

Abstract
Conflicting reports exist regarding the effects of interleukin-10 (IL-10) on mesangial cells. There have been reports of both proliferative and antiproliferative effects, and both proinflammatory and anti-inflammatory effects of IL-10 on mesangial cells. However, the potential for IL-10 to affect glomerulonephritis characterized by mesangial proliferation is not known. To test the hypothesis that IL-10 would limit experimental mesangial proliferative glomerulonephritis, IL-10 was administered to rats in which mesangial proliferative glomerulonephritis was induced by administration of anti-Thy 1 antibody. Compared to control treated rats, IL-10 treated rats showed less proliferation, with fewer cells in glomeruli. Glomerular cellular proliferation was reduced, assessed by the numbers of cells within glomeruli expressing either proliferating cell nuclear antigen (PCNA) or bromodeoxyuridine. Glomerular macrophage influx (but not the proportion of glomerular macrophages that were PCNA positive) was reduced by IL-10 administration. There was no significant reduction in glomerular α-smooth muscle actin staining. IL-10 treatment resulted in reduced renal IL-1β mRNA expression and reduced glomerular ICAM-1 expression, but renal expression of MCP-1 and osteopontin mRNA was unaltered. This study demonstrates that in experimental mesangial proliferative glomerulonephritis IL-10 diminishes inflammatory cell recruitment and mesangial cell proliferation. The effects of IL-10 in inhibiting mesangial cell proliferation are likely to be due to a combination of direct effects of IL-10 on mesangial cells and effects mediated by macrophages.
Keywords: interleukin-10, glomerulonephritis, macrophage, mesangial cell
INTRODUCTION
Interleukin-10 (IL-10) is a cytokine that is produced by a number of cell types, including T cells, macrophages [1] and resident tissue cells such as keratinocytes and mesangial cells [2]. IL-10 inhibits macrophage functions and diminishes macrophage and/or neutrophil mediated injury in a number of experimental models of innate and adaptive immune responses [3–8]. It has well- characterized inhibitory effects on T cells, particularly the T helper 1 (Th1) subset [9], antigen presenting cells [10] and macrophages [11], both directly and via inhibition of the secreted products of Th1 cells.
Despite these documented effects on T cell subsets and its effects in inhibiting macrophage effector functions, the effects of IL-10 on mesangial cell proliferation and expression of pro- and anti-inflammatory mediators are not well understood. There have been conflicting reports as to the effects of IL-10 on mesangial cells [2,12,13]. Mesangial cells can develop a macrophage-like phenotype, suggesting that IL-10 should have anti-inflammatory effects in this cell. The presence of IL-10 in lesions of human and experimental proliferative glomerulonephritis (GN) was predicted to exert a counter-regulatory role [14,15] and recent studies in mice genetically deficient in IL-10 support this contention [8]. However, it is unclear from the current literature whether IL-10 has proliferative or antiproliferative effects on mesangial cells or whether IL-10 enhances or inhibits the synthesis and secretion of proinflammatory mediators from mesangial cells [2,12,13,16].
Based on this evidence, we tested the hypothesis that IL-10 would inhibit mesangial proliferative GN. To address the hypothesis, IL-10 was administered to rats with mesangial proliferative GN (anti-Thy 1 GN) induced by polyclonal antithymocyte antibody. The degree of histological injury, cellular proliferation and the expression of proinflammatory mediators of injury was assessed in rats that had received vehicle alone and compared with disease in IL-10 treated rats.
MATERIALS AND METHODS
Experimental design
Anti-Thy 1 GN was initiated in 6-week-old male outbred Sprague–Dawley rats weighing 150–200 g, by i.v. injection of 1 ml /100 g of polyclonal rabbit-antirat thymocyte serum per rat (day 0). Rats were killed 72 h after the initiation of GN (day 3). Rats that were treated with IL-10 (n = 6) received recombinant murine IL-10 (specific activity 6·3 × 107 U /mg; Schering-Plough Research Institute, Kenilworth, NJ, USA) at a dose of 50 μg / 100 g /day in sterile PBS i.p. beginning 2 h after disease induction. As previous studies have demonstrated maximal glomerular binding of anti-Thy 1 antibody 1 h after injection [17], IL-10 treatment was commenced 2 h after anti-Thy 1 antiserum to avoid potentially affecting the deposition of the disease initiating antibody. Further doses were administered at day 1 and day 2. Control treated rats (Ctrl, n = 6) received the same volume of sterile PBS at the same time-points. Both groups of rats received bromodeoxyuridine (BrdU) at a dose of 50 mg/kg i.p. 3 h before the end of the experiment. Each result represents the mean of the six animals with GN from each group. Normal rats without disease (n = 6) provided baseline measurements. Histological assessments were performed on coded slides. The significance of differences between IL-10 treated rats and control treated rats with GN was determined by the Mann–Whitney U-test.
Assessment of histological injury
Kidney tissue was fixed in Bouin’s fixative, embedded in paraffin and 3 μm tissue sections were cut and stained with periodic acid-Schiff (PAS). Cell nuclei from a minimum of 50 consecutive glomeruli was assessed to determine the average total cell number for each animal, expressed as cells per glomerular cross section (c /gcs).
Immunohistochemistry using paraffin embedded sections
Paraffin sections (3 μm) were stained using three-layer immunohistochemical techniques. Cellular proliferation was assessed using antibodies against proliferating cell nuclear antigen (PCNA, mouse anti-PCNA, 1 in 100, PC-10, Dako, Glostrup, Denmark), BrdU (mouse anti-BrdU, 1 in 100, Bu20a, Dako), macrophages (mouse anti-rat-ED1, 1 in 50, American Type Culture Collection, Manassas, VA, USA) and α-smooth muscle actin (mouse antihuman α-smooth muscle actin, 1 in 200, 1A4, Dako). Sections were dewaxed, hydrated and microwave-treated (0·01 m Tri-sodium citrate, 10 min), then incubated with 10% normal rabbit serum in 5% BSA, followed by the primary antibody either for 1 h (room temperature) or overnight (4°C). After blocking endogenous peroxidase, sections were incubated with rabbit antimouse immunoglobulin (1 in 50, Dako) for 1 h and then either mouse peroxidase antiperoxidase (1 in 100, Dako) or mouse alkaline phosphatase, anti-alkaline phosphatase (1 in 100, Dako) for 1 h. Either 3,3-diaminobenzidine (DAB, Sigma Chemical Co., St Louis, MO, USA) forming a brown reaction product (for PCNA, ED-1 and α-smooth muscle actin) or fast blue BB salt (Sigma) that results in a blue reaction product (for BrdU) were used as substrates. Sections were counterstained with either PAS or haematoxylin. For double labelling PCNA (fast blue) and ED-1 (DAB)], sections were stained first for PCNA, followed by a further microwaving step to denature bound antibody [18]. Omission of the primary antibodies served as negative controls. A minimum of 50 consecutive glomeruli was assessed to determine the average number of cells per glomerulus staining positive for each animal (c/gcs). To assess semiquanititatively the accumulation of α-smooth muscle actin, the following scale was used [19] (0, no staining in the glomerulus; 1, 1–25% of glomerular tuft involved; 2, 25–50% involved; 3, 50–75% involved; 4, >75% involved).
Immunohistochemistry for glomerular ICAM-1 protein
Kidney tissue was fixed in periodate lysine paraformaldehyde for 4 h, washed in 7% sucrose solution, then frozen in liquid nitrogen. Mouse anti-rat ICAM-1 (1 in 400, 1A 29, R&D Systems, Minneapolis, MN, USA) was the primary antibody used. The immunohistochemical techniques and secondary antibody steps used were similar to those for paraffin sections, except sections were not microwave treated. The intensity of immunoperoxidase staining was assessed semiquantitatively (0–3 +). Sections in which only some glomeruli were positive were graded as 0·5 (i.e. ±).
Northern blot analysis for MCP-1 and osteopontin mRNA
Total RNA was prepared from control and IL-10-treated rat kidneys using the TRIzol® isolation reagent (Gibco BRL, Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s instructions. Following isolation, 15 μg of total RNA from each sample was resolved on 1% agarose gels containing 0·24 m formaldehyde. RNA was transferred to Genescreen Plus membranes (NEN Life Science Products, Boston, MA, USA), fixed using a stratalinker (Stratagene, La Jolla, CA, USA) and prehybridized in a solution containing 5 × SSPE, 10 × Denhardt’s solution, 0·1% SDS, 100 μg/ml sheared herring sperm DNA and 50% formamide. The MCP-1 insert was labelled with a 32P-dATP (NEN Life Science) and added to fresh buffer at 2 × 106 counts/ml and filters were hybridized overnight. Filters were washed in 2 × SSPE/0·1% SDS; 42°C; 15 min, 1 × SSPE /0·1% SDS; 42°C; 15 min and 0·1 × SSPE/0·1% SDS; 42°C; 15 min, wrapped in plastic and exposed to X-ray film at −70°C. Filters were then re-hybridized with a 1·6-kb human osteopontin insert (kindly provided by Dr M Gillespie, St Vincent’s Institute of Medical Research, Melbourne, Australia), stripped and hybridized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). MCP-1, osteopontin and GAPDH expression were quantified from autoradiographs. Bands were analysed by densitometry (Molecular Dynamics Computing Densitometer, Model 300 A, Sunnyvale, CA, USA) using ImageQuant Software 3·0. mRNA levels for MCP-1 or osteopontin were calculated as a ratio of the optical density units for MCP-1 or osteopontin to that of GAPDH.
RT-PCR for IL-1β mRNA
cDNA synthesis and PCR were performed as previously described [20]. Custom oligonucleotides were designed to detect and amplify rat IL-1β cDNA at an annealing temperature of 60°C (forward primer: 5′-CTGCAGCTGGAGAGTGTGG-3′; reverse primer: 5′-CATCCCATACACACGGACAACTAG-3′). The specificity of products was confirmed by Southern blot detection using a 32P-labelled internal oligonucleotide probe (5′-TGAGTCTGCACAGTTCCCCAAC-3′). RT-PCR and Southern blot detection of GAPDH cDNA was performed as previously described using primers GAPDH-1, 4 and 5 [20]. Densitometry values for IL-1β were normalized to GAPDH readings.
RESULTS
Effects of IL-10 on glomerular histology and mesangial cell proliferation
Control treated rats with anti-Thy 1 GN showed glomerular hypercellularity and mesangial cell proliferation at day 3 of disease (Figs 1a and 2a). Total glomerular cell numbers were increased (normal rat no GN] 46 ± 2 c/gcs, Ctrl GN 67 ± 3 c/gcs) and a number of cells were positive for the markers of cellular proliferation PCNA (normal rat 0·8 ± 0·2 c/gcs, Ctrl GN 18 ± 2 c/gcs; Figs 1b and 2c) and BrdU (Ctrl GN 4·8 ± 0·4 c/gcs; Fig. 1c). Administration of IL-10 diminished the severity of mesangial proliferative GN (Figs 1 and 2b,d). Total glomerular cell numbers were significantly reduced by IL-10 treatment (IL-10 GN 56 ± 2 c/gcs, P = 0·02; Fig. 1a). Consistent with this observation, PCNA + cells (IL-10 GN 11 ± 2 c/gcs, P = 0·04; Figs 1b and 2d) and BrdU + cell numbers (IL-10 GN 2·9 ± 0·5 c/gcs, P = 0·03; Fig. 1c) were decreased by IL-10 treatment, demonstrating that IL-10 inhibited mesangial cell proliferation in this model. However, while IL-10 appeared to reduce α-smooth muscle actin expression in glomeruli, this result did not reach statistical significance (Ctrl GN 1·7 ± 0·2 score 0–4 +], IL-10 GN 1·2 ± 0·2, P = 0·18; Figs 2e,f and 3).
Fig. 1.
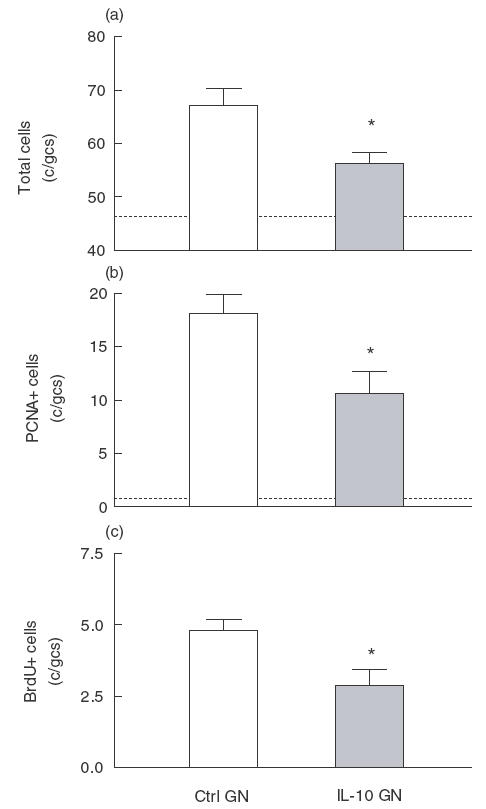
Analysis of glomerular hypercellularity and glomerular cell proliferation in control and IL-10 treated rats with mesangial proliferative GN. Dotted lines represent values for normal rats without GN. (a) Glomerular hypercellularity was reduced in rats with GN that were given IL-10 (IL-10 GN) compared with control treated rats (Ctrl GN). (b) The average numbers of cells per glomerular cross-section (c/gcs) positive for PCNA was reduced in IL-10 treated rats. (c) Cellular proliferation, as measured by the incorporation of BrdU into cells undergoing DNA synthesis, was reduced in rats with GN that had received IL-10. *P < 0·05 versus control rats with GN.
Fig. 2.
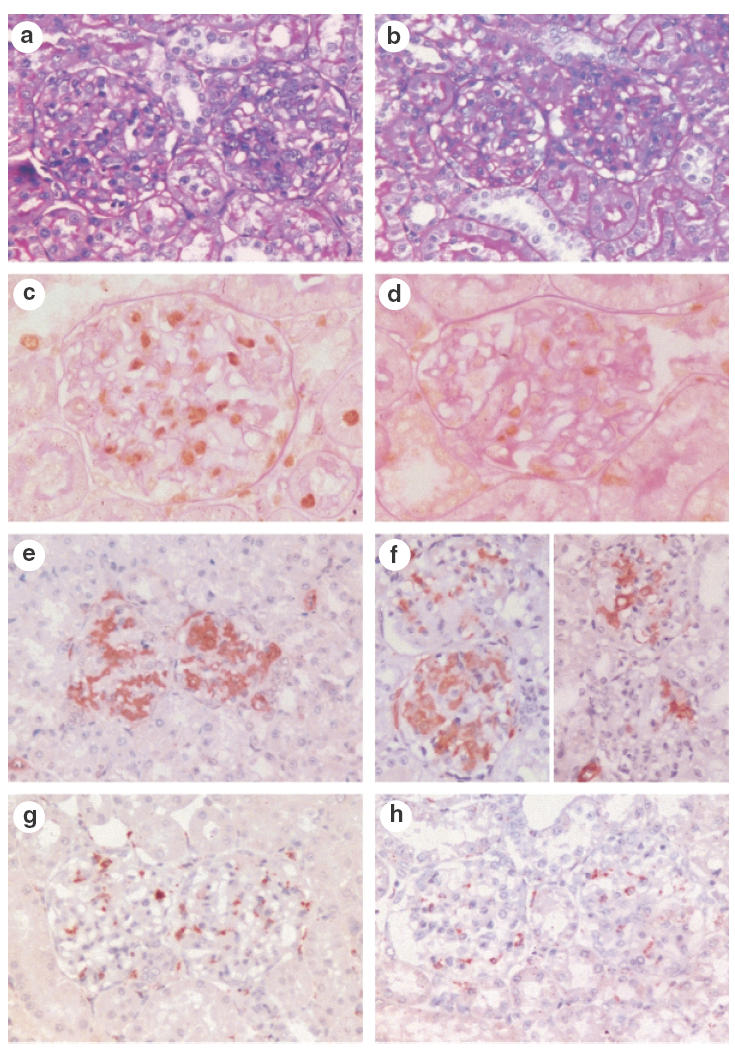
Glomerular histology in control treated rats with mesangial proliferative GN (a, c, e, g) and IL-10 treated rats with GN (b, d, f, h). Glomeruli from control rats with GN showed proliferative GN (a), the severity of which was reduced in IL-10 treated rats (b). Proliferating cells were plentiful in control rats with GN, and could be identified by immunohistochemistry for PCNA (c, brown reaction product). The number of PCNA + cells was reduced by IL-10 treatment (d, brown reaction product). Glomerular α-smooth muscle actin expression was not reduced significantly by IL-10 treatment (Ctrl GN, e and IL-10 GN, f). Glomerular ED1 + macrophages were reduced by IL-10 treatment (Ctrl GN, g and IL-10 GN, h, brown reaction product). Panels (a) and (b) were stained with PAS. (c–h) Immunoperoxidase with DAB substrate and PAS counterstain in panels (c) and (d), haematoxylin counterstain in (e–h). Magnifications: a, b, g, h and e × 300, f× 350, c × 500, d × 600.
Fig. 3.
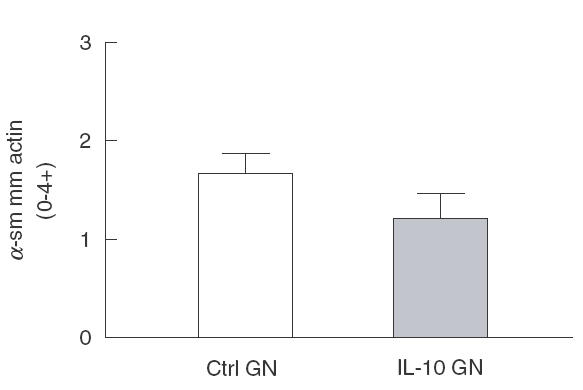
Semiquantitative assessment of the expression of ±-smooth muscle actin in glomeruli of control and IL-10 treated rats with mesangial proliferative GN, showing a trend towards reduced ±-smooth muscle actin expression in IL-10 treated rats with GN (P = 0·18)
Effects of IL-10 on macrophage recruitment and proliferation
Rats with anti-Thy 1·1 GN developed a moderate influx of macrophages by day 3 of disease, which was significantly inhibited by IL-10 treatment (normal rat no GN]: 0·8 ± 0·1 c/gcs, Ctrl GN 8·2 ± 0·4 c/gcs, IL-10 GN 4·5 ± 1·0, P = 0·02; Figs 2g,h and 4a). PCNA +/ED-1 + cells were observed within glomeruli of control treated rats with GN (Ctrl GN 3·0 ± 0·2 c/gcs; Fig. 4b), consistent with previous reports suggesting that macrophages proliferate within glomeruli in this model [21]. IL-10 treatment reduced the absolute number of proliferating macrophages within glomeruli (PCNA +/ED-1 + cells: IL-10 GN 1·6 ± 0·4, P = 0·03; Fig. 4b). This reduction of 1·4 c/gcs PCNA + cells in IL-10 treated rats accounts for only a small proportion of the reduction in total proliferating cells (PCNA +) observed. This finding, together with the similar proportion of macrophages found to be PCNA + in both groups (Ctrl GN 37 ± 2% of ED1 + cells PCNA +, IL-10 GN 37 ± 7% of ED1 + cells PCNA +) implies that it is unlikely that any potential inhibition of macrophage proliferation by IL-10 plays a significant role in the reduction of injury observed in IL-10 treated rats.
Fig. 4.
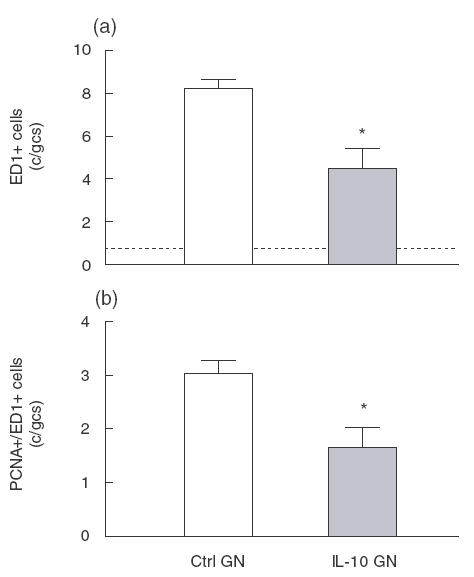
Macrophage accumulation and proliferation in glomeruli of rats with anti-Thy 1 GN, showing (a) fewer ED1 + macrophages in glomeruli of IL-10 treated rats with GN, and (b) a proportional reduction in the number of proliferating (PCNA +/ED1 +) macrophages in rats treated with IL-10. *P < 0·05 versus control rats with GN.
Effects of IL-10 on IL-1β mRNA in experimental mesangial proliferative GN
As the proinflammatory cytokine and mesangial growth factor IL-1β has been shown to be a relevant mediator in experimental mesangial proliferative GN [22], mRNA expression of IL-1β was measured using RT-PCR, compared to the housekeeping gene GAPDH. Compared with control treated rats with anti-Thy 1 GN, IL-10 treated rats expressed lower levels of IL-1β mRNA in their kidneys (Ctrl GN 1·00 ± 0·04, IL-10 GN 0·59 ± 0·11 IL-1β:GAPDH ratio, referenced to a value of 1 for Ctrl GN rats], P = 0·04; Fig. 5), consistent with the known effects of IL-10 on IL-1β and the previously published studies on the role of IL-10 in mesangial cells in vitro[2].
Fig. 5.
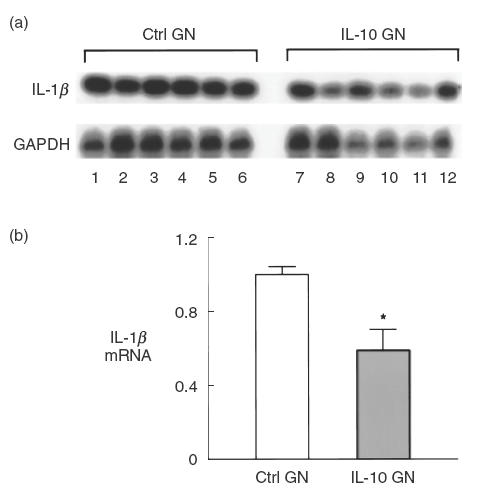
IL-1β mRNA expression of control treated and IL-10-treated rats with mesangial proliferative GN. (a) Representative Southern blot analysis of renal IL-1β cDNA from control rats with GN and from IL-10 treated rats with GN. Lanes 1–6 represent cDNA from kidneys of control treated rats with anti-Thy 1 GN and lanes 7–12 from IL-10 treated rats with GN. (b) Reduction in IL-1β mRNA in IL-10 treated rats. Densitometry values for IL-1β were normalized to GAPDH readings, Southern blot analysis was performed on four occasions and the results expressed as the mean densitometry values of these four separate experiments. Values represent fold expression of IL-1β mRNA, with control rats with anti-Thy 1 GN defined as having a value of 1. *P = 0·04 versus control rats with GN.
Effects of IL-10 on ICAM-1, osteopontin and MCP-1 in anti-Thy 1GN
ICAM-1 is important in leucocyte adhesion, is expressed by mesangial cells [23] and its expression is up-regulated in inflammatory renal injury, including human mesangial proliferative GN [24]. Glomeruli from control treated rats with mesangial proliferative GN expressed ICAM-1 protein, assessed by immunohistochemistry and semiquantitiated by scoring of the intensity of immunoperoxidase staining (Fig. 6a,b). Expression was also observed in periglomerular and interstitial areas, and in proximal tubular cells. ICAM-1 expression was reduced by IL-10 treatment (Ctrl GN 1·5 ± 0·1, IL-10 GN 1·0 ± 0·1 P < 0·01, Fig. 6). However, IL-10 administration had no effect on whole kidney mRNA expression for the chemokine MCP-1 or for osteopontin, suggesting that IL-10 does not affect these components of the inflammatory response in this model of GN. There was no significant difference in the osteopontin/GAPDH ratios between control GN and IL-10 treated rats with GN (Ctrl GN 0·47 ± 0·10, IL-10 GN 0·57 ± 0·17). Similarly, IL-10 did not affect MCP-1 mRNA expression in rats with mesangial proliferative GN for Ctrl GN (MCP-1/GAPDH ratio) 0·11 ± 0·06, IL-10 GN 0·15 ± 0·06].
Fig. 6.
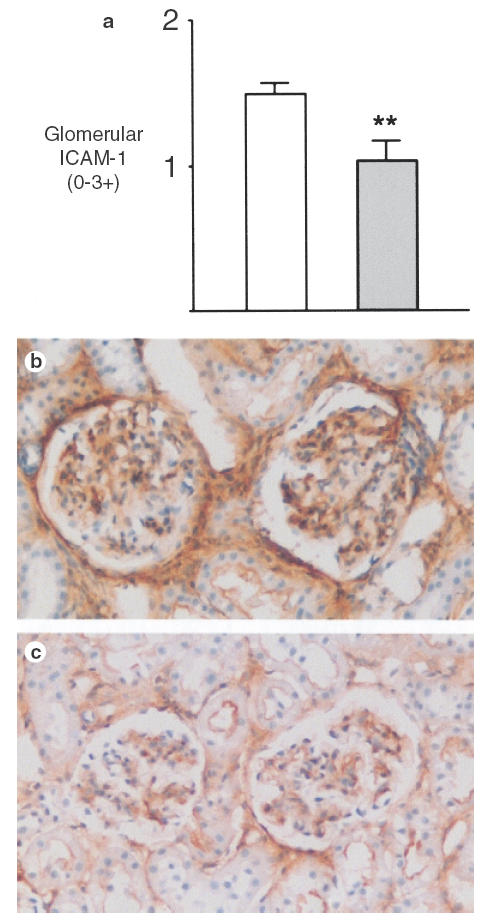
Glomerular ICAM-1 protein expression in control and IL-10 treated rats with GN. The mean intensity of ICAM-1 expression is reduced in glomeruli of IL-10 treated rats (a). (b) ICAM-1 expression in control treated rats with GN, with reduced expression in IL-10 treated rats (c). Immunoperoxidase with DAB substrate and haematoxylin counterstain, magnification × 300. **P < 0·01 versus control rats with GN.
DISCUSSION
These studies demonstrate that IL-10 inhibits mesangial cell proliferation in the anti-Thy 1 model of mesangial proliferative GN. In this well characterized and validated model, proliferation is induced by injection of a heterologous anti-Thy 1 antiserum that results in complement mediated lysis of mesangial cells [25]. The current studies show that in the presence of glomerular inflammation characterized by mesangial proliferation, the in vivo effects of IL-10 are to limit this proliferation. The administration of IL-10 beginning 2 h after the induction of disease inhibited proliferative GN, assessed by total glomerular cell numbers and the expression of two separate markers of cellular proliferation, PCNA and BrdU. Glomerular macrophage infiltration was reduced and there were fewer proliferating macrophages in glomeruli of IL-10 treated rats. However, the majority of cells expressing PCNA were ED1-, shown in other studies to be mesangial cells (expressing Thy 1 and α-smooth muscle actin) [26,27]. These observations, plus the fact that the reduction in proliferating macrophages observed (1·4 c/gcs) accounted for less than 20% of the total reduction (7·5 c/gcs) in PCNA + cells in glomeruli of IL-10 treated rats, demonstrate the inhibitory effect of IL-10 on mesangial cell proliferation in this lesion, although additional potential antiproliferative effects on other glomerular cell types cannot be ruled out. Although there was a trend towards reduced glomerular expression of α-smooth muscle actin, assessed by a previously published semiquantitative method, there was no statistically significant difference between control treated and IL-10 treated rats. This apparent dissociation between proliferation and α-smooth muscle actin expression has previously been observed in this model [19].
IL-10 is produced both by macrophages [28] and by mesangial cells [2,13], although its effects on the latter cell type have, until now, been unclear. IL-10 reduced lipopolysaccharide stimulated IL-1β and TNF-α release [2] and reduced IL-1β stimulated rat mesangial cell proliferation and ICAM-1 expression [12]. In contrast to these reports of antiproliferative and anti- inflammatory effects of IL-10 on mesangial cells, Chadban et al. found that exogenous IL-10 increased IFN-γ induced ICAM-1 expression in vitro[16] and stimulated mesangial cell proliferation [13]. Our in vivo studies of the effects of exogenous IL-10 in experimental mesangial proliferative GN support an anti- inflammatory and antiproliferative role for IL-10 in pathological mesangial cell proliferation.
IL-10 has been demonstrated in human GN [14] and in animal models of GN [15]. In an observational study of the anti-Thy 1 model of mesangial proliferative GN, IL-10 was present in proliferative lesions at day 6 [29]. While the current experiments do not directly address the effects of endogenous IL-10 on mesangial cells, they imply that the IL-10 found in this lesion is part of a counter-regulatory response to glomerular injury, as is the case in a murine model of crescentic GN [8].
The role of IL-10 has now been studied in a several models of GN. In some of these models the nature of the cognate (adaptive) immune response is an important determining factor in the degree of injury observed. In murine crescentic GN initiated by a planted antigen, IL-10 suppresses Th1 nephritogenic immune responses [30], and when given with IL-4 after the initiation of injury inhibits the effector arms of the immune response (CD4+ cells and macrophages) [6]. Endogenous IL-10 regulates Th1 responses and does not promote humoral injury in a similar model [8]. In contrast, IL-10 in murine lupus promotes humoral autoimmune renal injury and may in fact be important in the development of systemic autoimmunity that is the hallmark of this model [31]. IL-10 administration inhibited injury in passive anti-GBM GN [7]. In this model injury is uncoupled from cognate immune responses, despite the presence of macrophages, as disease is induced by passive administration of (autologous) rat anti-sheep globulin to rats given a subnephritogenic dose of sheep anti-rat GN.
In the anti-Thy 1 model, injury is initiated by heterologous antibody-induced, complement-mediated lysis of mesangial cells [25], with an additional role for bone marrow-derived elements, including platelets [32,33]. The results of this study are broadly in concordance with those in the passive anti-GBM GN model, although effects in that model were more profound [7]. Whether this reflects differences in the relative intensity of stimulus applied in the two models, differences in the timing of the initiation of IL-10 administration or differences in the magnitude of effects on mesangial cells compared to macrophages is not clear.
IL-10 clearly has antiproliferative effects on mesangial cells in this model. It is not clear to what extent these effects are due to direct anti-inflammatory and antiproliferative effects of IL-10 on mesangial cells, and to what extent the observed effects were due to effects mediated by macrophages. However, in vitro studies suggest that the down-modulatory effect of IL-10 on IL-1β, that can itself be produced by mesangial cells [2,22] and exert proliferative effects on mesangial cells [34], is likely to be relevant to the beneficial effects of IL-10 that were observed in these studies. The reduction in IL-1β assessed by RT-PCR could have been demonstrated more definitively by real time PCR, but is likely to be a real reduction, as it is consistent with previous effects of IL-10 in renal disease [2,7]. IL-1β is produced by macrophages, and ICAM-1 assists in macrophage recruitment. Both IL-1β and ICAM-1 were reduced by IL-10 in this study. While there is some evidence that macrophages play a role in this lesion [35], a definitive role for macrophages in this model remains to be established. As IL-10 has anti-inflammatory effects on both macrophages and on activated mesangial cells in culture, the inhibition of proliferation in this study is likely to reflect both direct and indirect (via macrophages) effects of IL-10.
The current studies examine the role of IL-10 in the glomerular response to injury by dissociating the effects of IL-10 from its role in the induction of and maintenance of the nephritogenic immune response. The model of mesangial proliferative GN used in these studies is not mediated by or characterized by the deposition of IgA in glomeruli. However, IL-10 has been shown to be protective in an IgA mediated model of pulmonary injury [5]. IgA nephropathy is the commonest from of mesangial proliferative GN in humans and IL-10 appears to synergize with TGF-β in inducing IgA synthesis [36]. Therefore, while IL-10 is likely to diminish glomerular injury in mesangial proliferative GN both by inhibition of macrophage function and by direct antiproliferative effects on mesangial cells, it is possible that these beneficial effects of systemically administered IL-10 could be nullified by enhanced IgA secretion and consequent deposition in glomeruli. In addition to effects on the systemic immune responses in mesangial proliferative GN, IL-10 may have effects on the resolution of injury in this model that have not thus far been studied.
In summary, these studies demonstrate that in experimental mesangial proliferative GN, systemic administration of IL-10 limits mesangial cell proliferation, at least in part by down- regulating proinflammatory cytokines such as IL-1β and by effects on ICAM-1. IL-10 is likely to have both direct and indirect (via macrophages) effects on mesangial cell proliferation.
Acknowledgments
These studies were supported by grants from the National Health and Medical Research Council of Australia (NH and MRC) and the Australian Kidney Foundation. Dr Tipping is an NH and MRC Senior Research Fellow. Recombinant IL-10 was a gift from the Schering-Plough Research Institute. The assistance of Mr S. Fraser, Ms J. Sharkey and Ms A. Wright is acknowledged.
REFERENCES
- 1.Mosmann TR. Properties and functions of interleukin-10. Adv Immunol. 1994;56:1–26. [PubMed] [Google Scholar]
- 2.Fouqueray B, Boutard V, Philippe C, et al. Mesangial cell-derived interleukin-10 modulates mesangial cell response to lipopolysaccharide. Am J Pathol. 1995;147:176–82. [PMC free article] [PubMed] [Google Scholar]
- 3.Daemen VA, van de Ven MW, Heineman E, et al. Involvement of endogenous interleukin-10 and tumor necrosis factor-alpha in renal ischaemia-reperfusion injury. Transplantation. 1999;67:792–800. doi: 10.1097/00007890-199903270-00003. [DOI] [PubMed] [Google Scholar]
- 4.Downing LJ, Strieter RM, Kadell AM, et al. IL-10 regulates thrombus-induced vein wall inflammation and thrombosis. J Immunol. 1998;161:1471–6. [PubMed] [Google Scholar]
- 5.Mulligan MS, Jones ML, Vaporciyan AA, et al. Protective effects of IL-4 and IL-10 against immune complex-induced lung injury. J Immunol. 1993;151:5666–74. [PubMed] [Google Scholar]
- 6.Kitching AR, Tipping PG, Huang XR, et al. Interleukin-4 and interleukin-10 attenuate established crescentic glomerulonephritis in mice. Kidney Int. 1997;52:52–9. doi: 10.1038/ki.1997.303. [DOI] [PubMed] [Google Scholar]
- 7.Huang XR, Kitching AR, Tipping PG, et al. Interleukin-10 inhibits macrophage induced glomerular injury: studies in passive anti-glomerular basement membrane glomerulonephritis. J Am. 1900;11:262–9. doi: 10.1681/ASN.V112262. [DOI] [PubMed] [Google Scholar]
- 8.Kitching AR, Tipping PG, Timoshanko JR, et al. Endogenous IL-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int. 1900;57:518–25. doi: 10.1046/j.1523-1755.2000.00872.x. 10.1046/j.1523-1755.2000.00872.x. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Elliott JF, Mosmann TR. IL-10 inhibits cytokine production, vascular leakage, and swelling during T helper 1 cell-induced delayed-type hypersensitivity. J Immunol. 1994;153:3967–78. [PubMed] [Google Scholar]
- 10.Fiorentino DF, Zlotnik A, Vieira P, et al. IL-10 acts on the antigen- presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–51. [PubMed] [Google Scholar]
- 11.Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor beta, interleukin-4, and interleukin-10. Ann NY Acad Sci. 1993;685:713–39. doi: 10.1111/j.1749-6632.1993.tb35934.x. [DOI] [PubMed] [Google Scholar]
- 12.Dai Z, Li X, Wang H. Inhibitory effect of interleukin-10 on inflammatory reaction in rat mesangial cells [in Chinese] Chung-Hua I Hsueh Tsa Chih [Chinese Med J] 1996;76:407–10. [PubMed] [Google Scholar]
- 13.Chadban SJ, Tesch GH, Foti R, et al. Interleukin-10 is a mesangial cell growth factor in vitro and in vivo. Lab Invest. 1997;76:619–27. [PubMed] [Google Scholar]
- 14.Niemir ZI, Ondracek M, Dworacki G, et al. In situ upregulation of IL-10 reflects the activity of human glomerulonephritides. Am J Kid Dis. 1998;32:80–92. doi: 10.1053/ajkd.1998.v32.pm9669428. [DOI] [PubMed] [Google Scholar]
- 15.Lakkis FG, Baddoura FK, Cruet EN, et al. Anti-inflammatory lymphokine mRNA expression in antibody-induced glomerulonephritis. Kidney Int. 1996;49:117–26. doi: 10.1038/ki.1996.16. [DOI] [PubMed] [Google Scholar]
- 16.Chadban SJ, Tesch GH, Foti R, et al. Interleukin-10 differentially modulates MHC class II expression by mesangial cells and macrophages in vitro and in vivo. Immunology. 1998;94:72–8. doi: 10.1046/j.1365-2567.1998.00487.x. 10.1046/j.1365-2567.1998.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto T, Wilson CB. Quantitative and qualitative studies of antibody-induced mesangial cell damage in the rat. Kidney Int. 1987;32:514–25. doi: 10.1038/ki.1987.240. [DOI] [PubMed] [Google Scholar]
- 18.Lan HY, Mu W, Nikolic-Paterson DJ, et al. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem. 1995;43:97–102. doi: 10.1177/43.1.7822770. [DOI] [PubMed] [Google Scholar]
- 19.Johnson RJ, Lombardi D, Eng E, et al. Modulation of experimental mesangial proliferative nephritis by interferon-gamma. Kidney Int. 1995;47:62–9. doi: 10.1038/ki.1995.7. [DOI] [PubMed] [Google Scholar]
- 20.Southby J, O’Keefe LM, Martin TJ, et al. Alternative promoter usage and mRNA splicing pathways for parathyroid hormone-related protein in normal tissues and tumours. Br J Cancer. 1995;72:702–7. doi: 10.1038/bjc.1995.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tesch GH, Nikolic-Paterson DJ, Lan HY. Do macrophages participate in mesangial cell proliferation? Nephrology. 1997;3:501–7. [Google Scholar]
- 22.Tesch GH, Lan HY, Atkins RC, et al. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151:141–50. [PMC free article] [PubMed] [Google Scholar]
- 23.Denton MD, Marsden PA, Luscinskas FW, et al. Cytokine-induced phagocyte adhesion to human mesangial cells: role of CD11/CD18 integrins and ICAM-1. Am J Physiol. 1991;261:F1071–9. doi: 10.1152/ajprenal.1991.261.6.F1071. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa T, Yorioka N, Ito T, et al. Precise ultrastructural localization of endothelial leukocyte adhesion molecule-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 in patients with IgA nephropathy. Nephron. 1997;75:54–64. doi: 10.1159/000189500. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto T, Wilson CB. Complement dependence of antibody-induced mesangial cell injury in the rat. J Immunol. 1987;138:3758–65. [PubMed] [Google Scholar]
- 26.Johnson RJ, Iida H, Alpers CE, et al. Expression of smooth muscle phenotype by rat mesangial cells in immune complex nephritis. Alpha-smooth muscle actin is a marker of mesangial cell proliferation. J Clin Invest. 1991;87:847–58. doi: 10.1172/JCI115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Floege J, Eng E, Young BA, et al. Heparin suppresses mesangial cell proliferation and matrix expansion in experimental mesangioproliferative glomerulonephritis. Kidney Int. 1993;43:369–80. doi: 10.1038/ki.1993.55. [DOI] [PubMed] [Google Scholar]
- 28.Moore KW, O’Garra A, de Waal Malefyt R, et al. Interleukin-10. Annu Rev Immunol. 1993;11:165–90. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 29.Chadban SJ, Nikolic-Paterson DJ. Interleukin-10: is it good or bad for the kidney? Nephrology. 1998;4:331–8. [Google Scholar]
- 30.Tipping PG, Kitching AR, Huang XR, et al. Immune modulation with interleukin-4 and interleukin-10 prevents crescent formation and glomerular injury in experimental glomerulonephritis. Eur J Immunol. 1997;27:530–7. doi: 10.1002/eji.1830270226. [DOI] [PubMed] [Google Scholar]
- 31.Ishida H, Muchamuel T, Sakaguchi S, et al. Continuous administration of anti-interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J Exp Med. 1994;179:305–10. doi: 10.1084/jem.179.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lianos E, Bresnahan BA, Pan C. Mesangial cell immune injury. Synthesis, origin and role of eicosanoids. J Clin Invest. 1991;88:623–31. doi: 10.1172/JCI115347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson RJ, Pritzl P, Iida H, et al. Platelet–complement interactions in mesangial proliferative nephritis in the rat. Am J Pathol. 1991;138:313–21. [PMC free article] [PubMed] [Google Scholar]
- 34.Lovett DH, Szamel M, Ryan JL, et al. Interleukin 1 and the glomerular mesangium. I. Purification and characterization of a mesangial cell-derived autogrowth factor. J Immunol. 1986;136:3700–5. [PubMed] [Google Scholar]
- 35.De Heer E, Prodjosudjadi W, Davidoff A, et al. Control of monocyte influx in glomerulonephritis in transplanted kidneys in the rat. Lab Invest. 1998;78:1327–37. [PubMed] [Google Scholar]
- 36.Defrance T, Vanbervliet B, Briere F, et al. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med. 1992;175:671–82. doi: 10.1084/jem.175.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]