

Abstract
The triad of small vessel vasculitides (SVV) comprise Wegener's granulomatosis (WG), microscopic polyangiitis (MPA) and Churg–Strauss syndrome (CS). All three are associated with presence of circulating IgG antineutrophil cytoplasm antibodies (ANCA) which target autoantigens contained, primarily, within neutrophil azurophilic granules. The widely accepted model of pathogenesis suggests that ANCA activate cytokine-primed neutrophils within the microvasculature, leading to by-stander damage to endothelial cells, and rapid escalation of inflammation with recruitment of mononuclear cells. Activation may be initiated, in vitro, by the coligation of the PR3 or MPO antigen, translocated to the cell surface, and FcγRIIa/FcγRIIIb receptors. This suggests that the IgG subclass profile of ANCA and, possibly, its glycosylation status could influence the inflammatory mechanisms activated. The glycosylation status of total IgG isolated from the sera of patients with WG (13), MPA (6) and CSS (1) was determined by analysis of the released oligosaccharides. A deficit in IgG galactosylation is demonstrated for all patient samples, compared to controls. The mean percentage values for the agalactosylated (G0) oligosaccharides were 57% (SD ± 9·71), 47% (SD ± 4·25) and 28% (SD ± 4·09) for WG, MPO and control samples, respectively. The G0 levels for polyclonal IgG isolated from the sera of both WG and MPA patients were significantly increased compared to controls (P < 0·0001). The major glycoform present therefore is agalactosylated (G0) IgG. In previous studies the G0 glycoform of IgG has been shown to bind and activate mannan binding lectin, and hence to activate the complement cascade, and to facilitate mannose receptor binding and the uptake of IgG complexes by macrophages and dendritic cells. Both of these activities could impact on the processing and presentation of self-antigens in autoimmune disease.
Keywords: ANCA-associated vasculitis, Fc glycosylation, G0 IgG, galactosylation
INTRODUCTION
Systemic vasculitis is an inflammatory disease characterized by inflammation within and around blood vessel walls. Certain forms of active vasculitis are strongly correlated with the presence of IgG antineutrophil cytoplasmic antibodies (ANCA) [1], with specificity for constituents of neutrophil azurophilic granules, that are thought to be involved in the pathogenesis of ANCA– associated vasculitis [2,3]. The accepted model of pathogenesis suggests that ANCA activate cytokine-primed neutrophils within the microvasculature, leading to by-stander damage to endothelial cells and a rapid escalation of inflammation with recruitment of mononuclear cells [3–5]. The target antigens for ANCA are primarily proteinase-3 (PR3-ANCA) or myeloperoxidase (MPO-ANCA) [1,6]. The specificity of ANCA can be demonstrated in ELISA and by immunofluorescent staining of alcohol fixed neutrophils; the PR3 antigen giving a diffuse granular staining of the cytoplasm (cANCA) while MPO-ANCA typically gives a perinuclear pattern (pANCA) [1]. ANCA can be shown to activate primed neutrophils, in vitro, with the generation of superoxide anion and release of inflammatory mediators [7–9]. Activation is initiated by the coligation of the PR3 or MPO antigen, translocated to the cell surface, and FcγRIIa/FcγRIIIb receptors [10–12]. Of the four IgG subclasses only IgG1 and IgG3 have been reported to activate both receptors, however, there are reports apparently instancing neutrophil activation by IgG2 and/or IgG4 ANCA [13,14]. In the course of a re-investigation of the role of individual IgG subclasses and their glycosylation status in ANCA activation we generated deglycosylated forms of ANCA expecting to ablate FcγRIIa and FcγRIIIb mediated responses [15] (manuscript in preparation). The oligosaccharide profile of total IgG preparation was analysed prior to and after deglycosylation. The results revealed a very marked deficit in galactosylation of polyclonal IgG isolated from the sera of patient with WG, CSS and MPA.
Human IgG antibodies are multifunctional adapter molecules that bind antigen, through the Fab regions, to form immune complexes that activate multiple effector mechanisms through interaction of the Fc with effector ligands, e.g. FcγRI, FcγRII, FcγRIII, the neonatal Fc receptor (FcγRn), the C1q component of complement, mannan binding lectin (MBL) and the mannose receptor (MR) [15]. Potential pathogens have evolved strategies to frustrate and/or evade the activation of these protective mechanisms by elaborating Fc binding proteins that neutralize or compete for Fc effector sites, e.g. Staphylococcus protein A (SpA), Streptococcus protein G (SpG) and virus encoded FcγR.
The IgG-Fc region is a homodimer comprised of interchain disulphide bonded hinge regions, glycosylated CH2 domains, bearing N-linked oligosaccharide at asparagine 297 (Asn-297) and noncovalently paired CH3 domains [16,17]. Effector mechanisms mediated through FcγRI, FcγRII, FcγRIII and C1q are severely compromised or ablated for aglycosylated or deglycosylated forms of IgG [15]. Multiple non–covalent interactions between the oligosaccharide and the protein result in a reciprocal influences of each on the conformation of the other [16,17]. Variable attachment of outer arm sugars (sialic acid, galactose, fucose and bisecting N-acetylglucosamine) to a heptasaccharide GlcNac2Man3GlcNac2 core structure results in the generation of heterogeneous array of IgG glycoforms. Random association of differently glycosylated heavy chains generates hundreds of different IgG glycoforms. These glycoforms can differ in their efficacy of effector function activation [18,19].
It is established that the profile of oligosaccharides expressed within polyclonal IgG varies with age, gender and over the course of pregnancy [20–22]. The oligosaccharide profile of polyclonal IgG has also been shown to vary for patients with certain inflammatory diseases, e.g. rheumatoid arthritis (RA), systemic lupus erythematosis (SLE) tuberculosis (TB) and Crohn's disease (CD) [23–26] and may be an indicator of disease activity and outcome. In these patients, a significant reduction in the galactosylation of IgG-Fc is observed, with a consequent increase in the level of oligosaccharides devoid of galactose (G0), as opposed to oligosaccharides with one or two galactose residues (G1 & G2 oligosaccharides) [27]. The terminal sugar residues of G0 IgG are N-acetylglucosamine which, when presented in appropriate arrays, may bind and activate the complement cascade through MBL [28] and/or the MR facilitating immune complex uptake, processing and antigen presentation by dendritic cells [29]. In the present report we show that polyclonal IgG isolated from the sera of patients with acute Wegener's granulomatosis or microscopic polyangiitis exhibit a profound disregulation of IgG galactosylation.
PATIENTS AND METHODS
Sample selection
All patients had been admitted with acute disease and were plasmaphoresed within 24–48h of admission. The first litre of plasma exchange fluid was retained, aliquoted and stored at −20°C. A large collection of plasma-derived sera from vasculitis patients, that fulfilled the Chapel Hill consensus conference definitions [30] was screened to select a panel with either PR3-ANCA or MPO-ANCA activity only. The clinical features are shown in Table 1. The panel was comprised of sera from 14 patients with PR3-ANCA, 6 with MPO-ANCA and 6 healthy controls. Thirteen of the patients with PR3-ANCA were diagnosed as suffering from active Wegener's granulomatosis (6 female, 7 male, mean female age 66·8 years, mean male age 57·9 years), the remaining PR3-ANCA patient was diagnosed with Churg– Strauss syndrome (female, age 35 years). All patients with MPO-ANCA were diagnosed with microscopic polyangiitis (3 females and 3 males, mean female age 68·5 years, mean male age). The controls group comprised 2 females and 4 males (Mean male age 38·8 years, Mean female age 63·5 years).
Table 1.
Clinical data of ANCA patient's studied
| Patient | Age | Sex | Diagnosis | URT (inc Ear) | Lung | Kidney | Eye | Heart | Skin | Joints | PNS | CNS | Gut | Unwell |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 72 | F | WG | P | P | P | + | |||||||
| 2 | 77 | F | WG | P | P | P | P | + | ||||||
| 3 | 73 | M | WG | P | P | P | + | |||||||
| 4 | 49 | M | WG | P | P | P | P | + | ||||||
| 5 | 84 | F | WG | P | P | + | ||||||||
| 6 | 66 | F | WG | P | P | P | + | |||||||
| 7 | 41 | F | WG | P | P | P | P | P | + | |||||
| 8 | 61 | F | WG | P | P | P | P | P | + | |||||
| 9 | 73 | M | WG | P | P | + | ||||||||
| 10 | 61 | M | WG | P | P | P | P | + | ||||||
| 11 | 55 | M | WG | P | P | P | + | |||||||
| 12 | 62 | M | WG | P | P | P | P | + | ||||||
| 13 | 32 | M | WG | P | P | P | P | + | ||||||
| 14 | 35 | F | CSS | P | P | P | P | + | ||||||
| 15 | 74 | M | MPA | P | P | + | ||||||||
| 16 | 68 | F | MPA | P | P | + | ||||||||
| 17 | 58 | F | MPA | P | P | + | ||||||||
| 18 | 63 | M | MPA | P | P | + | ||||||||
| 19 | 17 | F | MPA | P | P | P | P | + | ||||||
| 20 | 50 | M |
URT, Upper respiratory tract; PNS, Peripheral nervous system; CNS, Central nervous system; P, organs affected at presentation; + additional symptoms, e.g. general malaise
Indirect immunofluorescence (IIF)
Ethanol-fixed neutrophil slides (Binding Site, Birmingham, UK) were incubated with test sample, diluted 1 in 20 with sterile PBS, in a humidified environment at room temperature for 30min Following 3 × 5 minute washes with PBS, the slides were air dried and FITC conjugated antihuman IgG,A,M antibody (Binding Site), diluted 1 in 20 with PBS, was added. The slides were incubated in a humidified environment for 30min, followed by 3 × 5 minute washes with PBS, allowed to air dry before developing with 0·022m 1,4-Diazabicyclo [2,2,2]octane (DABCO)-glycerol, mounted using coverslips and viewed using fluorescence microscopy.
Anti-PR3 and anti-MPO ELISA
Samples were tested for PR3-ANCA and MPO-ANCA using Bind-azyme anti-PR3 and anti-MPO ELISA kits (Binding Site, Birmingham, UK). The ELISA was performed according to the manufacturer instructions. Briefly, standards, controls and samples, diluted 1:50 with PBS-0·05%Tween, were added to wells of the coated plates. Following incubation at room temperature (30min) the plates were washed 3 times with 300μl of 1 × kit wash buffer and 100μl of antihuman IgG conjugate was added and incubated for 30min at room temperature. The plates were washed again 3 times with 300μl of 1 × kit wash buffer and 100μl of 3,3′,5,5′ tetramethylbenzidine (TMB) substrate added per well. The plate was incubated at room temperature for 30min and 100μl of 3M phosphoric acid stopping solution added to each well. The optical density of the plate was read within 30min at 450nm.
IgG purification
Frozen serum was thawed, diluted 1:1 with PBS and 0·2μm filtered. Diluted serum was loaded onto a 2-ml protein G column (Amersham Biotech, Bucks, UK). The column was subsequently washed with a minimum of 10 column volumes of PBS. Bound IgG was eluted using 0·1m glycine/HCl, pH 2·6. The eluted fractions were collected into tubes containing 1m Tris Base, pH 9·0 (100μl Tris per 1ml of eluent). Eluted IgG was dialysed against PBS for a minimum of 12h at 4°C. The resulting IgG solution was concentrated using Vivaspin concentrators with a 10kD cut off and the protein concentration determined by spectroscopy at 280nm. The IgG was passed through a 0·2-μm filter into a sterile container and stored at 4°C. The total amount of IgG loaded on the column and that recovered was determined in the routine clinical immunology laboratory. All yields were >90%.
Carbohydrate analysis
Oligosaccharide analyses were conducted as described previously [31]; 10–15nmol of each IgG preparation was used for isolation of oligosaccharides. Following digestion with chymotrypsin and trypsin (1% w/w), the glycopeptide fraction was digested with 100–200 μU of glycoamidase (almond). The oligosaccharide was purified by passage over 1-ml columns of cation (Dowex 50WX8) and anion (Dowex 1) exchangers. The filtrate was evaporated to dryness, and reductively aminated with a fluorescent reagent, 2-aminopyridine, and sodium cyanoborohydride [32]. Pyridylaminated oligosaccharides were purified by gel filtration on a Sephadex G-15 column with 10mm of ammonium bicarbonate. The pyridylaminated oligosaccharide mixture was divided into two aliquots. The first aliquot was analysed for sialic acid content by HPLC on a DEAE-SPW TSK gel column (7·5 × 75 mm; Tosoh Corporation, Tokyo, Japan) [31]. Each oligosaccharide fraction resolved by the DEAE column was evaporated in vacuo without desalting and subjected to further HPLC on an ODS-silica column (Shimpack CLC-ODS, 6 × 150 mm: Shimadzu, Kyoto, Japan). The second aliquot of pyridylaminated oligosaccharide mixture was treated with 20 –40 mU of sialidase from Arthrobucter ureaficiens (Nacalai Tesque, Kyoto, Japan) and applied directly to the ODS-silica column. All subsequent analytic procedures, including chromatographic conditions, have been reported previously [33].
RESULTS
IIF and ELISA
Indirect immunofluorescence results were reported as cANCA, pANCA or negative. Following analysis by IIF, antigen specificity of the ANCA was confirmed by ELISA for anti-PR3 and anti-MPO antibodies. Of the 20 patients tested, 14 were PR3-ANCA positive and 6 were MPO-ANCA positive. The sera in the panel were also tested for the presence of antielastase and antilactoferrin antibodies, by Western blot analysis, and shown to be negative.
IgG N-glycan analysis
Examples of the neutral oligosaccharide profiles obtained for normal IgG, and IgG isolated from PR3-ANCA and MPO-ANCA positive sera are shown in Fig. 1 and structures for the neutral and sialylated oligosaccharides are shown in Fig. 2. The profile of oligosaccharides released from all patient derived IgGs was dominated by agalactosylated structures. The quantitative values for oligosaccharides released from the IgG of WG and CSS patients are presented in Table 2. The neutral oligosaccharides released for the IgGs of patients with MPA are presented in Table 3 and neutral oligosaccharides released for the IgGs of the controls are presented in Table 4. The G0 oligosaccharide values for all patients and controls are summarized in Fig. 3. and a summary of the occurrence of neutral (G0, G1 & G2), mono- and di-sialylated oligosaccharides isolated from polyclonal IgG of sera from patients with WG, MPA and controls is presented in Table 5. The G0 content of IgG isolated from Wegener's granulomatosis PR3-ANCA positive sera and Microscopic polyangiitis MPO-ANCA positive sera were significantly increased compared to controls; WG, t = 7·8, P < 0·0001; MPA, t = 8·2, P < 0·0001. The 95% confidence intervals were 22–38% higher for PR3-ANCA positive polyclonal IgG and 14–25% higher for MPO-ANCA positive polyclonal IgG compared to controls.
Fig. 1.
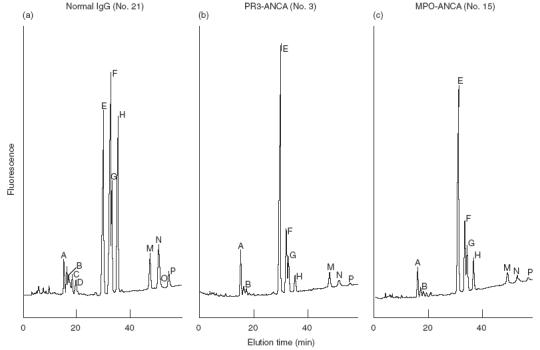
Elution profiles for (a) Normal IgG, (b) PR3-ANCA and (c) MPO-ANCA.
Fig. 2.
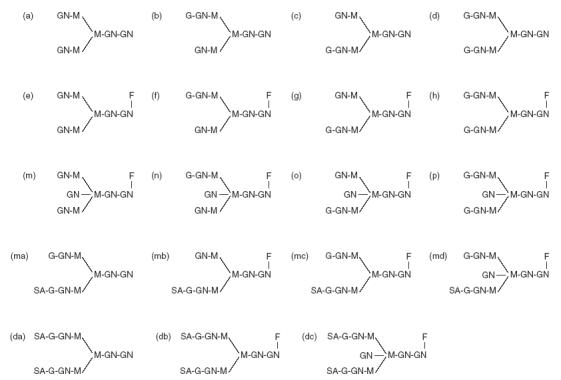
Structures of neutral (a–p), monosialylated (ma–md) and disialylated (da–dc) oligosaccharides released from IgG.
Table 2.
Glycoform content of PR3-ANCA IgG samples
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0·4 | 0·2 | 0·5 | 0·6 | 0·5 | 0·6 | 0·5 | 0·6 | 0·4 | 0·4 | 0·3 | 0·5 | 0·3 | 0·3 |
| B | 0·1 | 0·1 | 0·1 | 0·1 | 0·1 | 0·2 | 0·1 | 0·2 | 0·2 | 0·1 | 0·1 | 0·1 | 0·1 | 0·1 |
| C | ||||||||||||||
| D | ||||||||||||||
| E | 70·2 | 38·8 | 49·7 | 52·9 | 56·7 | 57·8 | 48·9 | 45·3 | 54·3 | 47·3 | 42·1 | 63·0 | 47·2 | 35·4 |
| F | 8·6 | 13·4 | 13·9 | 13·5 | 11·1 | 14·5 | 15·5 | 15·2 | 17·9 | 15·0 | 13·9 | 10·9 | 15·5 | 19·0 |
| G | 5·8 | 8·1 | 7·4 | 8·9 | 7·8 | 8·3 | 6·9 | 7·7 | 6·8 | 11·4 | 10·2 | 6·8 | 11·0 | 13·0 |
| H | 2·6 | 6·3 | 4·4 | 3·8 | 3·6 | 3·7 | 5·2 | 5·5 | 4·4 | 5·3 | 5·5 | 2·4 | 7·4 | 10·1 |
| M | 4·9 | 5·4 | 4·6 | 3·1 | 6·6 | 4·6 | 6·3 | 3·0 | 2·8 | 3·1 | 5·8 | 4·4 | 1·7 | 3·7 |
| N + O | 1·9 | 3·9 | 2·3 | 2·2 | 2·9 | 2·3 | 3·9 | 2·2 | 2·0 | 1·9 | 3·4 | 1·8 | 1·6 | 3·4 |
| P | 0·7 | 1·8 | 0·9 | 0·7 | 0·6 | 0·5 | 1·1 | 0·6 | 0·9 | 0·9 | 0·8 | 0·5 | 0·9 | 1·3 |
| Ma | 0·1 | 0·4 | 0·7 | 0·5 | 0·4 | 0·1 | 0·3 | 0·6 | 0·3 | 0·5 | 0·4 | 0·3 | 0·4 | 0·4 |
| Mb | 1·0 | 2·2 | 2·7 | 3·2 | 2·4 | 1·7 | 1·7 | 3·0 | 2·2 | 3·4 | 4·0 | 2·9 | 3·3 | 2·3 |
| Mc | 1·4 | 4·5 | 4·3 | 3·9 | 2·8 | 2·3 | 3·5 | 6·6 | 3·9 | 4·5 | 5·0 | 2·7 | 5·7 | 4·5 |
| Md | 0·5 | 5·1 | 1·3 | 1·7 | 1·3 | 0·1 | 1·8 | 2·4 | 1·4 | 1·5 | 2·5 | 1·0 | 1·1 | 1·5 |
| Da | 0·5 | 1·8 | 4·2 | 2·0 | 1·4 | 1·1 | 1·0 | 1·7 | 0·6 | 1·6 | 1·2 | 0·9 | 1·0 | 1·6 |
| Db | 0·9 | 3·6 | 1·9 | 1·5 | 0·8 | 1·2 | 1·7 | 3·3 | 1·2 | 1·8 | 2·1 | 0·9 | 1·8 | 2·0 |
| Dc | 0·4 | 4·4 | 1·1 | 1·4 | 1·0 | 1·0 | 1·6 | 2·1 | 0·7 | 1·3 | 2·7 | 0·9 | 1·0 | 1·4 |
| Total | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 |
Table 3.
Glycoform content of MPO-ANCA IgG samples
| 15 | 16 | 17 | 18 | 19 | 20 | |
|---|---|---|---|---|---|---|
| A | 0·5 | 0·4 | 0·5 | 0·4 | 0·3 | 0·3 |
| B | 0·2 | 0·1 | 0·1 | 0·2 | 0·1 | 0·1 |
| C | ||||||
| D | ||||||
| E | 39·5 | 48·2 | 40·0 | 38·0 | 41·4 | 42·7 |
| F | 17·9 | 16·6 | 14·2 | 17·9 | 14·0 | 15·6 |
| G | 8·5 | 10·6 | 7·5 | 9·1 | 9·1 | 8·9 |
| H | 6·6 | 6·2 | 5·5 | 8·5 | 6·5 | 5·2 |
| M | 5·8 | 5·1 | 3·8 | 3·1 | 2·7 | 5·5 |
| N + O | 3·5 | 2·8 | 3·1 | 2·7 | 2·6 | 3·7 |
| P | 1·0 | 0·7 | 1·0 | 0·9 | 0·7 | 0·8 |
| Ma | 0·5 | 0·2 | 1·0 | 0·2 | 0·7 | 0·4 |
| Mb | 2·6 | 2·1 | 2·5 | 2·5 | 3·0 | 2·6 |
| Mc | 4·5 | 3·7 | 6·4 | 6·7 | 8·0 | 4·4 |
| Md | 2·3 | 0·9 | 2·4 | 0·3 | 1·9 | 2·5 |
| Da | 1·7 | 0·7 | 5·8 | 3·3 | 2·8 | 2·0 |
| Db | 2·5 | 1·0 | 3·7 | 3·5 | 4·8 | 2·6 |
| Dc | 2·4 | 0·7 | 2·5 | 2·7 | 1·4 | 2·7 |
| Total | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 |
Table 4.
Glycoform content of control IgG samples
| 21 | 22 | 23 | 24 | 25 | 26 | |
|---|---|---|---|---|---|---|
| A | 0·5 | 0·6 | 0·5 | 0·4 | 0·4 | 0·5 |
| B | 0·5 | 0·3 | 0·4 | 0·4 | 0·3 | 0·5 |
| C | 0·2 | 0·3 | 0·2 | 0·3 | 0·4 | 0·3 |
| D | 0·7 | 1·0 | 0·4 | 1·6 | 0·3 | |
| E | 24·2 | 28·4 | 22·7 | 19·8 | 19·7 | 20·3 |
| F | 22·4 | 17·3 | 20·4 | 20·6 | 20·4 | 19·4 |
| G | 7·2 | 14·3 | 11·1 | 13·1 | 10·6 | 12·8 |
| H | 12·7 | 12·8 | 13·8 | 20·3 | 16·0 | 14·2 |
| M | 5·8 | 3·6 | 3·6 | 2·5 | 4·0 | 2·5 |
| N + O | 8·2 | 3·4 | 5·4 | 3·0 | 5·2 | 3·6 |
| P | 2·1 | 2·0 | 1·8 | 1·1 | 1·8 | 1·3 |
| Ma | 0·2 | 0·3 | 0·6 | 0·4 | 0·6 | 0·8 |
| Mb | 1·8 | 3·4 | 2·8 | 2·8 | 2·8 | 4·0 |
| Mc | 6·1 | 7·6 | 8·1 | 9·5 | 8·8 | 8·7 |
| Md | 2·1 | 1·8 | 1·1 | 2·4 | 2·1 | |
| Da | 0·8 | 2·0 | 1·5 | 0·8 | 1·0 | 3·9 |
| Db | 1·4 | 2·5 | 2·1 | 2·3 | 2·0 | 2·3 |
| Dc | 3·1 | 1·2 | 2·2 | 1·2 | 2·0 | 2·5 |
| Total | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 | 100·0 |
Fig. 3.
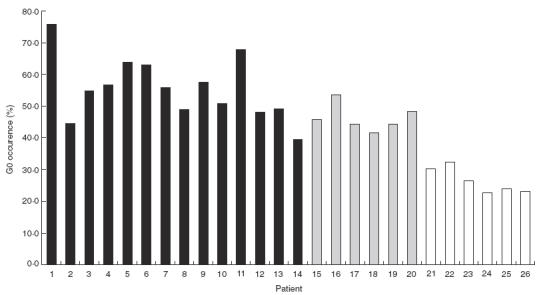
Occurence of the G0 glycoform in ANCA patient's and control IgG. Results are expressed as the percentage of the G0 glycoform present in patients and control IgG. ▪ patients with PR3-ANCA diagnosed as with Wegener's granulomatosis or Churg–Strauss syndrome (patient 14);  patients with MPO-ANCA diagnosed with Microscopic polyangiitis; □ the healthy controls.
patients with MPO-ANCA diagnosed with Microscopic polyangiitis; □ the healthy controls.
Table 5.
Occurrence of neutral (G0, G1 & G2), mono- and disialylated-oligosaccharides released from IgG isolated from sera of patients with Wegener's granulomatosis, Microscopic polyangiitis and controls
| G0 | G1 | G2 | Monosialylated | Disialylated | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean (%) | Range (%) | SD | Mean (%) | Range (%) | Mean (%) | Range (%) | Mean (%) | Range (%) | Mean (%) | Range (%) | |
| WG | 57 | 44–76 | 9·7 | 25 | 16–28 | 6 | 3–8 | 9 | 3–13 | 5 | 2–10 |
| MPA | 47 | 42–54 | 4·3 | 28 | 25–30 | 7 | 6–9 | 10 | 7–19 | 8 | 2–12 |
| Control | 28 | 23–33 | 4·1 | 37 | 35–38 | 17 | 15–22 | 13 | 10–16 | 6 | 4–9 |
DISCUSSION
Anti-neutrophil cytoplasmic antibodies (ANCA) associated with Wegener's granulomatosis and microscopic polyangiitis are characteristically of the IgG isotype; although the presence of IgM and IgA ANCA has been reported [34,35]. The accepted model of pathogenesis suggests that neutrophil activation by ANCA results from coligation of the PR3 or MPO antigen and FcγRIIa/FcγRIIIb receptors. It is widely held that these receptors are activated only by immune complexes with human IgG1 or IgG3 subclass antibodies [15], however, claims for a pathogenic role for IgG2 and/or IgG4 ANCA have been made [13].
It has been shown that F(ab′)2 fragments of ANCA that cannot coligate FcγRIIa and/or FcγRIIIb are not activating. From previous studies we anticipated that deglycosylated ANCA would also be nonactivating since FcγRII and FcγRIII binding is ablated for deglycosylated IgG1 and IgG3 (15, 18). To test this hypothesis we generated deglycosylated IgG isolated from the serum of a WG patient. To confirm deglycosylation we analysed the oligosaccharide profiles of the IgG before and after the deglycosylation procedure. Unexpectedly, the neutral oligosaccharides released from the native IgG were predominantly agalactosylated (G0). To determine whether this was a general phenomenon for polyclonal IgG isolated from ANCA positive sera we isolated IgG from the serum of 13 patients with WG, 6 with MPA, 1 with Churg–Strauss syndrome and 6 healthy controls. A gross disregulation of galactosylation is evident for all patient samples, as reflected in the G0 values, Tables 2, 3 and 5 and Fig. 3. It is of interest to note that patients 19 and 13 were 17 and 32 years of age, respectively, at the time of plasmaphoresis; thus, there is no evidence of an age related difference in G0 values for the patient panel. The high G0 values are reflected in low G1 (B + F + G + N + O) and G2 (D + H + P) values for the WG and MPA samples, compared to controls, Table 5. The G1 and G2 values for the Churg-Strauss sample are within the normal range. No obvious difference in the levels of mono- and di-sialylated oligosaccharides released from polyclonal IgG is evident for patients and controls.
Previous studies have shown that c. 10–20% of human IgG molecules may bear oligosaccharides attached within the variable regions of either the heavy or light chains. These oligosaccharides are seen to be more accessible to glycosyltransferases than are the Fc oligosaccharides and consequently are more highly galactosylated and sialylated. It is reasonable to presume that the monosialylated and disialylated oligosaccharides released from these polyclonal IgG samples were attached to variable regions. Similarly, variable region glycosylation would account for a significant proportion of the G1 and G2 oligosaccharides released. The corollary of this rationalization is that for the patient samples an overwhelming majority of IgG-Fc regions bear G0 oligosaccharides on each heavy chain.
The finding of a deficit in galactosylation of polyclonal IgG isolated from the sera of WG and MPA patients is similar to that observed in RA, SLE and TB. In attempting to understand possible impacts on PR3-ANCA and MPO-ANCA neutrophil activation, we may presume that the oligosaccharide profile of the specific antibody populations are similar to those observed for total IgG. Given this caveat a number of interesting possibilities may be considered. IgG glycoforms bearing G0 oligosaccharides present terminal N-acetylglucosamine (GlcNAc) residues to the environment. It has been demonstrated that arrays of GlcNAc presented by human IgG can bind MBL and activate the lectin pathway of complement [28]. It may be anticipated that antigen/antibody complexes generated in vivo may also have potential to bind and activate MBL – possibly dependent on the epitope density, antigen/antibody ratio, etc., resulting in the generation of an inappropriate pro-inflammatory response. Interestingly, Garred et al.[36] suggested that allelic variants of MBL may lead to a reduced clearance of G0 IgG complexes and play a significant role in the onset of early RA. The mannose receptor has also been shown to bind arrays of GlcNAc presented by IgG antibody immune complexes, resulting in their uptake by antigen presenting macrophages and dendritic cells [29]. Interaction of IgG immune complexes with both MBL and MR could provide a route for antigen driven maturation of an immune response to self-antigens.
In a mouse model of RA G0 anticollagen II antibodies have been reported to be more pathogenic than the fully galactosylated (G2) antibodies [40]. It has been shown that removal of terminal galactose from human IgG increased the uptake of soluble IgG mediated by the mannose receptor on macrophages and dendritic cells [29]. It was suggested that this could provide a novel pathway by which Abs or Ag-Ab complexes can be taken into dendritic cells and macrophages, and potentially generate epitopes recognized by T cells. This could have a particular relevance for autoimmune disorders characterized by high levels of G0 IgG.
In the present study the polyclonal IgG samples analysed were all from patients with acute disease. As in other diseases cited therefore disregulated galactosylation is coincident with profound inflammation. It remains to be seen whether it initiates an autoimmune response or exacerbates an existing one. Evidence bearing on this question could come from analysis of IgG isolated from the sera of patient at first presentation with symptoms that may indicate the onset of vasculitis together with subsequent follow-up. It would also be of interest to analyse samples from patients who have achieved remission. Patients with rheumatoid arthritis experience remission of disease symptoms during pregnancy and a coincident increase in IgG galactosylation is observed [20–22]. It would be of obvious interest to investigate changes that may occur for patients with vasculitis.
Acknowledgments
A. Ben-Smith and M. Holland thank the Medical Research Council for financial support. Dr Peter Nightingale (Wolfson Computer Centre) is thanked for assistance with statistical analysis.
REFERENCES
- 1.Savige J, Gillis D, Benson E, et al. International consensus statement on testing and reporting of anti-neutrophil cytoplasmic antibodies (ANCA) Am J Clin Path. 1999;111:507–13. doi: 10.1093/ajcp/111.4.507. [DOI] [PubMed] [Google Scholar]
- 2.Jennette JC, Falk RJ. Pathogenic potential of anti-neutrophil cytoplasmic autoantibodies. Laboratory Invest. 1994;70:135–7. [PubMed] [Google Scholar]
- 3.Harper L, Savage COS. Pathogenesis of ANCA-associated systemic vasculitis. J Path. 2000;190:349–59. doi: 10.1002/(SICI)1096-9896(200002)190:3<349::AID-PATH524>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 4.Harper L, Ren Y, Savill J, Adu D, Savage COS. Antineutrophil cytoplasmic antibodies induce reactive oxygen-dependent dysregulation of primed neutrophil apoposis and clearance by macrophages. Am J Path. 2000;157:211–20. doi: 10.1016/S0002-9440(10)64532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hewins P, Tervaert JWC, Savage COS, Kallenberg CGM. Is Wegeners granulomatosis an autoimmune disease? Current Op Rheum. 2000;12:3–10. doi: 10.1097/00002281-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Goldschmeding R, van der Schoot CE, Huinink DT, Hack CE, van den Ende ME, Kallenberg CGM, von dem Borne AEGKr. Wegener's granulomatosis autoantibodies identify a novel diisopropylfluorophosphate binding-protein in the lysosomes of normal human-neutrophils. J Clin Invest. 1989;84:1577–87. doi: 10.1172/JCI114335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic antibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. PNAS USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keogan MT, Esnault VLM, Green AJ, Lockwood CM, Brown DL. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clinical & Experimental Immunology. 1992;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release in human neutrophils. J Am Soc Neph. 1996;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 10.Csernok E, Ernst M, Schmitt W, Baintoni DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clinical & Experimental Immunology. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radford DJ, Lord JM, Savage COS. The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm antibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clinical & Experimental Immunology. 1999;118:171–9. doi: 10.1046/j.1365-2249.1999.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Smith A, Dove SK, Martin A, Wakelam MJO, Savage COS. Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signalling cascades: comparison with conventional Fcγ receptor ligation. Blood. 2001;98:1448–55. doi: 10.1182/blood.v98.5.1448. [DOI] [PubMed] [Google Scholar]
- 13.Jayne DRW, Weetman AP, Lockwood CM. IgG subclass distribution of autoantibodies to neutrophil cytoplasmic antigens in systemic vasculitis. Clinical & Experimental Immunology. 1991;84:476–81. [PMC free article] [PubMed] [Google Scholar]
- 14.Mellbye OJ, Mollnee TE, Sleittevoll Steen L. IgG subclass distribution and complement activation ability of autoantibodies to neutrophil cytoplasmic antigens (ANCA) Clin Immunol Immunopath. 1994;70:32–9. doi: 10.1006/clin.1994.1007. [DOI] [PubMed] [Google Scholar]
- 15.Jefferis R, Lund J, Pound JD. IgG-Fc mediated effector functions. molecular definition of interaction sites for effector ligand and the role of glycosylation. Immunol Rev. 1998;163:59–76. doi: 10.1111/j.1600-065x.1998.tb01188.x. [DOI] [PubMed] [Google Scholar]
- 16.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from staphylococcus aureus at 2·9- and 2·8-Å resolution. Biochem. 1981;20:2361–70. [PubMed] [Google Scholar]
- 17.Sonderman P, Huber R, Oosthuizen V, Jacob U. The 3·2-Å crystal structure of the human IgG1 Fc fragment-FcgRIII complex. Nature. 2000;406:267–73. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 18.Mimura Y, Sondermann P, Ghirlando R, Lund J, Young SP, Goodall M, Jefferis R. The role of oligosaccharide residues of IgG1-Fc in FcγIIb binding. J Biol Chem. 2001;276:45539–47. doi: 10.1074/jbc.M107478200. [DOI] [PubMed] [Google Scholar]
- 19.Davies J, Jiang L, Pen L-Z, LaBarre MJ, Anderson D, Reff M. Expression of GTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies of altered glycoforms leads to an increase in ADCC thro′ higher affinity for FcRIII. Biotechn Bioeng. 2001;74:288–94. [PubMed] [Google Scholar]
- 20.Yamada E, Tsukamoto Y, Sasaki R, Yagyu K, Takahashi N. Structural changes of immunoglobulin G oligosaccharides with age in healthy human serum. Glycoconjugate J. 1997;14:401–5. doi: 10.1023/a:1018582930906. [DOI] [PubMed] [Google Scholar]
- 21.Kibe T, Fujimoto S, Ishida C, Togari H, Okada S, Nakagawa H, Tsukamoto Y, Takahashi N. Glycosylation and placental transport of immunoglobulin G. J Clin Biochem Nutr. 1996;21:57–63. [Google Scholar]
- 22.Alavi A, Arden N, Spector TD, Axford JS. Immunoglobulin G glycosylation and clinical outcome in rheumatoid arthritis during pregnancy. J Rheum. 2000;27:1379–85. [PubMed] [Google Scholar]
- 23.Rademacher TW, Parekh RB, Dwek RA. Glycobiology. Ann Rev Biochem. 1988;57:785–838. doi: 10.1146/annurev.bi.57.070188.004033. [DOI] [PubMed] [Google Scholar]
- 24.Axford J. Glycobiology and medicine: an introduction. J Roy Soc Med. 1997;90:260–4. doi: 10.1177/014107689709000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–6. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 26.Watson M, Rudd PM, Bland M, Dwek RA, Axford JS. Sugar printing rheumatic diseases. Arth Rheum. 1999;42:1682–90. doi: 10.1002/1529-0131(199908)42:8<1682::AID-ANR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Parekh RB, Dwek RA, Sutton BJ, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature. 1985;316:452–7. doi: 10.1038/316452a0. [DOI] [PubMed] [Google Scholar]
- 28.Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med. 1995;1:237–43. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- 29.Dong X, Storkus WJ, Salter RD. Binding and uptake of agalactosyl IgG by mannose receptor on macrophages and dendritic cells. J Immunol. 1999;163:5427–34. [PubMed] [Google Scholar]
- 30.Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Arth Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi N, Nakagawa H, Fujikawa K, Kawamura Y, Tomiya N. Three-dimensional elution mapping of pyridylaminated N-linked neutral and sialyl oligosaccharides. Ann Bioch. 1995;226:139–46. doi: 10.1006/abio.1995.1201. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto S, Hase S, Fukuda S, Sano O, Ikenaka T. Structures of the sugar chains of interferon-y produced by human myelomonocyte cell line HBL-38. J Biochem. 1989;105:1034–9. doi: 10.1093/oxfordjournals.jbchem.a122703. [DOI] [PubMed] [Google Scholar]
- 33.Tomiya N, Awaya J, Kurono M, Endo S, Arata Y, Takahashi N. Analyses of N-linked oligosaccharides using a two-dimensional mapping technique. Anal Biochem. 1988;171:73–90. doi: 10.1016/0003-2697(88)90126-1. [DOI] [PubMed] [Google Scholar]
- 34.Jayne DRW, Jones SJ, Severn A, Shaunak S, Murphy J, Lockwood CM. Severe pulmonary haemorrhage and systemic vasculitis in association with circulating anti-neutrophil antibodies of IgM class only. Clin Neph. 1989;32:101–6. [PubMed] [Google Scholar]
- 35.Segelmark M, Wieslander J. IgG subclasses of anti-neutrophil cytoplasm autoantibodies (ANCA) Neph Dial Transplant. 1993;8:696–702. doi: 10.1093/ndt/8.8.696. [DOI] [PubMed] [Google Scholar]
- 36.Garred P, Madsen HO, Marquart H, et al. Two edged role of mannose binding lectin in rheumatoid arthritis: a cross sectional study. J Rheumatol. 2000;27:26–34. [PubMed] [Google Scholar]
- 37.Cockwell P, Savage COS. Role of leukocytes in the immunopathogenesis of ANCA-associated glomerulonephritis. Nephron. 2000;85:287–306. doi: 10.1159/000045679. [DOI] [PubMed] [Google Scholar]
- 38.Kallenberg CGM, Brouwer E, Weening JJ, Tervaert JWC. Anti-neutrophil cytoplasmic antibodies. current diagnostic and pathophysiological potential. Kidney Int. 1994;46:1–15. doi: 10.1038/ki.1994.239. [DOI] [PubMed] [Google Scholar]
- 39.Axford J, Mackenzie L, Lydyard PM, Hay FC, Isenberg DA, Roitt IM. Reduced B-cell glycosyltransferase activity in rheumatoid arthritis. Lancet. 1987;2:1486–8. doi: 10.1016/s0140-6736(87)92621-3. [DOI] [PubMed] [Google Scholar]
- 40.Rademacher TW, Williams P, Dwek RA. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc Natl Acad Sci USA. 1994;91:6123–7. doi: 10.1073/pnas.91.13.6123. [DOI] [PMC free article] [PubMed] [Google Scholar]