

Abstract
β7 Integrins have been shown to have an important role in the localization of T cells to the intestine. Utilizing two different experimental mouse models of inflammatory bowel disease (IBD), this study was undertaken to determine if β7 integrin expression is critical for T cell localization to the intestine and colitis pathogenesis. Transfer of CD4+ CD45RBhigh cells into immunodeficient mice results in colitis. To examine the role of β7 integrins, donor cells were obtained from β7 integrin gene-deficient animals and disease induction was examined following transfer into severe combined immunodeficiency (SCID) mice. Additionally, β7 integrin gene-deficient animals were crossed to IL-2-deficient mice and the onset of spontaneous colitis that normally occurs in IL-2-deficient animals was examined. No differences in the onset or severity of spontaneous colitis was noted in animals that were deficient in both β7 integrin and IL-2. In contrast, the onset of colitis in recipients of T cells from β7 integrin-deficient donors was delayed significantly. In mice receiving β7 integrin negative cells, the initial lack of colitis appeared to correlate with fewer numbers of CD3+β7 integrin –/– donor lymphocytes present in the host colon. The eventual development of disease, however, was associated with increased numbers of donor β7 integrin –/– lymphocytes. These results show that β7 integrin expression is not absolutely required for T cell localization to the intestine and colitis pathogenesis.
Keywords: β7 integration, colitis, T cells, trafficking
INTRODUCTION
Enhanced trafficking of lymphocytes to the intestine, or expansion of cells in that site, appears to be a key feature of inflammatory disorders involving the gastrointestinal tract such as Crohn's disease and ulcerative colitis [1,2]. The β7 integrin family of molecules plays a major role in the homing and retention of T cells within the gastrointestinal mucosa [3]. The β7 chain associates with the α4 subunit to form the α4β7 integrin (LPAM-1). The α4β7 integrin binds to MAdCAM-1, the mucosal vascular addressin present on high endothelial venules. Expression of α4β7 by activated T cells is thought to facilitate lymphocyte homing to Peyer's patches and to the lamina propria [4,5]. The β7 integrin chain also combines with the αΕ chain to form the αΕβ7 integrin. This integrin is present on intestinal intraepithelial lymphocytes (IEL), and is believed to mediate the localization or retention of IEL to the basolateral aspect of intestinal epithelial cells by binding to E-cadherin [6,7]. The critical role that β7 integrins have in mediating lymphocyte localization within the gastrointestinal lymphoid tissue (GALT) has been demonstrated by the generation of β7 integrin gene-deficient mice [8]. These mice have a normal peripheral immune compartment in the spleen and lymph nodes, but have a significantly reduced number of lymphocytes in the Peyer's patches as well as in the intraepithelial and lamina propria compartments of the intestine [8]. Similarly, in animals deficient in the αE chain of the αEβ7 integrin, the number of lymphocytes is reduced in both the intraepithelial and lamina propria compartments of the intestine [7]. The near absence of lymphocytes within the GALT of β7 integrin-deficient animals emphasizes the importance of this molecule for localization of lymphocytes in the gut mucosa under normal non-pathological conditions. However, the extent to which β7 integrins participate in T cell localization during inflammatory situations is less known. Studies using monoclonal antibodies directed against the α4 integrin, MAdCAM-1, or αΕβ7 molecules suggest that interruption of the interaction between mucosal homing receptors with their ligands can attenuate the development of disease in experimental models of intestinal inflammation [9–11]. Because results from antibody studies have some limitations, we utilized an alternate strategy to investigate whether β7 integrin expression by T cells is necessary for the development of experimental inflammatory bowel disease (IBD). We took advantage of two immunologically mediated mouse models of colitis that differ in the mode of induction of disease. One model involves the transfer of sorted CD4+ T cells to immunodeficient SCID mice [12–14]. In this system, peripheral CD4+ cells from non-immunized healthy mice are purified on the basis of CD45RB levels prior to adoptive transfer. The animals that receive the CD4+ CD45RBhigh (CD45RBhigh) T cells develop colitis and weight loss over a 6–8-week period. Donor CD4+ cells that are found in the intestines of recipient animals have revealed that they express high levels of β7 associated integrins such as α4β7 and αΕβ7 [15]. Because only a small percentage of donor CD4+ cells obtained from the spleen express β7 integrins, the increased levels found on mucosal T cells recovered from the intestine of recipient animals suggests that β7 integrins are important for T cell engraftment within the gut in this transfer system [15].
The other model of IBD we utilized takes advantage of the spontaneous colitis that develops in IL-2 deficient mice. IL-2 is secreted by activated T cells and in an autocrine fashion supports T cell proliferation as well as the differentiation of B cells, macrophages and NK cells. IL-2-deficient mice start developing IBD at the age of 6 weeks [16]. The IBD is restricted to the colon, which exhibits almost continuous inflammation. The pathogenesis of IBD in IL-2-deficient mice involves thymic changes by which CD4+ T cells are abnormally directed by IL-12 toward Th1-type T cells that can induce colitis [17]. To investigate how the absence of β7 integrin expression by T cells affects disease pathogenesis in these experimental systems of IBD, we carried out a series of studies. First, purified CD4+ lymphocytes obtained from β7 integrin-deficient animals were transferred to SCID mice. We determined if there was a difference in disease outcome in comparison to recipients of purified CD4+ cells from β7 integrin-positive mice. Secondly, β7 integrin knockout mice were crossed with IL-2 deficient mice to determine if animals that lack both IL-2 and β7 integrin still developed IBD.
MATERIALS AND METHODS
Animals and maintenance
β7 Integrin negative mice were provided by Dr Norbert Wagner, Institute for Genetics, University of Cologne, Germany. They were rederived at the University of California, Los Angeles, transgenic mouse facility by caesarean section and maintained at the UCLA vivarium in ventilated cage racks (Thoren Caging Systems, Hazelton, PA, USA) under specific pathogen-free (SPF) conditions. C57BL/6 (B6) and B6 SCID mice of both sexes were initially obtained from the Jackson laboratories, and bred within the UCLA facility. They were maintained in ventilated cage racks, and given autoclaved food and acidified water. Donor mice were always female and were used between 6 and 12 weeks of age. Recipient mice were of both sexes and were used between 8 and 12 weeks. IL-2-deficient mice were obtained from Horak [18] and crossed to the β7 integrin-deficient mice. The genotype for β7 as well as for IL-2 was determined by PCR. These mice were kept in a conventional animal facility at the University of Cologne. All experiments involving animals were performed in accordance with federal and institutional regulations.
Preparation and transfer of T-cell populations
Spleens were removed from donor mice and teased into single cell suspensions in PBS. Erythrocytes were removed by hypotonic lysis. After washing, the cells were stained with FITC-conjugated anti-CD4 clone RM 4–4 (PharMingen, San Diego, CA, USA) and PE-conjugated anti-CD45RB clone 16 A (PharMingen). The cells were sorted on FACStar (Becton-Dickinson, San Jose, CA, USA) in the UCLA Flow Cytometry Core Facility (Jonsson Comprehensive Cancer Center). Lymphocytes that stained brightly with both anti-CD4 and anti-CD45RB MoAbs were collected and designated CD4+ CD45RBhigh. CD4+ CD45RBlow and intermediate staining cells were discarded. Sorted populations were at least 96–98% pure on postsort analysis. Sorted donor lymphocytes, 4–5 × 105, were resuspended in 200 μl of sterile PBS and injected intraperitoneally in recipient B6 SCID mice. Animals were weighed initially, then weekly thereafter. They were observed for clinical signs of illness: ruffled fur, diarrhoea and hunched-over appearance.
Isolation of intraepithelial and lamina propria lymphocytes
Small and large intestines were removed from SCID recipients of CD45RBhigh cells and placed in Petri dishes containing HBSS without Ca+2 and Mg+2 (Gibco, Bethesda, MD, USA). Each intestine was cleaned carefully from its mesentery, opened longitudinally, flushed of fecal contents, and then cut into 0·5-cm pieces. The intestinal tissue was transferred to 250 ml Erlenmeyer flasks and shaken three separate times at 200 r.p.m. for 20 min at 37°C in HBSS without Ca+2 and Mg+2 containing 1 mm DTT (Sigma Chemical Co., St Louis, MO, USA). After each 20-min incubation the tissue suspensions were passed through a 60-μm nylon mesh (Fisher) into 50-ml conical tubes and the cells pelleted by centrifugation at 1200 r.p.m. The cell pellets were resuspended in 20% Percoll (Pharmacia, Piscataway, NJ, USA) made with complete RPMI 1640 (UCLA Media Center, Los Angeles, CA, USA), layered over a discontinuous 40%/70% Percoll gradient, and centrifuged at 900 g for 25 min. Cells from the 40%/70% interface were collected, washed and resuspended in complete RPMI 1640 media supplemented with 5% HIFCS (Irvine Scientific). This purified cell suspension constituted the IEL population. In order to isolate lamina propria lymphocytes (LPL), the remaining intestinal tissue was minced using a razor blade, transferred to 250-ml Erlenmeyer flasks and shaken for 60 min at 37°C in complete RPMI 1640 supplemented with 5% FCS containing protease at 1·5 mg/ml (Sigma). The tissue suspensions were collected, passed through nylon mesh and the cells pelleted by centrifugation at 1400 r.p.m. The cells were resuspended in 20% Percoll in RPMI as before, layered over a discontinuous 40%/70% Percoll gradient, centrifuged and processed as described above for the preparation of IEL. Viable lymphocytes were then enumerated on the basis of trypan blue extinction by light microscopy using a haemocytometer.
Flow cytometric analysis of lymphocyte populations
Isolated IEL and LPL were resuspended in PBS staining buffer containing 2% BSA and 0·02% NaN3 and placed in 96-well round-bottom plates. Pretitred MoAb directly conjugated to Fluorescein isothyocyanate (FITC), phycoerythrin (PE) or biotin were added to cell suspensions at 4°C and incubated on ice for 20 min. After staining with the primary antibody, samples were washed twice in PBS, followed by centrifugation at 1200 r.p.m. After the final wash, tricolour-conjugated streptavidin was added as secondary staining reagent for the biotinylated MoAb. After MoAb staining and washing, all samples were fixed in PBS containing 1% paraformaldehyde and 0·02% NaN3. The samples were stored at 4°C prior to flow cytometric analysis. All MoAbs used to characterize IEL and LPL populations were directly conjugated and included: Biotinylated CD3ɛ or CD3ɛ-PE clone 145–2C11, integrin β7 chain-FITC or PE clone M293 and streptavidin tricolor (Caltag, San Francisco, CA, USA). Flow cytometric analysis was performed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) in the UCLA Flow Cytometry Core Facility (Jonsson Comprehensive Cancer Center). Lymphocytes were distinguished on the basis of forward and side scatter and, if possible, at least 5000 gated events were acquired.
Preparation of tissue for histopathology analysis
For analysis of intestinal samples from the CD4 CD45RBhigh transfer studies, approximately 5-mm segments were taken from the proximal and distal segments of the large intestine and fixed in 10% formalin. For the IL-2/β7 integrin double-deficient mice, the intestines were dissected en bloc, fixed in 4% formaldehyde containing 1% acetic acid. Fixed tissue was embedded in paraffin. Deparaffinated sections were stained with haematoxylin/eosin.
RESULTS
Weight loss is significantly delayed following the transfer of β7 integrin −/− CD4+ CD45RBhigh T cells to SCID mice
The adoptive transfer of CD4+ CD45RBhigh cells to SCID mice results in the development of colitis within 6–10 weeks [12,15,19]. Disease progression can be monitored by measuring the degree of weight loss and assessing the animals for the development of diarrhoea, hunched-over appearance and ruffled fur [15]. SCID recipients of the wild-type CD4+ CD45RBhigh cells developed disease as described previously [15]. Within 3–4 weeks of cell transfer they began to lose weight followed by the development of diarrhoea and hunched-over appearance, the characteristic systemic signs of illness. By 10 weeks the animals had lost nearly 20% of their original weight. In contrast, mice that had received sorted CD4+ CD45RBhigh cells from β7 integrin –/– mice did not lose weight during this time period (Fig. 1). They appeared well and did not manifest the typical signs of disease. The difference in weight loss observed at 7 weeks between the animals that received wild-type versusβ7 integrin –/– CD4+ CD45RBhigh cells was statistically significant with a P-value of 0·0004. Interestingly, at 20 weeks post-transfer, the recipients of β7 integrin –/– cells began to show signs of illness such as ruffled fur and loose stool. This was associated with the onset of weight loss progressing to approximately a 15% decline in their original weight by 25 weeks. These results indicate that when pathogenic CD4+ CD45RBhigh cells are obtained from β7 integrin –/– mice, disease development in recipient animals is greatly delayed. It should be noted that control SCID mice maintained in our colony for this length of time did not lose weight spontaneously (data not shown).
Fig. 1.
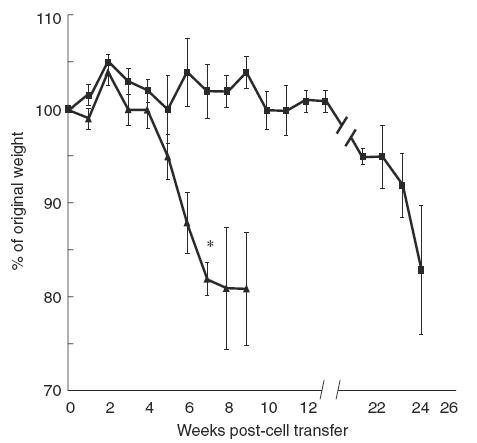
Weight loss is delayed when disease is induced by the transfer of CD4+ CD45RBhigh T cells from β7 integrin −/− mice. Donor lymphocytes were prepared as described and injected into recipient animals. The results show the change in weight as a percentage of original weight at day 0, the time of injection. Each data point represents the mean and the standard error for the indicated number of mice in each group: recipients of wild-type cells (N = 5), recipients of β7 integrin −/− cells (N = 4). Statistical analysis was performed using Student's two-tailed t-test to compare the two groups at 7 weeks. * indicates P = 0·0004. ▪, β7 Integrin −/−; ▴, wild-type.
Colonic inflammation in SCID recipients of β7 integrin −/− cells is delayed
To determine if the weight change correlated with evidence of inflammatory changes in the colon of the mice that had received β7 integrin −/− CD4+ CD45RBhigh T cells, we examined intestinal tissue sections from recipient mice at 9 and 25 weeks following transfer. Microscopic analysis of colon tissue from SCID recipients of wild-type CD4+ CD45RBhigh T cells showed that these animals had the typical histopathological hallmarks of disease (Fig. 2b). These include the presence of a significant inflammatory infiltrate, profound mucin depletion and epithelial hyperplasia. In contrast, examination of colonic tissue from mice that received β7 integrin –/– CD4+ CD45RBhigh T cells 9 weeks following cell transfer showed no significant inflammatory changes (Fig. 2c), a time-point when animals that had received wild-type cells already show severe mucosal changes. At 25 weeks, however, large intestinal sections from SCID mice that had received β7 integrin –/– cells showed the same inflammatory features that occurred in recipients of wild-type CD4+ CD45RBhigh T cells at 9 weeks. These results show that significant inflammatory changes do occur in the colon of mice that had received β7 integrin –/– donor cells, but the onset is much later in comparison to animals that had received wild-type cells.
Fig. 2.
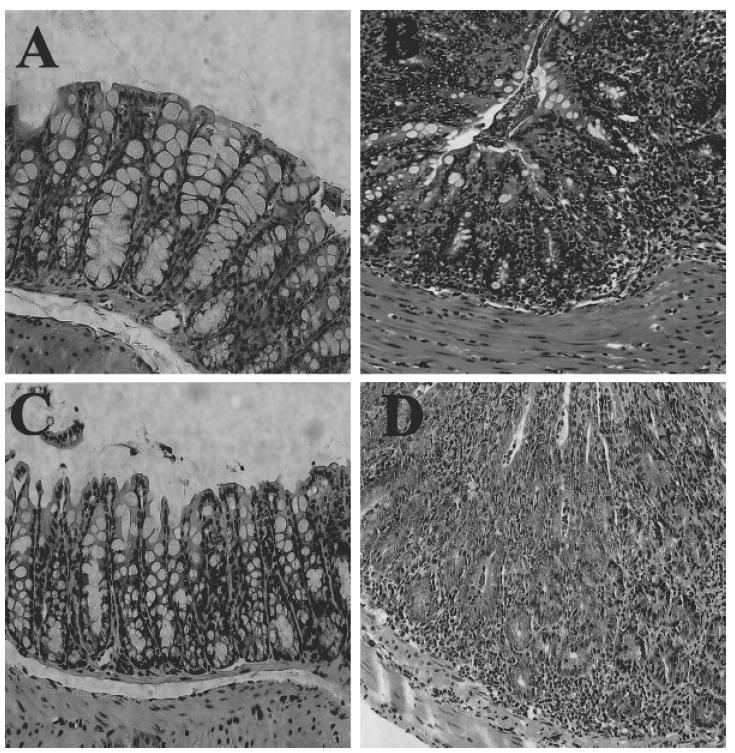
Delayed colonic inflammation following transfer of β7 −/− T cells. Colonic sections were obtained post mortem, fixed in formalin and stained with haematoxylin–eosin. Each panel is a representative section of the large intestine from animals in the following experimental groups. (a) Normal B6 SCID mouse without induced colitis. (b) Severe colonic inflammation at 9 weeks following transfer of wild-type CD4+ CD45RBhigh T cells. (c) Absence of colonic inflammation at 9 weeks following transfer of CD4+ CD45RBhigh T cells from β7 integrin −/− mice. (d) Severe colonic inflammation at 25 weeks following transfer of CD4+ CD45RBhigh T cells from β7 integrin −/− mice (magnification 40×).
Donor β7 integrin –/– CD4+ CD45RBhigh cells are not efficient in populating the intestine of SCID mice
The delay in colitis development following transfer of CD4+ CD45RBhighβ7 integrin –/– T cells to SCID mice suggested that the absence of β7 integrins may impact on the ability of the transferred T cells to populate the intestine of recipient animals. To examine this further, we isolated lymphocytes from the intraepithelial and lamina propria compartments of the small and large intestine and analysed them for expression of CD3 and β7 integrin by flow cytometry. We reasoned that the great majority of donor cells in the SCID recipient could be identified from host cells by the presence of CD3 and the absence of β7 integrin expression. Representative examples of flow cytometry profiles from individual animals are shown in Fig. 3. In agreement with our earlier results [15], SCID recipients of wild-type cells examined at 9 weeks contain a large percentage of donor CD3+β7 integrin+ cells present in the intraepithelial and lamina propria compartments of both the small and large intestine (Fig. 3a). In contrast, when the transferred T cells lack β7 integrins, there were substantially fewer donor CD3+ lymphocytes recovered from the intraepithelial and lamina propria compartment at 9 weeks post-transfer (Fig. 3b). At 25 weeks, however, there was a marked increase in the percentage of CD3+β7-negative cells present in the intraepithelial and lamina propria compartments of the small and large intestine of recipient animals (Fig. 3c). These results also demonstrate that the extent of repopulation of the IEL and LPL compartments by transferred β7 integrin –/– cells was similar when examined at 9 and 25 weeks. In some cases small numbers of CD3+β7+ cells were also observed. Because disease took nearly 5–6 months to develop, the CD3+β7+ population most probably represent ‘leaky’ host cells that can sometimes be observed in older SCID mice [20,21]. The presence of CD3−β7+ cells were also noted, particularly in those cases where there were few donor-derived CD3+ T cells. CD3− cells within the IEL compartment have been described previously in both immune-deficient and immune-competent mice [22]. β7 Integrin expression by these cells in Fig. 3b confirms that they are of host origin.
Fig. 3.
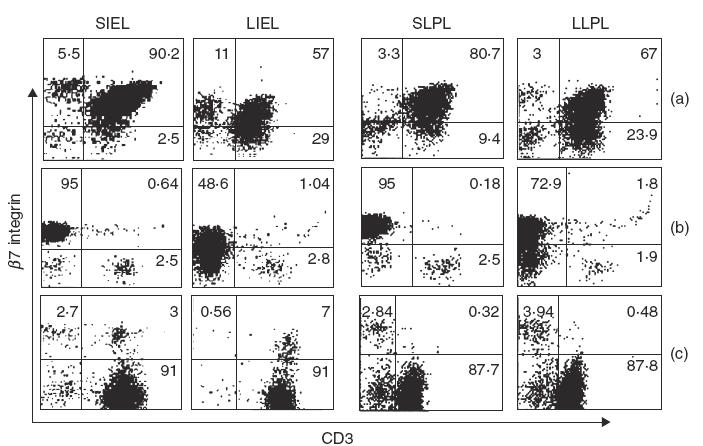
Donor β7 integrin −/− CD3+ lymphocytes are inefficient in populating the intestine of recipient SCID mice. IEL and LPL were isolated from the intestine of SCID hosts at the indicated times following cell transfer. CD3+β7+/+ and β7−/− integrin lymphocytes were enumerated by staining with anti-CD3ɛ-PE, and β7 integrin-FITC and analysed by flow cytometry. The two left-hand columns show small intestine IEL (SIEL) and large intestine IEL (LIEL) while the two right-hand columns show small intestine LPL (SLPL) and large intestine LPL (LLPL). (a) CD3+β7+/+ lymphocytes isolated from the SCID intestine at 9 weeks following transfer of β7+/+ CD4+ CD45RBhigh T cells. (b) CD3+β7−/− lymphocytes isolated from the SCID intestine at 9 weeks following transfer of β7−/− CD4+ CD45RBhigh T cells. (c) CD3+β7−/− lymphocytes isolated from the SCID intestine at 25 weeks following transfer of β7−/− CD4+ CD45RBhigh T cells. The data presented are from a representative experiment of four mice.
We enumerated the number of recovered CD3+ from the IEL compartments of the small and large intestine at 9 and 25 weeks from a number of animals following the transfer of either β7 integrin +/+ or –/– CD4+ CD45RBhigh cells. Figure 4 depicts the results of this analysis. At 9 weeks the average number of small intestinal CD3+ IEL recovered from recipients of β7 integrin –/– cells was 4 × 104 cells, compared to 4 × 106 from recipients of wild-type T lymphocytes. This difference is statistically significant with a P-value of 0·03. By 25 weeks the average number of CD3+ IEL isolated from recipients of β7 integrin –/– cells increased to 2·5 × 106 cells. This increase is statistically significant when compared to the number of cells recovered at 9 weeks, with a P-value of 0·003, but it was not significantly different when compared to recipients of wild-type cells at 9 weeks. Similarly, in the large IEL compartment, a significant reduction in the number of CD3+ cells was noted in mice that had received β7 integrin –/– lymphocytes compared to recipients of wild-type CD4+ CD45RBhigh cells. At 9 weeks the average number of CD3+ cells isolated from the IEL compartment of the large intestine was 8·5 × 104, compared to 1 × 106 in the control group. This difference is statistically significant with a P-value of 0·0029. At 25 weeks, when the mice that received β7 integrin –/– CD4+ CD45RBhigh cells showed weight loss and histological evidence of colonic inflammation, the average number of CD3+ cells isolated from the IEL compartment increased markedly to 1 × 106 cells. In summary, these findings indicate that CD4+ CD45RBhigh cells that lack β7 integrins are impaired in their ability to populate efficiently both the small and large intestine following adoptive transfer. After some time, however, increased numbers of these cells are found there, indicating that β7 integrin –/– cells are ultimately capable of entering, expanding and/or residing within the intestine. These data also suggest that in order for disease pathogenesis to progress, a certain threshold number of pathogenic cells is probably needed within the host colon.
Fig. 4.
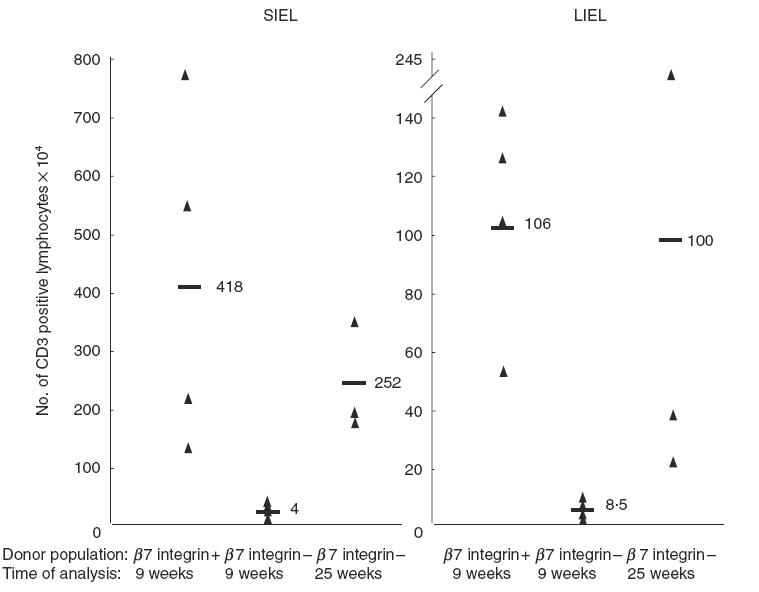
Fewer β7 integrin −/− cells populate the intestine of SCID mice compared to the recipients of wt β7 integrin +/+ cells at 9 weeks. Mucosal lymphocytes were isolated from the IEL compartment of the small and large intestine of recipient mice for each experimental group at the indicated times. CD3+β7 integrin +/+ and −/− lymphocytes were enumerated by staining with anti-CD3ɛ-PE, and β7 integrin-FITC and analysed by flow cytometry. Isolated lymphocytes recovered from recipients of β7integrin −/− T cells were considered donor in origin if they stained positively for CD3 and negatively for β7 integrin. Lymphocytes from recipients of wild-type cells were considered donor in origin if they stained positively for both CD3 and β7 integrin. The data represent the analysis of the following number of animals in their respective experimental groups: recipients of wild-type CD4+ CD45RBhigh cells analysed at 9 weeks (N = 4), recipients of β7 integrin −/− cells analysed at 9 weeks (N = 4), recipients of β7 integrin −/− cells analysed at 25 weeks (N = 3). Statistical analysis was performed using the t-test. For the small intestine: β7 integrin +/+ versusβ7 integrin −/− at 9 weeks, P = 0·03; β7 integrin +/+ at 9 weeks versusβ7 integrin −/− at 25 weeks, P = 0·40; β7 integrin −/− at 9 weeks versus β7 integrin −/− at 25 weeks, P = 0·0031. For large intestine: β7 integrin +/+ versusβ7 integrin −/− at 9 weeks, P = 0·0029; β7 integrin +/+ at 9 weeks versusβ7 integrin −/− at 25 weeks, P = 0·91, β7 integrin −/− at 9 weeks versusβ7 integrin −/− at 25 weeks, P = 0·17.
Colitis development is unaltered in IL-2/β7 integrin double-deficient mice
Colitis induction in the SCID transfer model requires the transfer and engraftment of donor lymphocyte populations. Analysis of colitis development where lymphocytes are not adoptively transferred but arise de novo may provide additional insight into the requirement for β7 integrins in lymphocyte localization within the gut during inflammatory processes. To accomplish this, we mated β7 integrin –/– mice with IL-2 knockout mice predisposed to the development of spontaneous colitis. IL-2/β7 integrin double-deficient mice were obtained at the expected Mendelian ratio from intercrossing mice that were heterozygous for both mutations (data not shown). IL-2-deficient mice heterozygous for the β7 integrin mutation express wild-type levels of β7 integrin (IL-2–/–β7+/– mice) and were therefore used as controls. More than five animals in each group were analysed. Control mice as well as IL-2/β7 integrin double-deficient mice developed normally for 3–4 weeks. Starting at 6 weeks of age all IL-2-deficient mice, irrespective of their β7 integrin genotype, developed colitis characterized by diarrhoea, weight loss and rectal prolapse. The histopathological examination of colonic sections taken from 12-week-old individual animals revealed pronounced hyperplasia of the mucosa, infiltration of the lamina propria and submucosa with lymphocytes and plasma cells in all mice. Ulcerations, dysplastic crypts and loss of goblet cells were also evident (data not shown).
DISCUSSION
This study utilized two different mouse experimental models of IBD to examine the role of β7 integrin expression on the ability of lymphocytes to localize to the gut associated lymphoid system and mediate inflammation. Despite lacking an important molecule for T cell localization to the GALT, deletion of β7 integrin did not alter the development and the course of spontaneous colitis in IL-2-deficient mice. In contrast, disease development in the CD4+ CD45RBhigh transfer model of colitis was delayed significantly. The onset of weight loss, systemic signs of illness and colonic inflammation occurred much later in the adoptive transfer system. Enumeration of the mucosal lymphocytes in the intestine of recipient animals correlated with the observed course of disease progression. At 9 weeks after transfer, recipients of pathogenic CD4+ CD45RBhigh cells from β7 integrin –/– mice had only a few CD3+β7− cells that could be recovered from their intestines. At 25 weeks, however, there was a marked increase in the number of donor lymphocytes, indicating that β7 integrin –/– CD4+ T cells were ultimately successful in populating the SCID host intestine. It is unclear from these results, however, if the increase in cell numbers observed in the intestine at 25 weeks reflects predominantly an increased entry of β7 integrin –/– cells or if it represents an increase in number of a population of donor cells already present in the intestine that expand under the influence of pro-inflammatory cytokines. It is possible that both processes are active.
The differences in the requirement for β7 integrins in these two models of IBD may reflect initial differences in the number of pathogenic T cells at the site of disease. In the IL-2/β7 integrin double-deficient mice, there should be a steady production of thymus derived cells that are potentially pathogenic, as well as the localization of some of these T cells to the intestine by β7 integrin-independent mechanisms. In support of this is the previous report showing that β7 integrin –/– mice have only a marked reduction in the number of IELs, not their complete absence [8]. The presence of these residual resident lymphocytes in the gut of IL-2/β7 integrin-deficient animals may be sufficient to allow colitis progression to occur, particularly in the context of the generalized autoimmune disorder accompanying the IL-2 deficiency [16]. Once the inflammatory cascade is initiated other cells may then be recruited to the site of inflammation through these alternate pathways. In the adoptive transfer system, however, a relatively small bolus of T cells is transferred, and many of these cells are probably caught in capillary beds and eliminated. Therefore, the actual number of pathogenic T cells transferred might be small, consistent with our previous finding of an oligoclonal T cell population in diseased recipients [23]. These donor lymphocytes probably require initial activation, homing to the gut, and subsequent expansion within the intestinal compartment in order to mediate colitis pathogenesis. The site of initial activation is not known, but some work favours an extra intestinal site with subsequent migration to the intestine [24]. Our results indicate, however, that this multi-step process of lymphocyte migration is impaired in the absence of β7 integrins. Previous studies have shown that β7 integrin –/– lymphocytes have a significant limitation in their ability to bind to HEVs of Peyer's patches in vitro and to migrate into Peyer's patches in vivo[8,25]. This limitation was less pronounced when similar studies were performed using mesenteric lymph nodes (MLN), suggesting that factors unique to a specific anatomical site are likely to direct lymphocyte adhesion and entry. In this context, it is interesting to note that in β7 integrin/l-selectin double-deficient mice, migration to MLNs is abrogated [26].
While there were differences in the kinetics of disease development with these two models, the results indicate none the less that β7 integrin expression is not crucial for T cell localization to the intestine and colitis progression. Under these circumstances, alternate pathways such as LFA-1/ICAM-1, VLA-4/VCAM-1 and l-selectin/MAdCAM-1or GlyCAM–1 interactions may compensate for the lack of β7 integrin and facilitate lymphocyte entry into the gut.
Monoclonal antibodies to α4β7 or αΕβ7 integrins ameliorate the colitis in IL-2-deficient mice kept in specific pathogen-free conditions in which the disease develops only following immunization [11]. Similarly, treatment with monoclonal antibodies to β7 integrin and MAdCAM-1 have been shown to ameliorate established colitis in the CD45RBhigh transfer model [10]. These results differ to some extent from the findings obtained in our studies, and probably reflect the inherent complexities associated with inhibiting the process of intestinal inflammation in experimental models. Some of these factors include the timing of antibody administration, the antibody's specificity and the duration of the biological effect. In the aforementioned studies, blockade of β7 integrin function by antibody is implemented at disease induction, and its effect on disease pathogenesis is evaluated only for a short period of time. These factors may limit the time available for the colitis-inducing cells to adjust to alternate adhesion pathways. Similarly, there may be a compensatory increase in the expression of some integrins or adhesion molecules in the β7 integrin deficient mice, although so far there is no evidence in favour of this. In studies in which there is treatment of established disease, the duration of the therapeutic effect may not be sustained. As suggested from the studies described herein, alternate homing and adhesion pathways are capable of allowing T cells to localize to the intestine under proinflammatory conditions.
Acknowledgments
We thank Dr Hilde Cheroutre and Ms Katherine Williams for rederivation of the β7−/− integrin mice. Grant support was received from the Crohn's and Colitis Foundation of America (to BCS), the Deutsche Forschungsgemeinschaft (WA 1127/1–2) and the Bennigsen-Foerder research award by the state of Nordrhein-Westfalen, Germany (to NW), NIH grant PO1 DK46763 (to MK), a VA Career Development Award (to RA), and the Blinder Foundation.
REFERENCES
- 1.Yacyshyn BR, Lazarovits A, Tsai V, Matejko K. Crohn's disease, ulcerative colitis, and normal intestinal lymphocytes express integrins in dissimilar patterns. Gastroenterology. 1994;107:1364–71. doi: 10.1016/0016-5085(94)90538-x. [DOI] [PubMed] [Google Scholar]
- 2.Souza HSCC, Elia J, Spencer TT. MacDonald,Expression of lymphocyte-endothelial receptor-ligand pairs, alpha4beta7/ MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut. 1999;45:856–63. doi: 10.1136/gut.45.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bargatze RF, Jutila MA, Butcher EC. Distinct roles of l-selectin and integrins alpha 4 beta 7 and LFA-1 in lymphocyte homing to Peyer's patch-HEV in situ: the multistep model confirmed and refined. Immunity. 1995;3:99–108. doi: 10.1016/1074-7613(95)90162-0. [DOI] [PubMed] [Google Scholar]
- 4.Holzmann B, Weissman IL. Integrin molecules involved in lymphocyte homing to Peyer's patches. Immunol Rev. 1989;108:45–61. doi: 10.1111/j.1600-065x.1989.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 5.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–6. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 6.Cepek KL, Parker CM, Madara JL, Brenner MB. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J Immunol. 1993;150:3459–70. [PubMed] [Google Scholar]
- 7.Schon MPA, Arya EA, Murphy CM, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol. 1999;162:6641–9. [PubMed] [Google Scholar]
- 8.Wagner N, Lohler J, Kunkel EJ, et al. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 1996;382:366–70. doi: 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- 9.Podolsky DKR, Lobb N, King CD, et al. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J Clin Invest. 1993;92:372–80. doi: 10.1172/JCI116575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler DJ. Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J Immunol. 1997;158:2099–106. [PubMed] [Google Scholar]
- 11.Ludviksson BR, Strober W, Nishikomori R, Hasan SK, Ehrhardt RO. Administration of MoAb against alpha E beta 7 prevents and ameliorates immunization-induced colitis in IL-2–/– mice. J Immunol. 1999;162:4975–82. [PubMed] [Google Scholar]
- 12.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 SCID mice. Int Immunol. 1993;5:1461–71. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 13.Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–62. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 14.Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell- mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aranda R, Sydora BC, McAllister PL, et al. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol. 1997;158:3464–73. [PubMed] [Google Scholar]
- 16.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene [see comments] Cell. 1993;75:253–61. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 17.Ehrhardt RO, Ludviksson B. Induction of colitis in IL2-deficient-mice: the role of thymic and peripheral dysregulation in the generation of autoreactive T cells. Res Immunol. 1997;148:582–8. doi: 10.1016/s0923-2494(98)80153-3. [DOI] [PubMed] [Google Scholar]
- 18.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–4. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 19.Morrissey PJK, Charrier S, Braddy D, Liggitt, Watson JD. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178:237–44. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev Immunol. 1991;9:323–50. doi: 10.1146/annurev.iy.09.040191.001543. [DOI] [PubMed] [Google Scholar]
- 21.Bosma MJ. B and T cell leakiness in the scid mouse mutant. Immunodefic Rev. 1992;3:261–76. [PubMed] [Google Scholar]
- 22.Rocha BD, Guy-Grand, Vassalli Extrathymic P. T cell differentiation. Curr Opin Immunol. 1995;7:235–42. doi: 10.1016/0952-7915(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 23.Matsuda JL, Gapin L, Sydora BC, et al. Systemic activation and antigen-driven oligoclonal expansion of T cells in a mouse model of colitis. J Immunol. 2000;164:2797–806. doi: 10.4049/jimmunol.164.5.2797. [DOI] [PubMed] [Google Scholar]
- 24.Lefrancois L, Parker CM, Olson S, et al. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response [published erratum appears in J Exp Med 190 (9): following 1362] J Exp Med. 1999;189:1631–8. doi: 10.1084/jem.189.10.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunkel EJ, Ramos CL, Steeber DA, et al. The roles of l-selectin, beta 7 integrins, and P-selectin in leukocyte rolling and adhesion in high endothelial venules of Peyer's patches. J Immunol. 1998;161:2449–56. [PubMed] [Google Scholar]
- 26.Wagner N, Lohler J, Tedder TF, Rajewsky K, Muller W, Steeber DA. l-selectin and beta7 integrin synergistically mediate lymphocyte migration to mesenteric lymph nodes. Eur J Immunol. 1998;28:3832–9. doi: 10.1002/(SICI)1521-4141(199811)28:11<3832::AID-IMMU3832>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]