

Abstract
The aim of the present study was to analyse in rats the ability of C-ANCA-positive IgG fraction in triggering inflammatory response on pulmonary tissue. Wistar rats (n = 18) were injected via the the internal jugular vein with 20 mg of total C-ANCA-positive IgG fraction isolated from serum of three different Wegener's granulomatosis patients obtained before therapy. Similarly, control rats were treated with IgG fraction from two rheumatoid arthritis patients (n = 7), IgG from six normal human sera (n = 15) or saline (n = 18), respectively. Animals were sacrificed after 24h of injection for histological analysis of the lungs. Vasculitis and inflammatory infiltrate were consistently absent in rats injected with rheumatoid arthritis IgG or saline and in 14/15 of normal IgG treated animals. In contrast, marked vasculitis was observed in all 18 animals injected with C-ANCA-positive IgG fraction. The histological features were characterized by the presence of a perivascular pleomorphic cellular sheath, particularly around small vessels, endothelial adherence and diapedesis of polymorphonuclear leucocytes and presence of granuloma-like lesions. A dose–response relationship was observed between protein concentration of C-ANCA IgG sample and the intensity of the inflammatory response in the animals. In addition, IgG fraction with undetectable C-ANCA, obtained from one patient in remission after treatment, was not able to reproduce the pulmonary tissue alterations induced by its paired IgG that was positive for C-ANCA taken before therapy. The experimental model described herein may be useful to characterize more effectively the pathogenic mechanism of C-ANCA in Wegener's disease.
Keywords: antineutrophil cytoplasmic antibodies (C-ANCA), Wegener's granulomatosis, vasculitis
INTRODUCTION
Wegener's granulomatosis (WG) is a systemic vasculitis characterized by the predominant involvement of upper and lower respiratory tracts, which can be associated in most cases with kidney failure. Pulmonary disease, including pleuritis, cough and haemoptysis, is the outstanding feature of WG and occurs in 45% at onset to 87% throughout the course of the disease [1]. Parenchymal cellular infiltrates and presence of multiple pulmonary nodules have been revealed by computerized tomography of the chest. In addition, pulmonary biopsy specimens show a marked inflammatory process including vasculitis, granulomatous lesions and necrosis [2].
Laboratory findings in untreated patients may include general abnormalities as leucocytosis, thrombocytosis, elevated erythrocyte sedimentation rate and elevated C-reactive protein. The more remarkable finding concerning WG serology and possibly pathophysiology is the association with the classic antineutrophil cytoplasmic antibodies, C-ANCA. These autoantibodies which yield a diffuse granular cytoplasmic immunofluorescence pattern on ethanol fixed neutrophils, have a predominant specificity for neutrophil proteinase 3 (PR3), a component of cytoplasmic azurophyl granules [3,4]. The presence of C-ANCA in the circulation, although not exclusive to WG, suggests strongly a diagnosis for it. These autoantibodies occur in a frequency varying from 90% in clinically active disease to 40% in WG remission [5].
In addition to the well-documented diagnostic potential of these antibodies their direct participation in triggering and sustaining the vascular disease activity has also been proposed. Evidence for the direct involvement of PR3-specific C-ANCA in WG pathogenesis comes from their prognostic value. Indeed, it has been shown that the titre of C-ANCA fluctuates following clinical relapses or remissions of the disease [6,7]. Circumstantial evidence that ascribes a putative pathogenic role for C-ANCA-induced vasculitis in WG includes extensive in vitro and ex-vivo studies. In fact, the ability of these antibodies to potentiate activation of primed neutrophils [8,9] has been demonstrated to promote oxidative burst and neutrophil degranulation, resulting in the release of injurious products [10]. In addition, C-ANCA induce increased adherence of neutrophils to endothelium by up-regulating cell adhesion molecules expression [11]. Accumulation of neutrophil granule enzymes adjacent to endothelium, causing localized endothelial damage [12,13], would account for the involvement of C-ANCA in vasculitis pathogenesis.
However, there is only a little indirect in vivo proof that C-ANCA are involved in vascular injury [14] and the precise pathophysiology of WG still remains to be elucidated [15]. The establishment of an appropriate experimental model will certainly provide clues to a better understanding of those mechanisms and/or factors involved in the disease pathogenesis. The aim of the present study was therefore to analyse, through the development of an experimental model, the ability of human C-ANCA-positive IgG fraction to induce vascular alterations in lungs of rats.
MATERIALS AND METHODS
Patients
Three patients, female, mean age 44 years old, with recent diagnosis of active Wegener's granulomatosis were selected for this study. At inclusion, serum was obtained at the time of WG diagnosis prior to treatment and stored at −20°C. The diagnosis of WG was based on the presence of classic clinical symptoms and typical histopathological findings. These patients fulfilled the criteria for the disease as defined by the American College of Rheumatology [16]. Patients were characterized clinically by the predominance of upper and lower respiratory tract involvement. Cavitating lesions and presence of nodules on computerized axial tomographic scan and on chest X-ray were observed in lungs. In addition histological analysis of open lung and/or of nasal sinuses/laryngeal biopsy specimens was performed, providing additional support for WG diagnosis. Furthermore, all three patients presented a normal renal evaluation. The control group consisted of sera from two patients with active rheumatoid arthritis fulfilling the American Rheumatism Association (ARA) criteria [17], both with RF ≥ 400 IU/ml and elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Additionally, sera from four healthy volunteers of laboratory staff were also included as negative control. The protocol was approved by the local Ethical Committee.
Detection of antineutrophil antibodies
All sera were tested at twofold serial dilutions starting at 1:20 by indirect immunofluorescence technique (IIF) on ethanol-fixed cytospins of human freshly isolated granulocytes, as described elsewhere [18]. Antibody binding was determined by a fluorescein-isothiocyanate conjugated goat antihuman immunoglobulins (Sigma, Chemical Co., St Louis, MO, USA).
Detection of anti-PR3 and anti-MPO antibodies
C-ANCA IgG antibody specificity to proteinase 3 (PR3) was confirmed by ELISA [19,20] using commercially available sensitized microplates with purified protein (Diastat Anti-PR3, Shield Diagnostics, Dundee, UK). Similarly, all serum samples were tested by ELISA for antimyeloperoxidase antibody (Diastat Anti-MPO, Shield Diagnostics). Serum samples were tested at 1/100 dilution.
Reactivity of human C-ANCA-positive sera to rat neutrophils
Polymorphonuclear cells were isolated from freshly drawn blood of Wistar rats by density gradient centrifugation on a Fycoll-Hypaque gradient. Sera were tested at 1:20 dilution by IIF on ethanol-fixed cytospin cells. Antibody binding was determined by using goat antihuman IgG conjugated to fluorescein-isothiocyanate (Sigma Chemical Co., St. Louis, MO, USA).
Analysis of antinuclear and antiphospholipid autoantibodies
Sera were analysed by IIF on HEP-2 cells for the presence of antinuclear antibodies and by ELISA for IgG and IgM anticardiolipin antibodies as described [21]. No reactivity was detected with any of WG sera and normal control.
Detection of circulating immune complexes
Circulating immune complexes (CIC) were investigated in all WG sera by capture ELISA using anti-C1q and anti-C3c specific antibodies (Sigma, Chemical Co.). Binding above 35 μg/ml equivalent of aggregate human gamma globulin, was considered positive for CIC. No serum tested had detectable CIC.
IgG isolation
Total IgG fraction from serum of the three WG patients, two RA patients and six normal individuals was purified by ion exchange chromatography on DEAE column and assayed for C-ANCA activity before use. Total protein concentration was determined by Lowry's method after checking samples on SDS-PAGE for purity. Isolated IgG fraction from all sera were stored in samples at −70°C and repeated freezing and thawing were avoided. Endotoxin (≥5 EU/ml) was detected in all IgG preparations (C-ANCA and controls) as evaluated by commercially available Limulus amebocyte assay (Charles River Laboratories, Endosafe®, Charleston, SC, USA) because protein handling was not performed under apyrogenic conditions. Titration of samples (double dilution) revealed no difference in endotoxin levels between C-ANCA-positive fractions and all control samples.
Protocol for IgG injection in rats
Anaesthetized Wistar rats (pentobarbital sodium, 30 mg/kg i.p.), female/male, weighting 250 g, received a single dose of 20 mg of C-ANCA-positive IgG fraction (n = 18) through an indwelling internal jugular vein catheter in order to access the pulmonary circulation directly. Similarly, control groups were injected with RA IgG (n = 7) and normal IgG (n = 15). Saline control (n = 18) was included systematically in every experiment. In a dose–effect assay, groups of rats received a single injection of 6, 12 and 20 mg of C-ANCA-positive IgG fraction from one single blood donor. After injection, catheter was removed from jugular vein and surgical incision was sutured.
Pulmonary histological analysis
Twenty-four hours after IgG injection, animals were anaesthetized with thiopental sodium (50 mg/kg i.p.), tracheotomized and a polyethylene cannula (1·7 mm i.d.) inserted. The tracheal cannula was connected to a small-animal ventilator (Harvard 683, HarvardApparatus Co., South Natick, MA, USA) adjusted to deliver a tidal volume of 10 ml/kg of body weight at a respiratory frequency of 80 breaths/min. A wide bilateral anterior thoracotomy was accomplished in order to expose both lungs and a positive pressure of 5 cm H2O was applied to the expiratory circuit to maintain an appropriate functional residual capacity. The trachea was occluded at the end of the expiration and the animals were exsanguinated. Lungs were rapidly removed and immediately frozen by immersion in liquid nitrogen and then fixed in Carnoy's fixative at −70°C (ethanol:choloroform:acetic acid, 60:30:10%). After 24 h, the concentration of ethanol was progressively increased (70, 80, 90 and 100%, respectively, 1h each solution, at −20°C) and lungs were kept in 100% ethanol for 24h at 4°C. Distal areas of the lungs were selected, embedded in paraffin and 5 μm thick histological sections were obtained and stained with haematoxylin and eosin.
Slides were analysed by two independent investigators in a blinded manner. The severity of lesion was scored according to the presence of inflammatory infiltrate: absent (−), sparse (1+); moderate (2+) and intense (3+).
RESULTS
C-ANCA reactivity of serum samples
At the time of the first bleeding, all three sera from WG patients were positive for C-ANCA by indirect immunofluorescence (IIF). C-ANCA titre was low for patients 1 and 2 (1:20) and moderate/high for patient 3 (1:400) as measured by IIF. The specificity of this cytoplasmic reactivity was confirmed by uniformly IgG binding to proteinase 3 (PR3) on ELISA. Moreover, a consistent negative binding to myeloperoxidase (MPO) was observed with these sera. Serum sample from patient 3 obtained after immunosuppressive treatment with cyclophosphamide was negative for C-ANCA and PR3. Control sera were uniformly negative for these antibodies. Additionally, binding of human C-ANCA to rat neutrophil was demonstrated by the moderate reactivity of patient 3 serum to this substrate, displaying a cytoplasmic immunofluorescent pattern, whereas a weak staining was observed with the other two WG patients’ sera (Fig. 1).
Fig. 1.
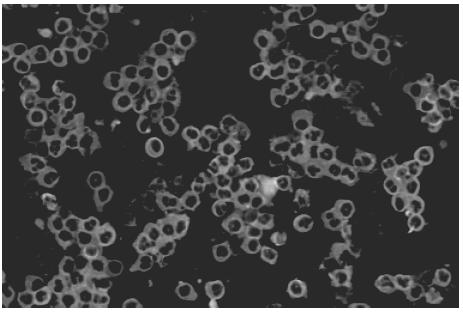
Binding of human C-ANCA-positive serum to ethanol fixed neutrophil from blood of Wistar rats by indirect immunofluorescence (original magnification ×20).
Pulmonary histopathological findings followed human C-ANCA IgG injection in rats
A remarkable inflammatory cell infiltrate, predominantly perivascular, was observed in all 18 animals injected with C-ANCA IgG samples (Table 1). Inflammatory response was intense in seven animals (39%) and moderate in 11 (61%) (Table 1). As illustrated in Fig. 2a, the prominent feature was the presence of a cellular sheath particularly around small vessels. This cellular infiltrate consisted of neutrophils, monocytes, lymphocytes, eosinophils and plasmocytes as revealed by the higher magnification microscopic analysis (Fig. 2b,c). Interestingly, adherence and diapedese were consistently observed particularly in venules and capillaries (Fig. 2c).
Table 1.
Effect of C-ANCA-positive IgG fraction from patients with active Wegener's granulo-matosis on pulmonary tissue of rats
| Pulmonary vasculitis | ||||
|---|---|---|---|---|
| C-ANCA IgG from patient | No. of animals injected | Pos (%) | Intensity of inflammatory response*(n) | Granuloma-like formation Pos (%) |
| 1 | 3 | 3(100) | 2+(3) | 0 |
| 2 | 7 | 7(100) | 2+(4) | 3(43) |
| 3+(3) | ||||
| 3 | 8 | 8(100) | 2+(4) | 4(50) |
| 3+(4) | ||||
Rats were injected via intrajugular vein with a single dose of 20mg of human C-ANCA-positive IgG fraction. Lungs were removed 24h later for histological analysis.
Inflammatory infiltrate score: (−) absent; (1+) scarce; (2+) moderate; (3+) intense.
Fig. 2.
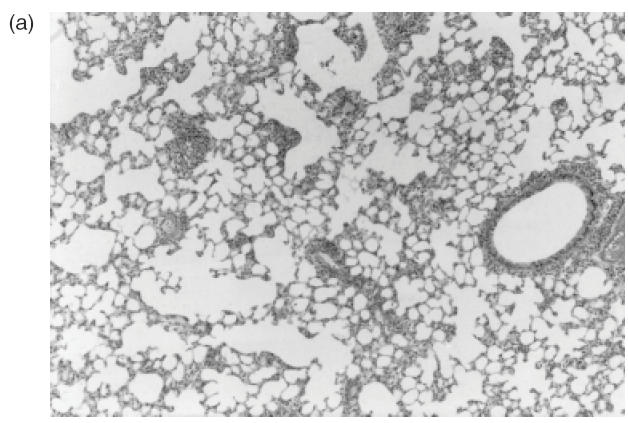
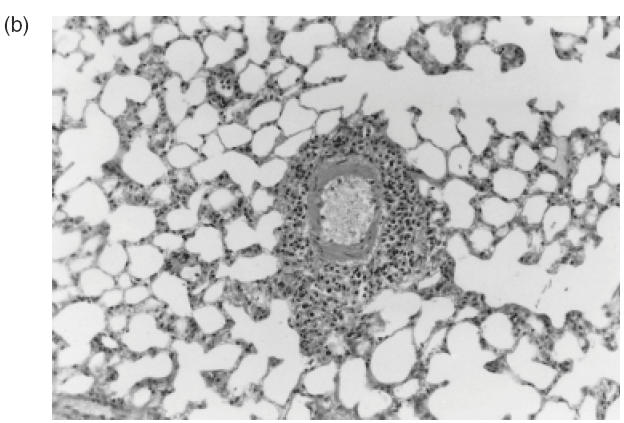
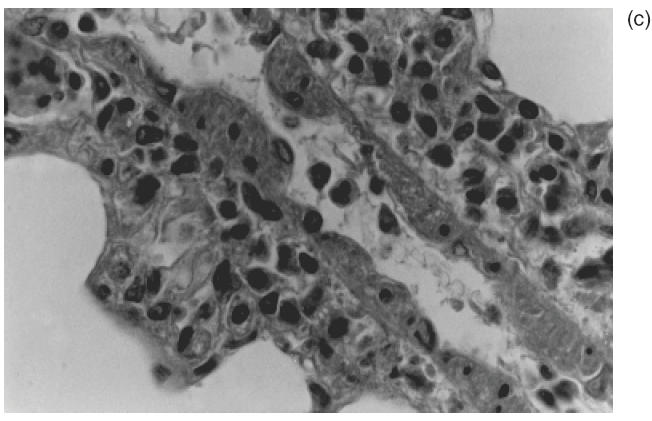
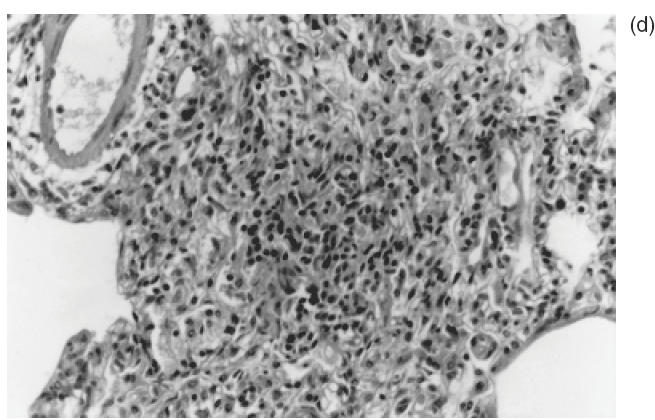
Representative histophatological changes in pulmonary tissue of Wistar rats after 24h of the intrajugular injection of human C-ANCA IgG fraction. (a–d) Haematoxylin and eosin-stained cryosections. (a) Presence of cellular sheath particularly around the small vessels throughout the pulmonary tissue (original magnification ×20). (b) Pleomorphic cellular composition of the perivascular infiltrate included neutrophils, monocytes, lymphocytes, eosinophils and plasmocytes (original magnification ×40). (c) Furthermore, adherence of inflammatory cells to the vessel wall was also found (original magnification ×40). (d) Granuloma-like formation of histiocytes concentrically distributed (original magnification ×100).
Fibrinoid necrosis of vessel wall and alveolar haemorrhage were absent in all lung tissue specimens analysed. No association of C-ANCA titre and the intensity of the inflammatory cells infiltrate was observed. Indeed, the frequency and the magnitude of the response were similar in rats injected with IgG samples from a low-titre C-ANCA patient 2 or high antibody-titre patient 3 (Table 1). Its noteworthy that a granulomatous-like formation consisting of histiocytes concentrically distributed, without the multi-nucleated giant cell was observed in 7/18 (39%) of the pulmonary tissue specimens (Fig. 2d). Similar to the cellular infiltrate, there was no association of the frequency of granulomatous-like formation in the lungs and C-ANCA titre in test sera.
Pulmonary histological findings in control groups of rats
The histological analysis of lungs of all seven animals injected with 20 mg of two different RA IgG and 14/15 (95%), which received IgG isolated from four normal individuals, revealed uniformly a consistent absence of perivascular inflammatory cell infiltrate (Table 2; Fig. 3a,b). However, one of four animals injected with normal IgG sample 1 presented an unexpected moderate perivascular infiltrate (data not shown). All other three animals injected with the same normal IgG sample 1 failed to show any inflammatory cell infiltrate. Tissue features compatible with chronic obstructive pulmonary disease was the only alteration observed in the control group probably due to the non-pathogen-free facilities of animals housing. No evidence of perivascular inflammation was found in all 18 animals receiving saline solution (data not shown).
Table 2.
Effect of control human IgG fractions on pulmonary tissue of rats
| Pulmonary vasculitis | ||||
|---|---|---|---|---|
| Control IgG | No.of animals injected | Pos (%) | Intensity of inflammatory response* (n) | Granuloma-like formation Pos (%) |
| NL 1 | 4 | 1(25) | −(3) | 0(0) |
| 2+(1) | ||||
| NL 2 | 3 | 0(0) | − | 0(0) |
| NL 3 | 3 | 0(0) | − | 0(0) |
| NL 4 | 5 | 0 (0) | − | 0(0) |
| RA 1 | 6 | 0(0) | − | 0(0) |
| RA 2 | 1 | 0(0) | − | 0(0) |
Rats were injected via intrajugular vein with a single dose of 20mg of normal human IgG (NL) and rheumatoid arthritis IgG (RA). Lungs were removed 24h later for histological analysis.
Inflammatory infiltrate score: (−) absent; (1+) scarce; (2+) moderate; (3+) intense.
Fig. 3.

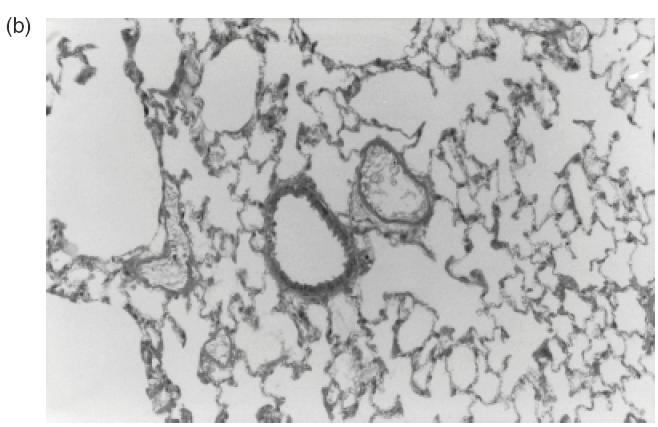
Representative sections of pulmonary tissue of Wistar rats after 24h of the intrajugular injection of normal human IgG. (a,b) Haematoxylin and eosin-stained cryosections. Lack of perivascular cellular infiltrate in lung tissue was detected in all animals injected with normal IgG (original magnification ×20 in (a) and ×40 in (b)).
C-ANCA IgG dose-dependent pulmonary inflammatory response
Two additional IgG concentrations, 6 and 12 mg from patient 3 with high titre C-ANCA, from whom sufficient material from the same bleeding was available, were tested in a dose–response experiment. Animals showed a more intense and frequent pulmonary inflammatory vasculitis when higher doses of the immunoglobulin preparation were injected (Table 3). Indeed, granuloma-like formation and grade 3 + inflammation process were detected only in those rats treated with 20 mg of C-ANCA IgG. In contrast, just one-fourth of animals injected with the lowest concentration (6 mg of protein) presented moderate inflammatory cell infiltrate but without evidence of granuloma-like formation. Attempts to concentrate this particular fraction further in order to inject 40 mg of protein were unsuccessful, due to the spontaneous aggregation of the IgG protein.
Table 3.
Dose–response effect of C-ANCA-positive IgG fraction from patient 3 on pulmonary tissue of rats
| Pulmonary vasculitis | ||||
|---|---|---|---|---|
| Dose of IgG (mg of protein) | No.of animals injected | Pos(%) | Intensity of inflammatory response * (n) | Granuloma-like formation Pos (%) |
| 6 | 4 | 1(25) | 2+(1) | 0(0) |
| 12 | 2 | 2(100) | 2+(2) | 0(0) |
| 20 | 8 | 8(100) | 2+(4) | 4(50) |
| 3+(3) | ||||
Groups of rats received via intrajugular vein a single dose of 6, 12 and 20 mg of C-ANCA-positive IgG fraction from WG patient 3. Lungs were removed 24 h later for histological examination.
Inflammatory infiltrate score: (−) absent; (1+) scarce; (2+) moderate; (3+) intense.
C-ANCA requirement for the experimental induction of pulmonary vasculitis
The involvement of C-ANCA antibodies on the pulmonary vascular abnormalities observed in this experimental model was suggested by the comparison of the ability of IgG sample with undetectable C-ANCA obtained from patient 3 during remission of WG after immunosuppressive therapy, with its paired IgG, positive for the antibody, taken originally from the same patient during active disease. C-ANCA-negative IgG sample was no longer able to reproduce signs of vascular pulmonary involvement as those induced by the matched C-ANCA-positive IgG (Table 4).
Table 4.
Ability of paired IgG fractions obtained from one single patient during active WG and in remission of the disease to induce pulmonary vasculitis in rats
| Pulmonary vasculitis | |||||
|---|---|---|---|---|---|
| WG disease status | C-ANCA (IIF) | No. of animals injected | Pos(%) | Intensity of inflammatory response* (n) | Granuloma-like ormation Pos (%) |
| Active | Positive | 7 | 7(100) | 2+(4) | 3(43) |
| 3+(3) | |||||
| Remission | Negative | 6 | 0(0) | − | 0(0) |
Rats were injected via intrajugular vein with a single dose of 20mg of IgG isolated from WG patient 3 during active disease and after immunosuppressive therapy (disease in remission). Lungs were removed 24h later for histological analysis. IIF: indirect immunofluorescence.
Inflammatory infiltrate: (−) absent; (1+) scarce; (2+) moderate; (3+) intense.
DISCUSSION
The results of this experimental model provide evidence that systemic injection of human C-ANCA-positive IgG fraction is able to induce in Wistar rats a pulmonary vasculitis, a finding which may contribute to the understanding of the mechanisms involved in the pathogenesis of human Wegener's granulomatosis. In fact, 24h after a single injection of C-ANCA IgG we have observed a typical venulitis with pleomorphic cellular infiltrate of lymphocytes, eosinophils, neutrophils and hystiocytes, defining a perivascular sheath very similar to the inflammatory lesion observed in human disease, although the characteristic fibrinoid necrosis of vessel wall was not detected [22]. Interestingly, we have also encountered granuloma-like formation in roughly one-third of the lung tissues analysed. It should be emphasized that the true granuloma formation is a complex sequence of immunological events, requiring access of helper T cells to the tissue and formation of giant cells probably by the fusion of histiocytes in response to a prolonged stimulation, and therefore should not be expected in the study design of the present work [23].
Supporting a direct role for C-ANCA in the pathogenesis of the pulmonary involvement of WG in humans, many studies based on bronchoalveolar lavage of these patients [24] have shown that PR3 C-ANCA are produced locally in the bronchial associated lymphoid tissue (BALT) [25] and in response to infections, particularly by Staphylococci [26].
This may represent a mechanism by which an infectious agent could trigger WG and the reasoning for the use of antistaphylococcal antibioticotherapy to protect patients and reduce the relapse frequency of the disease. Interestingly, it has been demonstrated that bacterial lipolysaccaharide augment the effects of hypoxia and inflammation in human pulmonary endothelial cells [27]. We speculated therefore that the presence of endotoxin in our samples may have aggravated the histological alterations induced by C-ANCA-positive IgG fraction.
Additionally, the non-pathogen-free housing conditions of Wistar rats in our study may have predisposed them to develop upper respiratory infections, which would result in an increased production of proinflammatory cytokines such as interleukin-1 (IL-1), IL-8 and alpha-tumour necrosis factor (TNF-α). These cytokines could, subsequently, induce granule stored PR3 translocation to the surface plasma membrane of polymorphonuclear neutrophils (PMN). In fact, in vitro data have shown that priming of neutrophils by proinflammatory cytokines leads to a time-dependent cell surface expression of PR3 enzyme which becomes accessible to circulating C-ANCA [28].
Alternatively, surgical stress due internal jugular vein dissection under general anaesthesia could be an additional factor for PMN priming by cytokines in our model. In this regard, it has been demonstrated increased levels of IL-1 and TNF-α in animals and in humans followed surgical procedures [29].
Importantly, the non-specific effect of IgG from patients with other active inflammatory disease in PMN priming was not sufficient to reproduce the histological lesions observed with C-ANCA IgG.
The option to work with human C-ANCA IgG autoantibody instead of anti-PR3 monoclonal antibody was based on the fact that most of the known monoclonal antibodies with this specificity are of the IgM class and lack true C-ANCA activity as they are not directed to the PR3 active enzyme site [30]. In fact, C-ANCA autoantibodies recognize conformational epitopes on PR3 [31] and interfere with enzyme activity [32]. Furthermore, the present demonstration that human C-ANCA IgG binds to rat neutrophil cytoplasm, and previous proof that PR3 molecule is phylogenetically conserved and is expressed in rodents [33–35], validate the use human C-ANCA in the current model. Possible controversies of PR3 C-ANCA binding to rat PMN [36] are most probably explained by the lower sensitivity of this substrate because rat neutrophil granules are less dense than humans [37], and by the known heterogeneity on the recognition of PR3 molecule by C-ANCA-positive sera on different methods, as demonstrated previously [31].
This model extends the observations of previous studies which have suggested that active immunization of mice with WG-derived C-ANCA IgG emulsified in complete Freund's adjuvant, produced either non-bacterial microabscesses or perivascular lymphocytic infiltration in lungs after 8 months of observation [38,39]. In this study the authors have described a process which involves the generation of idiotypic/anti-idiotypic network resulting in the production of a mouse antibody that has identical binding characteristic of human C-ANCA. We provided further evidence that a short period is also able to induce the histopathological changes of lung vasculitis with a single injection of human C-ANCA into the pulmonary circulation of Wistar rats supporting a direct pathogenic role of C-ANCA. Moreover, the dose-dependent effect observed herein clearly reinforces this possibility. Furthermore, evidence for a pathogenic potential of this antibody is also provided by the inability of the IgG fraction of a clinically inactive WG patient (C-ANCA negative) to induce the severe histopathological pulmonary lesions observed with the same patient IgG (C-ANCA positive) during the active phase of the disease.
The search for an appropriate experimental model which is capable to reproduce a definite lung vasculitis in animals, as presented in this paper, may have important implications for the comprehension of WG and other autoimmune diseases triggered by infectious agents. Moreover, the proposed animal model may lead to new insights into therapeutic approach in Wegener's granulomatosis.
REFERENCES
- 1.Fauci AS, Haynes BF, Katz P, et al. Wegener's granulomatosis. prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983;98:76–85. doi: 10.7326/0003-4819-98-1-76. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Hoffman GS, Leavitt RY, et al. Surgical pathology of the lung in Wegener's granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol. 1991;15:315–33. doi: 10.1097/00000478-199104000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Csernok E, Lüdemann J, Gross WL, et al. Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am J Pathol. 1990;137:1113–20. [PMC free article] [PubMed] [Google Scholar]
- 4.Braun MG, Csernok E, Gross WL, et al. Proteinase 3, the target antigen of anticytoplasmic antibodies circulating in Wegener's granulomatosis. Immunolocalization in normal and pathologic tissues. Am J Pathol. 1991;139:831–8. [PMC free article] [PubMed] [Google Scholar]
- 5.Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clinical & Experimental Immunology. 1993;91:1–12. doi: 10.1111/j.1365-2249.1993.tb03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen Tervaert JW, Vander Woude FJ, Fauci AS, et al. Association between active Wegener's granulomatosis and anticytoplasmic antibodies. Arch Intern Med. 1989;149:2461–5. doi: 10.1001/archinte.149.11.2461. [DOI] [PubMed] [Google Scholar]
- 7.Radu AS, Bueno C, Bonfa ESDO, et al. Anticorpos contra o citoplasma de neutrófilos nas vasculites sistêmicas. Rev Hosp Clín Fac Med S Paulo. 1993;48:264–71. [PubMed] [Google Scholar]
- 8.Keogan MT, Esnault VLM, Green AJ, et al. Activation of normal neutrophils by antineutrophil cytoplasm antibodies. Clinical & Experimental Immunology. 1992;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouwer E, Huitema MG, Mulder AHL, et al. Neutrophil activation in vitro and in vivo in Wegener's granulomatosis. Kidney Int. 1994;45:1120–31. doi: 10.1038/ki.1994.149. [DOI] [PubMed] [Google Scholar]
- 10.Falk RJ, Terrell RS, Charles LA, et al. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayet WJ, Schwarting A, Orth T, et al. Antibodies to proteinase 3 mediate expression of vascular cell adhesion molecule-1 (VCAM-1) Clinical & Experimental Immunology. 1996;103:259–67. doi: 10.1046/j.1365-2249.1996.d01-626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savage COS, Pottinger BE, Gaskin G, et al. Autoantibodies developing to myeloperoxidase and proteinase 3 in systemic vasculitis stimulate neutrophil cytotoxicity toward cultured endothelial cells. Am J Pathol. 1992;141:335–42. [PMC free article] [PubMed] [Google Scholar]
- 13.Mayet WJ, Schwarting A, Meyer zum Büschenfelde KH. Cytotoxic effects of antibodies to proteinase 3 (C-ANCA) on human endothelial cells. Clinical & Experimental Immunology. 1994;97:458–65. doi: 10.1111/j.1365-2249.1994.tb06110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jennette JC, Ewert BH, Falk RJ. Do antineutrophil cytoplasmic autoantibodies cause Wegener's granulomatosis and other forms of necrotizing vasculitis? Rheum Dis Clin North Am. 1993;19:1–14. [PubMed] [Google Scholar]
- 15.Ewert BH, Jennette JC, Falk RJ. The pathogenic role of antineutrophil cytoplasmic autoantibodies. Am J Kidney Dis. 1991;18:188–95. doi: 10.1016/s0272-6386(12)80879-1. [DOI] [PubMed] [Google Scholar]
- 16.Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 17.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 18.Cohen Tervaert JW, Mulder L, Stegeman C, et al. Occurrence of autoantibodies to human leucocyte elastase in Wegener's granulomatosis and other inflammatory disorders. Ann Rheum Dis. 1993;52:115–20. doi: 10.1136/ard.52.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasmussen N, Sjölin C, Isaksson B, et al. An ELISA for the detection of anti-neutrophil cytoplasm antibodies (ANCA) J Immunol Meth. 1990;127:139–45. doi: 10.1016/0022-1759(90)90350-5. [DOI] [PubMed] [Google Scholar]
- 20.Westman KW, Selga D, Bygren P, et al. Clinical evaluation of a capture ELISA for detection of proteinase-3 antineutrophil cytoplasmic antibody. Kidney Int. 1998;53:1230–6. doi: 10.1046/j.1523-1755.1998.00873.x. [DOI] [PubMed] [Google Scholar]
- 21.Gharavi AE, Harris EN, Asherson RA, Hughes GRV. Anticardiolipin antibodies. Isotype distribution and phospholipid specificity. Ann Rheum Dis. 1987;46:1–6. doi: 10.1136/ard.46.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegener F. Über generalisierte, septische Gefäβerkrankungen. Verh Dtsch Ges Pathol. 1936;29:202–10. [Google Scholar]
- 23.Rasmussen N, Petersen J. Cellular immune responses and pathogenesis in c-ANCA positive vasculitides. J Autoimmun. 1993;6:227–36. doi: 10.1006/jaut.1993.1020. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman GS, Sechler JM, Gallin JI, et al. Bronchoalveolar lavage analysis in Wegener's granulomatosis. A method to study disease pathogenesis. Am Rev Respir Dis. 1991;143:401–7. doi: 10.1164/ajrccm/143.2.401. [DOI] [PubMed] [Google Scholar]
- 25.Baltaro RJ, Hoffman GS, Sechler JMG, et al. Immunoglobulin G antineutrophil cytoplasmic antibodies are produced in the respiratory tract of patients with Wegener's granulomatosis. Am Rev Respir Dis. 1991;143:275–8. doi: 10.1164/ajrccm/143.2.275. [DOI] [PubMed] [Google Scholar]
- 26.Stegeman CA, Cohen-Tervaert JW, Sluiter WJ, et al. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003. [DOI] [PubMed] [Google Scholar]
- 27.Ziesche R, Petkov V, Williams J, et al. Lipopolysaccharide and interleukin-1 augment the effects of hypoxia and inflammation in human pulmonary arterial tissue. Proc Natl Acad Sci USA. 1996;93:12478–83. doi: 10.1073/pnas.93.22.12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csernok E, Ernst M, Schmitt W, et al. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clinical & Experimental Immunology. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ono S, Mochizuki H. Cytokine production in surgical stress. Nippon Geka Gakkai Zasshi. 2000;101:582–7. [PubMed] [Google Scholar]
- 30.Davis JA, Peen E, Williams RC, et al. Determination of primary amino acid sequence and unique three-dimensional structure of WGH1, a monoclonal human IgM antibody with anti-PR3 specificity. Clin Immunol Immunopathol. 1998;89:35–43. doi: 10.1006/clin.1998.4582. [DOI] [PubMed] [Google Scholar]
- 31.Bini P, Gabay JE, Teitel A, et al. Antineutrophil cytoplasmic autoantibodies in Wegener's granulomatosis recognize conformational epitope(s) on proteinase 3. J Immunol. 1992;149:1409–15. [PubMed] [Google Scholar]
- 32.Griffith ME, Coulthart A, Pemberton S, et al. Anti-neutrophil cytoplasmic antibodies (ANCA) from patients with systemic vasculitis recognize restricted epitopes of proteinase 3 involving the catalytic site. Clinical & Experimental Immunology. 2001;123:170–7. doi: 10.1046/j.1365-2249.2001.01420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aveskogh M, Lutzelschwab C, Huang MR, et al. Characterization of cDNA clones encoding mouse proteinase 3 (myeloblastine) and cathepsin G. Immunogenetics. 1997;46:181–91. doi: 10.1007/s002510050260. [DOI] [PubMed] [Google Scholar]
- 34.Sturrock A, Franklin KF, Wu S, et al. Characterization and localization of the genes for mouse proteinase-3 (Prtn3) and neutrophil elastase. Cytogenet Cell Genet. 1998;83:104–8. doi: 10.1159/000015144. [DOI] [PubMed] [Google Scholar]
- 35.Belaaouaj A, Moog-Lutz C, Just J, et al. Genomic organization and chromosomal localization of mouse proteinase 3. Mamm Genome. 1999;10:210–2. doi: 10.1007/s003359900974. [DOI] [PubMed] [Google Scholar]
- 36.Esnault VLM, Mathieson PW, Thiriu S, et al. Autoantibodies to myeloperoxidase in Brown Norway Rats treated with mercuric chloride. Laboratory Invest. 1992;67:114–20. [PubMed] [Google Scholar]
- 37.Ringler DH, Dabich L. Hematology and clinical biochemistry. In: Baker HJ, Lindsey JR, Weisbroth SH, editors. The laboratory rat. New York: Academic Press; 1979. pp. 105–21. [Google Scholar]
- 38.Blank M, Tomer Y, Stein M, et al. Immunization with anti-neutrophil cytoplasmic antibody (ANCA) induces the production of mouse ANCA and perivascular lymphocyte infiltration. Clinical & Experimental Immunology. 1995;102:120–30. doi: 10.1111/j.1365-2249.1995.tb06645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomer Y, Gilburd B, Blank M, et al. Characterization of biologically active antineutrophil cytoplasmic antibodies induced in mice. Pathogenic role in experimental vasculitis. Arthritis Rheum. 1995;38:1375–81. doi: 10.1002/art.1780381004. [DOI] [PubMed] [Google Scholar]