechanism by which CIC in IE patients become deposited in tissue and thus initiate extracardiac injuries remains unknown. Under normal conditions, immune-complexes should be solubilized and cleared by phagocytes. It is well known that complement components of both alternative and classical pathways are required to prevent immune complex aggregation by inhibiting the formation of large immune complexes [37,38]. It is evident that in IE some factors are impeding the dissolution of these complexes, leading to their deposition in tissues. Different studies have suggested that rheumatoid factors, detectable in 18–60% of IE patients, block the binding sites on IgG for complement binding as well as for phagocytosis, preventing the clearance of CIC [24,39], despite effective complement activation. In our study, 30% of the IE patients were positive for RF; half of them were CIC positive and all of them showed elevated levels of complement activation. Also, once excessive amounts of IC become deposited in tissue, ongoing complement activation takes place and affects surrounding tissue, such as vascular endothelial cells, leading to their injury. The development of microvascular injury may be dependent on C5 activation and the action of activated neutrophils and platelets [40]. The chemotactic, aggregating and adhesion-promoting effects of C5a and C5adesArg may result in circulating neutrophil aggregates that will be trapped in the capilaries of different organs [41]. Activated neutrophils have been shown to damage pulmonary endothelial cells [42]. The direct effect of complement split products, such as C3a and C5a, and the terminal lytic complex C5b-9 as well as other mediators of inflammation on endothelial cells must also be considered to contribute to the development of microvascular injury in critically ill patients.
Our data support the hypothesis that complement activation may have an important role in the pathogenesis of the IE, since the patients who died from the illness showed elevated levels of C1rs-C1Inh and C3d (P = 0·02) when compared to the patients who recovered well. We therefore conclude that complement activation occurs in IE mainly through the classical pathway and is involved in the development of extracardiac manifestations and deterioration of the disease.
Acknowledgments
The authors are grateful to CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico).
REFERENCES
- 1.Stekelberg JM, Wilson WR. Risk factor for infective endocarditis. Infect Dis Clin North Am. 1993;7:1–20. [PubMed] [Google Scholar]
- 2.Johnson CM. Adherence events in pathogenesis of infective endocarditis. Infect Dis Clin North Am. 1993;7:21–36. [PubMed] [Google Scholar]
- 3.Durack DT, Beeson PB. Experimental bacterial endocarditis II. Survival of bacteria in endocardial vegetations. Br J Exp Pathol. 1972;53:50–3. [PMC free article] [PubMed] [Google Scholar]
- 4.Kauffmann RH, Thompson J, Valentijn RM, Daha RM, Leendert VA. The clinical implications and pathogenetic significance of circulating immune complexes in infective endocarditis. Am J Med. 1981;71:17–25. doi: 10.1016/0002-9343(81)90253-9. [DOI] [PubMed] [Google Scholar]
- 5.Bayer AS, Theofilopoulos AN. Imunopathogenetic aspects of infective endocarditis. Chest. 1990;97:204–12. doi: 10.1378/chest.97.1.204. [DOI] [PubMed] [Google Scholar]
- 6.Cabane J, Godeau P, Herrerman G, Acar J, Digeon M, Bach JF. Fate of circulating immune complexes in infective endocarditis. Am J Med. 1979;66:277–82. doi: 10.1016/0002-9343(79)90545-x. [DOI] [PubMed] [Google Scholar]
- 7.Bayer AS, Theophilopoulos NA, Tillmann DB, Dixon KJ, Guze LB. Use of circulating immune complex levels in the serodifferention of endocardic and nonendocarditic septicemias. Am J Med. 1979;66:58–62. doi: 10.1016/0002-9343(79)90482-0. [DOI] [PubMed] [Google Scholar]
- 8.Bayer AS, Theophilopoulos NA, Dixon FJ, Guze LB. Circulating immune complexes in infective endocarditis. N Engl J Med. 1976;30:1500–5. doi: 10.1056/NEJM197612302952703. [DOI] [PubMed] [Google Scholar]
- 9.Bayer AS, Theofilopoulos AN, Dixon FJ, Guze LB. Circulating immune complexes in experimental endocarditis. A monitor of therapeutic efficacy. J Infec Dis. 1979;139:1–8. doi: 10.1093/infdis/139.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Heffner JE. Extra cardiac manifestations of bacterial endocarditis. West J Med. 1979;131:85–91. [PMC free article] [PubMed] [Google Scholar]
- 11.Kee JB, Capner PM, Mombray JF. Nature of circulating immune complexes in infective endocarditis. J Clin Pathol. 1980;33:653–9. doi: 10.1136/jcp.33.7.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenzie PE, Hawke D, Woodroffe AJ, Thompson AJ, Seymour AE, Larkson AR. Serum and tissue immune complexes in infective endocarditis. J Clin Immunol. 1980;4:125–32. [PubMed] [Google Scholar]
- 13.Keslin MH, Messner RP, Willians RC, Albuquerque NM. Glomerulonephritis with subacute bacterial endocarditis. Immunofluorescent studies. Arch Intern Med. 1973;132:578–81. [PubMed] [Google Scholar]
- 14.Hugli TE. Structure and function of the anaphylatoxins. Springer Semin Immunopathol. 1984;7:193–219. doi: 10.1007/BF01893020. [DOI] [PubMed] [Google Scholar]
- 15.Lachmann PJ. Complement. In: McGee JOD, Isaacson PG, Wright NA, editors. Oxford textbook of pathology. Oxford: Oxford University Press; 1992. pp. 259–66. [Google Scholar]
- 16.Hänsch GM, Seitz M, Betz M. Effect of the late complement components C5b-9 on human monocytes. Release of prostanoids, oxygen radicals and of a factor inducing cell proliferation. Int Archs Allergy Appl Immunol. 1987;82:317–20. doi: 10.1159/000234216. [DOI] [PubMed] [Google Scholar]
- 17.Durack DT, Lukes AS, Bright DK. The Duke endocarditis service. New criteria for the diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Am J Med. 1994;96:200–9. doi: 10.1016/0002-9343(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 18.Mollnes TE, Lea T, Froland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent against a neoantigen of the complex. Scand J Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 19.Brandslund I, Siersted HC, Svehag SE, Teisner B. Double-decker rocket immunoeletrophoresis for direct quantitation of complement C3 split products with C3d specificities in plasma. J Immunol Methods. 1981;44:63–71. doi: 10.1016/0022-1759(81)90107-1. [DOI] [PubMed] [Google Scholar]
- 20.Cat R, Rosario NA, de Messias IJT, Resener TD, Kirschfink M. Evaluation of complement activation in premature newborn infants with hyaline membrane disease. Eur J Pediatr. 1993;52:205–8. doi: 10.1007/BF01956145. [DOI] [PubMed] [Google Scholar]
- 21.Pereira AB, Theophilopoulos NA, Dixon FJ. Detection and partial characterization of circulating immune complexes with solid-phase anti C3. J Immunol. 1980;125:763–70. [PubMed] [Google Scholar]
- 22.Willians RC, Kunkel HG. Rheumatoid factor, complement and conglutinin aberrations in patients with subacute bacterial endocarditis. J Clin Invest. 1962;41:666–75. doi: 10.1172/JCI104523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurwitz D, Quismorio FD, Friou GJ. Cryoglobulinemia in patients with infectious endocarditis. Clin Exp Immunol. 1975;19:131–5. [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr MA, Wilton E, Naama JK, Whaley K. Circulating immune complexes associated with decreased complement-mediated inhibition of immune precipitation in sera from patients with bacterial endocarditis. Clin Exp Immunol. 1986;63:359–66. [PMC free article] [PubMed] [Google Scholar]
- 25.Boulton-Jones JM, Sissons JGP, Evans DJ, Peters DK, Cutler RE. Renal lesions of subacute infective endocarditis. Br Med J. 1974;6:11–5. doi: 10.1136/bmj.2.5909.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutman RA, Striker GE, Gilliland BC, et al. The immune complex glomerulonephritis of bacterial endocarditis. Ann Intern Med. 1952;36:1086–89. doi: 10.1097/00005792-197201000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Willians LM, Burks AW, Steele RW. Complement function and clinical relevance. Ann Allergy. 1988;60:293–302. [PubMed] [Google Scholar]
- 28.Pocidalo MA, Gilbert G, Verroust P, et al. Circulating immune complexes and severe sepsis: duration of infection as the main determinant. Clin Exp Immunol. 1982;47:513–9. [PMC free article] [PubMed] [Google Scholar]
- 29.Enriquez MM, Reyes PA. Estudios immunologicos en fiebre reumatica activa y endocarditis infecciosa. Arch Inst Cardiol Mex. 1984;54:153–8. [PubMed] [Google Scholar]
- 30.Kirschfink M. The clinical laboratory: testing the complement system. In: Rother K, Hänsch GM, Till G, editors. The complement system. Springer; 1998. pp. 420–7. [Google Scholar]
- 31.Sindrey M, Barratt J, Hewitt J, Naish P. Infective endocarditis-associated glomerulonephritis in rabbits: evidence of a pathogenetic role for antiglobulins. Clin Exp Immunol. 1981;45:253–60. [PMC free article] [PubMed] [Google Scholar]
- 32.Davis JÁ, Weisman MH, Dail DH. Vascular disease in infective endocarditis – report of immune mediated events in skin and brain. Arch Intern Med. 1978;138:480–3. doi: 10.1001/archinte.138.3.480. [DOI] [PubMed] [Google Scholar]
- 33.Nast CC, Colodro IH, Cohen AH. Splenic immune deposits in bacterial endocarditis. Clin Immunol Immunopathol. 1986;40:209–13. doi: 10.1016/0090-1229(86)90023-1. [DOI] [PubMed] [Google Scholar]
- 34.Till GO, Friedl HP, Ward PA. Lung injury and complement activation: role of neutrophils and xantine oxidase. Free Radic Biol Med. 1991;10:379–86. doi: 10.1016/0891-5849(91)90046-6. [DOI] [PubMed] [Google Scholar]
- 35.Younger JG, Sasaki N, Delgado J, et al. Systemic and lung physiological changes in rats after intravascular activation of complement. J Appl Physiol. 2001;90:228–95. doi: 10.1152/jappl.2001.90.6.2289. [DOI] [PubMed] [Google Scholar]
- 36.Lukacs NW, Glovsky MM, Ward PA. Complement–dependent immune complex-induced bronchial inflammation and hyperreactivity. Am J Physiol Lung Cell Mol Physiol. 2001;280:L512–18. doi: 10.1152/ajplung.2001.280.3.L512. [DOI] [PubMed] [Google Scholar]
- 37.Schifferli JA, Bartolotti SR, Peters DK. Inhibition of immune precipitation by complement. Clin Exp Immunol. 1980;42:387–94. [PMC free article] [PubMed] [Google Scholar]
- 38.Miller GW, Nussenzweig V. A new complement function: solubulization of antigen–antibody aggregates. Proc Natl Acad Sci USA. 1975;72:418–22. doi: 10.1073/pnas.72.2.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin CNG, Peters DK, Walport MJ. Monoclonal rheumatoid factor–IgG immune complexes. Poor fixation of opsonic C4 and C3 despite efficient complement activation. Arthritis Rheum. 1988;31:99–107. doi: 10.1002/art.1780310114. [DOI] [PubMed] [Google Scholar]
- 40.Tvedten HW, Till GO, Ward PA. Mediators of lung injury in mice following systemic activation of complement. Am J Pathol. 1985;119:92–6. [PMC free article] [PubMed] [Google Scholar]
- 41.Craddock PR, Hammerschmidt D, White JG, Dalmosso AP, Jacobs HS. Complement (C5a)-induced granulocyte aggregation in vitro. J Clin Invest. 1977;60:260–3. doi: 10.1172/JCI108763. A possible mechanism of complement-mediated leukocytosis and leukopenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varani J, Fligiel SE, Till GO, Kungel RG, Ryan US, Ward PA. Pulmonary endothelial cell killing by human neutrophils. Possible involvement of hydroxyl radical. Lab Invest. 1985;53:656–63. [PubMed] [Google Scholar]