

Abstract
We have reported previously that circulating anti-Fas ligand (FasL) autoantibodies able to inhibit Fas/FasL-mediated apoptosis were present in patients with systemic lupus erythematosus (SLE). In the present study, we describe the epitopes recognized by these anti-FasL autoantibodies. Rabbit antihuman antibody, raised against a FasL fragment consisting of amino acids (aa) 103–179 (fragment 2·0), inhibited Fas/FasL-mediated apoptosis, whereas an antibody against a FasL aa 103–146 fragment (fragment 1·0) did not. This suggested that an epitope around aa 146–179 was important for Fas/FasLinteraction. Epitope mapping of anti-FasL autoantibodies using deletion mutants indicated that the epitope was located around aa 163–179. Three-dimensional molecular modelling of the Fas/FasLcomplex revealed that the aa 162–169 region was located on the outermost side of FasL, which suggested that the anti-FasL autoantibody would easily have access to the epitope. FasL point mutants involving aa positions 162–169 resulted in complete loss of apoptosis-inducing capability, which suggested that the aa 162–169 region was important for Fas/FasLinteraction. A synthetic FasL peptide consisting of aa 161–170 blocked the binding of anti-FasL autoantibodies to FasL fragment 2·0 (aa 103–179). The FasL aa 161–170 sequence was found to be highly homologous with aa sequences from several infectious agents. Synthetic peptides derived from some of these microorganisms cross-reacted with the epitope recognized by the autoantibodies, suggesting that several foreign infectious agent-derived proteins may share an epitope with human FasL. As lymphocytes from SLE patients aberrartly expressed FasL, it is possible that infection by one of several infectious agents may trigger cross-reactive antibody responses, after which aberrantly expressed endogenous FasL might induce the shift from a cross-reactive response to an authentic autoimmune response. Therefore, a combination of molecular mimicry and aberrant autoantigen expression may be important for the development of anti-FasL autoantibodies in SLE patients.
Keywords: apoptosis, epitope, human, molecular mimicry, tolerance
INTRODUCTION
Systemic lupus erythematosus (SLE) is considered to be a systemic autoimmune disease, and is accompanied by impaired T cell regulation and B cell hyperactivity, both of which are associated with autoantibody production and subsequent tissue damage caused by immune complexes [1–8]. Considerable evidence also suggests that the development of SLE has a strong genetic basis [9,10].
Defects in self-tolerance are thought to play an important role in the development and maintenance of the autoimmune response in human SLE [11–15]. Because a wide variety of autoantibodies are found in the sera from SLE patients, it is possible that defective self-tolerance mechanisms common to various autoantigen specificities are involved in SLE [16]. Indeed, autoreactive T cell clones established from the peripheral blood of SLE patients can provide help for B cells of various antigen specificities, including autoantigens [17].
Fas is a cell-surface protein belonging to the TNFR/nervegrowth factor receptor family [18], while Fas ligand (FasL) is a type II membrane protein of the TNF family acting as a death factor, transducing death signals to Fas-positive cells [19]. Mice with loss-of-function mutations of Fas and FasL suffer from autoimmune diseases [20–23]. Therefore, it is likely that the Fas/FasLsystem is involved in the clonal deletion of autoreactive T cells in the periphery, and has been linked to the activation-induced cell death of autoreactive T cells [20–23]. Thus, the Fas/FasLsystem is thought to play a pivotal role in the induction of apoptosis of Fas-expressing autoreactive lymphocytes, and therefore, the establishment and maintenance of peripheral tolerance [24–31].
It has been shown that membrane-bound Fas and FasL molecules can be secreted as soluble Fas (sFas) and FasL (sFasL), respectively, which can then modulate membrane Fas- and membrane FasL-mediated apoptosis [32,33]. Several reports have investigated the Fas/FasLpathway in SLE patients [34–43] and have demonstrated that sFas and sFasL levels are higher in SLE patients than in normal controls, suggesting that these soluble factors are involved in the pathophysiology of SLE [32–35]. We have shown previously that circulating anti-FasL autoantibodies present in approximately one-third of SLE patients were able to inhibit the Fas/FasL-mediated apoptosis of lymphocytes [44]. Even though it is not clear whether these autoantibodies are the primary cause of SLE, it is likely that once anti-FasL autoantibodies are present, normal apoptotic responses would be affected, further perturbing the normal immune response in SLE patients.
In order to further characterize anti-FasL autoantibodies in SLE, we conducted fine epitope mapping of anti-FasL autoantibodies that could potentially inhibit FasL-mediated apoptosis. The use of site-directed mutagenesis of FasL cDNA confirmed that the epitope recognized by the autoantibodies was a functionally important region of human FasL involved in the induction of apoptosis of Fas-expressing cells. We also found that the amino acid sequence of the epitope recognized by the autoantibodies was shared with proteins from several infectious agents, which suggested that a molecular mimicry mechanism might be involved in the development of the autoantibodies. In addition, lymphocytes from SLE patients aberrantly expressed FasL, which may lead to further stimulation of the autoantibody-secreting B cells.
PATIENTS AND METHODS
Patients
We studied 21 patients, all women, who fulfilled the 1982 revised criteria for the classification of SLE by the American Rheumatism Association [45]. The mean age ± s.d. of these patients was 35·1 ± 9·2 years (range 14–45). Fourteen healthy female volunteer blood donors (mean age ± s.d. = 36·2 ± 6·3 years, range 25–52), served as control subjects. Ten patients with rheumatoid arthritis (RA) (mean age ± s.d. = 58·4 ± 12·8 years, range 36–76) were also recruited as a disease control. All patients were unselected, unrelated Japanese.
Informed consent from each patient and ethical approval from the Human Studies Committee were obtained before commencement of the study.
Plasmid construction and expression of FasL
The prokaryotic expression vector pQE30 carrying the extracellular domain of human FasL cDNA (pQE30-FasL fragment 5) has been described previously [44]. pQE30-FasL fragment 5 encoded the whole extracellular domain of FasL, consisting of amino acids (aa) 103–281, tagged with 6 histidine residues. The following deletion mutants of the FasL extracellular domain were prepared, as shown in Fig. 1, using a PCR-based technique [46]; FasL fragment 1 (aa 103–146), FasL fragment 1·5 (aa 103–156), FasL fragment 1·8 (aa 103–163), FasL fragment 2 (aa 103–179), FasL fragment 3 (aa 103–219), FasL fragment 3·5 (aa 103–228), FasL fragment 3·8 (aa 103–236), FasL fragment 4 (aa 103–241), FasL fragment II (aa 180–281), FasL fragment III (aa 220–281), FasL fragment IV (aa 242–281), FasL fragment II− (aa 180–219). Sequences of the amplification primers are shown in Table 1. The PCR products were digested with Sph I and Sal I, and subcloned into the pQE30 vector (Qiagen, Tokyo, Japan). In-frame 6 × histidine-tagged FasL deletion-mutant constructs were used for protein expression. FasL-fragment proteins were purified using Ni-NTA agarose according to the manufacturer's instructions (Qiagen).
Fig. 1.
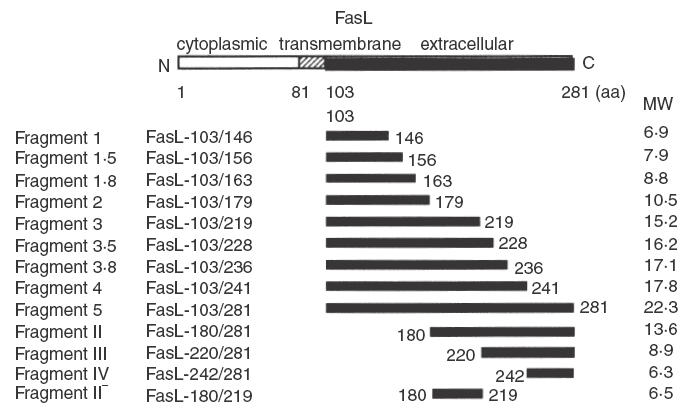
Schematic representation of wild-type FasL and FasL deletion-mutant proteins. FasL deletion-mutant proteins were produced in E. coli using the pQE30 bacterial expression vector. The aa numbers in each mutant FasL are indicated. MW represents the molecular weight (kDa) of the mutant FasL proteins tagged with 6 × histidine.
Table 1.
Oligonucleotide primers used for amplyfication of FasL fragments
| FasL fragment 1.0, 1.5, 1.8, 2.0, 3.0, 3.5, 3.8, and 4.0 | ||
| Sense | GGCGCATGCGAATTCCAGCTCTTCCACCTACAGAAGGAG | |
| Antisense | FasL fragment 1.0, | TAAGTCGACCTATCACACTTTCCTCAGCTCCTTTTTTTC |
| FasL fragment 1.5, | TAAGTCGACCTATCACCTTGAGTTGGACTTGCCTGTTAA | |
| FasL fragment 1.8, | TAAGTCGACCTATCATTCCCATTCCAGAGGCATGGACCT | |
| FasL fragment 2.0, | CACGTCGACCTATCAACCCTTCTTATACTTCACTCCAGA | |
| FasL fragment 3.0, | CAGGTCGACCTATCAGGGATACTTAGAGTTCCTCATGTA | |
| FasL fragment 3.5 | GTAGTCGACCTATCACTTCCCCTCCATCATCACCAGATC | |
| FasL fragment 3.8 | GTAGTCGACCTATCACCCAGTAGTGCAGTAGCTCATCAT | |
| FasL fragment 4.0, | GTAGTCGACCTATCAGCGGGCCCACATCTGCCCAGTAGT | |
| FasL fragment II, III and IV | ||
| Sense | FasL fragment II, | GGCGCATGCGAATTCGGCCTTGTGATCAATGAAACTGGG |
| FasL fragment III, | GGCGCATGCGAATTCCAGGATCTGGTGATGATGGAGGGG | |
| FasL fragment IV, | GGCGCATGCGAATTCAGCAGCTACCTGGGGGCAGTGTTC | |
| Antisense | GTGGTCGACTTAGAGCTTATATAAGCCGAAAAACG | |
| FasL fragment IIminus | ||
| Sense | GGCGCATGCGAATTCGGCCTTGTGATCAATGAAACTGGG | |
| Antisense | CAGGTCGACCTATCAGGGATACTTAGAGTTCCTCATGTA | |
A mammalian expression vector carrying the full-length wild-type human FasL cDNA (pME18S-FasL) was prepared previously [44]. FasL point mutants were created using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). Briefly, pME18S-FasL was used as a template, and oligonucleotide primers containing the desired mutations were extended during PCR cycling using PfuTurbo DNA polymerase (Stratagene). The amplification cycle consisted of 1 cycle of denaturation (95°C) for 1 min, followed by 12 cycles of denaturation (95°C) for 30 s, annealing for 1 min (55°C), and polymerization for 10 min (68°C). To select for the synthesized, non-methylated DNA containing the mutations, the resultant PCR products were treated with Dpn I, which is specific for methylated and hemimethylated DNA, and then transformed into Escherichia coli. The oligonucleotide primers used for creating point mutation constructs are shown in Table 2. Sequences of all constructs were confirmed by DNA sequencing.
Table 2.
Oligonucleotides* used to generate FasL point mutants for mammalian expression usingsite-derected mutagenasis
| E161S | CTCAAGGTCCATGCCTCTGTCATGGGAAGACACCTATGGAATTG |
| CAATTCCATAGGTGTCTTCCCATGACAGAGGCATGGACCTTGAG | |
| W162S | GGTCCATGCCTCTGGAATCGGAAGACACCTATGG |
| CCATAGGTGTCTTCCGATTCCAGAGGCATGGACC | |
| E163S | GGTCCATGCCTCTGGAATGGTCCGACACCTATGGAATTGTCC |
| GGACAATTCCATAGGTGTCGGACCATTCCAGAGGCATGGACC | |
| D164S | GCCTCTGGAATGGGAATCCACCTATGGAATTGTCCTGC |
| GCAGGACAATTCCATAGGTGGATTCCCATTCCAGAGGC | |
| T165S | GCCTCTGGAATGGGAAGACTCCTATGGAATTGTCCTGC |
| GCAGGACAATTCCATAGGAGTCTTCCCATTCCAGAGGC | |
| Y166S | GGAATGGGAAGACACCTCCGGAATTGTCCTGCTTTCTGG |
| CCAGAAAGCAGGACAATTCCGGAGGTGTCTTCCCATTCC | |
| G167S | GGAATGGGAAGACACCTATTCGATTGTCCTGCTTTCTGGAGTG |
| CACTCCAGAAAGCAGGACAATCGAATAGGTGTCTTCCCATTCC | |
| I168S | GGGAAGACACCTATGGATCCGTCCTGCTTTCTGGAGTG |
| CACTCCAGAAAGCAGGACGGATCCATAGGTGTCTTCCC | |
| V169S | GGGAAGACACCTATGGAATTTCGCTGCTTTCTGGAGTGAAG |
| CTTCACTCCAGAAAGCAGCGAAATTCCATAGGTGTCTTCCC | |
| L170S | GAAGACACCTATGGAATTGTCTCGCTTTCTGGAGTGAAGTATAAG |
| CTTATACTTCACTCCAGAAAGCGAGACAATTCCATAGGTGTCTTC | |
| L171S | GACACCTATGGAATTGTCCTGTCTTCTGGAGTGAAGTATAAG |
| CTTATACTTCACTCCAGAAGACAGGACAATTCCATAGGTGTC | |
| S172A | CCTATGGAATTGTCCTGCTTGCTGGAGTGAAGTATAAGAAGG |
| CCTTCTTATACTTCACTCCAGCAAGCAGGACAATTCCATAGG | |
| G173S | GGAATTGTCCTGCTTTCTAGTGTGAAGTATAAGAAGGGTGGC |
| GCCACCCTTCTTATACTTCACACTAGAAAGCAGGACAATTCC | |
| V174S | GGAATTGTCCTGCTTTCTGGATCGAAGTATAAGAAGGGTGGCC |
| GGCCACCCTTCTTATACTTCGATCCAGAAAGCAGGACAATTCC | |
| K175S | GGAATTGTCCTGCTTTCTGGAGTGAGTTATAAGAAGGGTGGCC |
| GGCCACCCTTCTTATAACTCACTCCAGAAAGCAGGACAATTCC | |
| Y176S | CCTGCTTTCTGGAGTGAAGTCGAAGAAGGGTGGCC |
| GGCCACCCTTCTTCGACTTCACTCCAGAAAGCAGG | |
| K177S | CCTGCTTTCTGGAGTGAAGTATTCGAAGGGTGGCCTTGTGATC |
| GATCACAAGGCCACCCTTCGAATACTTCACTCCAGAAAGCAGG | |
| K178S | GCTTTCTGGAGTGAAGTATAAGTCGGGTGGCCTTGTGATCAATG |
| CATTGATCACAAGGCCACCCGACTTATACTTCACTCCAGAAAGC | |
| G179S | GGAGTGAAGTATAAGAAGAGTGGCCTTGTGATCAATGAAAC |
| GTTTCATTGATCACAAGGCCACTCTTCTTATACTTCACTCC | |
| P206S | GGTCAATCTTGCAACAACCTGAGTCTGAGCCACAAGGTCTACATG |
| CATGTAGACCTTGTGGCTCAGACTCAGGTTGTTGCAAGATTGACC | |
| Y218R | GGTCTACATGAGGAACTCTAAGAGGCCCCAGGATCTGG |
| CCAGATCCTGGGGCCTCTTAGAGTTCCTCATGTAGACC |
All the point mutant FasL cDNAs were created to exchange respective aa for serine (except for S172A and Y218R that encode alanine and arginine, respectively) and the regions were underlined.
Production and biological activity of wild-type and mutant FasL proteins
Biological activity of wild-type and mutant FasL proteins was estimated by cytotoxicity against Jurkat-NU cells, which are susceptible to FasL-mediated apoptotic cell death [44]. The proteins were produced from COS cells transfected transiently by electroporation. After 52 h, the supernatants were harvested to collect sFasL. sFasL concentrations were estimated by both immunoblotting and ELISA as described below. Graded amounts (0–3·2 ng/ml) of wild-type and mutant sFasL were introduced into Jurkat-NU cell cultures [44] and the extent of apoptotic cell death was estimated by DNA staining with hypotonic propidium iodide (PI) [47]. The PI fluorescence of isolated nuclei was measured with a flow cytometer and the proportion of subdiploid DNA peaks in the DNA fluorescence histogram calculated to give the percentage of apoptotic cells [48].
In experiments to assess membrane FasL-mediated apoptosis, wild-type FasL cDNA-transfected COS cells were incubated with PBS, rabbit IgG or affinity purified rabbit anti-FasL fragment antibody [44]. Jurkat-NU cells were then co-cultured with the COS cells and cell death estimated by two-colour fluorescence analysis employing TUNEL staining with FITC (Boehringer, Mannheim, Mannheim, Germany) and OKT3-PE antibody (Ortho Pharmaceutical, Raritan, NJ, USA) [44].
Preparation of rabbit affinity-purified antihuman FasL deletion mutant antibodies
Recombinant FasL deletion-mutant proteins (fragments 1, 2, 3, 4 and 5) eluted from Ni-resin columns were further purified for immunization by preparative SDS-PAGE [44]. The gel-purified FasL deletion-mutant proteins were then used to immunize rabbits (New Zealand White) according to standard protocols [49]. Each of the anti-FasL deletion-mutant protein antibodies were affinity-purified using the same immobilized FasL deletion-mutant protein used for the immunization, as described previously [49].
SDS-PAGE and immunoblotting analysis
1 × 107 PBMC, purified lymphocytes or COS cells transfected with FasL expression vectors were lysed in 100 μl NP40 lysis buffer [49] containing protease inhibitors (1 mg/mlPMSF, 5 mm EDTA, 2 mg/ml aprotinin and 2 mg/ml leupeptin). Cell lysates and affinity-purified FasL deletion mutant proteins were electrophoresed on 4–20% or 12% SDS-PAGE gels. Proteins were electrotransferred onto polyvinylidene difluoride membranes (Millipore, Beetford, MA, USA). To assess relative protein amounts, SDS-PAGE gels and membranes were stained with Quick CBB and Ponceau-S (both from Wako, Osaka, Japan), respectively. Membranes were probed with rabbit antihuman FasL antibodies, followed by incubation with biotin-labelled antirabbit IgG antibodies and streptavidin–alkaline phosphatase. Anti-FasL autoantibodies in SLE patients were affinity-purified from patient sera [44], and were used as purified autoantibodies in the experiments. Proteins were visualized by chemiluminescence (Amersham, Tokyo, Japan). In experiments to address fine epitope mapping by the competitive inhibition method, membranes containing FasL fragment 2 were reacted with antibodies in the presence of various concentrations of synthetic peptide. We used dimethyl formamide (DMF) as solvent, because some peptides dissolved completely in water, but the others did not. Thereafter the membranes were developed as described above.
Comparative modelling of human FasL
A three-dimensional model of human FasL monomer was built by modeller (Molecular Simulations, San Diego, CA, USA), a program that implements comparative modelling by the satisfaction of spatial restraints [50]. The crystallographic structures of TNF-α and TNFR (Brookhaven Protein Data Bank entries, 1TNF and 1TNR) were used as suitable template structures. After modelling, three copies of each monomer model were superimposed onto the TNF-α trimer (1TNF). The trimer structures were optimized by 100 steps of conjugate gradient minimization using the charmm program (Molecular Simulations).
sFasL ELISA
sFasL in culture supernatants was estimated using the sFasL ELISA (Mochida Pharmaceutical, Tokyo, Japan). The anti-FasL MoAbs used in the ELISA were developed by immunization with a synthetic peptide consisting of the N-terminal region of sFasL (aa 126–145). FasL mutations used in this study were all between FasL aa 161 and 246, so that all FasL mutant proteins were recognized efficiently by the ELISA [51].
RESULTS
Characterization of binding epitopes of anti-FasL autoantibodies in patients with SLE
We have found previously that anti-FasL autoantibodies were present in the sera of approximately one-third of SLE patients [44], while no such autoantibodies were found in RA patients and normal individuals. These autoantibodies inhibited the membrane FasL- and sFasL-mediated apoptotic cell death of Fas-expressing T lymphocytes.
To characterize further these anti-FasL autoantibodies with inhibitory potential on Fas/FasL-mediated apoptosis, we performed fine epitope mapping experiments. We designed PCR primers to amplify various lengths of human FasL extracellular domain cDNA, and ligated the amplified products into the bacterial expression vector pQE30 (Fig. 1). The various FasL extracellular domain deletion-mutant proteins were purified and immunoblotted to test for those that reacted preferentially with anti-FasL autoantibodies from SLE patient sera. As a control, we developed rabbit anti-FasL antibodies, using the whole FasL extracellular domain (fragment 5) for immunization, and affinity purified the resultant antibodies. We found that purified SLE patient anti-FasL autoantibodies bound preferentially to FasL fragments 2, 3, 4 and 5, but not to fragment 1 (Fig. 2). The autoantibodies also reacted with FasL fragments II, III, IV and II− (data not shown). In contrast, anti-FasL antobodies from 3 rabbits immunized with FasL fragment 5 bound predominantly to fragments 4 and 5, but only reacted weakly with fragments 1, 2 and 3 (Fig. 2). Antihuman FasL fragment 5 antibody from two rats showed the same reaction pattern as for the rabbit antibodies (data not shown). These results indicated that one of the major epitopes of the SLE patient-derived autoantibodies was present within fragment 2 but not fragment 1, whereas the epitope for the rabbit antihuman FasL antibodies was contained within fragment 4. It also suggested that the major epitopes of antibodies induced by immunization and ‘spontaneously’ arising SLE autoantibodies were different.
Fig. 2.
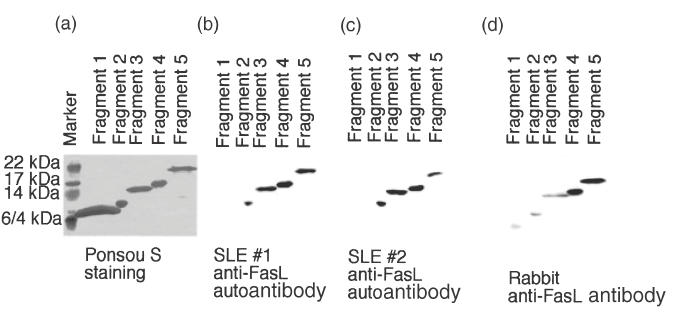
Epitope mapping of the anti-FasL autoantibodies in SLE patients. Recombinant human FasL fragments 1, 2, 3, 4 and 5 were produced and purified using Ni-NTA resin. The proteins were blotted onto polyvinylidene difluoride membranes, stained with Ponceau-S (a), and reacted with immunization-induced rabbit anti-FasL antibody (d) or anti-FasL autoantibodies from SLE patients (b,c). Data shown are representative of seven independent experiments. Note that the immunization-induced antihuman FasL whole extracellular domain (fragment 5) antibody from three rabbits showed the same reaction pattern to the FasL deletion-mutant proteins (d). Among 21 SLE patients studied in this experiment, anti-FasL autoantibodies were present in the seven patients. The seven SLE patients showed the same reaction pattern (b,c).
Development of anti-FasL fragment antibodies in rabbits and effects on Fas/FasL mediated apoptosis
We next developed antibodies against a panel of FasL fragment proteins, and examined which anti-FasL fragment antibody inhibited the FasL-mediated apoptotic cell death of Fas-expressing lymphocytes. The aim of this experiment was to define the epitope(s) recognized by those anti-FasL fragment antibodies with FasL-neutralizing potential.
To this end, wild-type FasL transfected COS cells were incubated with PBS, rabbit IgG or affinity purified rabbit anti-FasL fragment antibodies. Jurkat-NU cells, used as indicators of apoptosis, were then introduced into the COS cell cultures. Cell death was estimated by two-colour fluorescence analysis employing TUNEL staining with FITC and OKT3-PE antibody [44]. We found that anti-FasL fragments 2, 3, 4 and 5 antibodies inhibited FasL-mediated apoptosis (Fig. 3). However, affinity purified anti-FasL fragment 1 antibody did not inhibit apoptosis, even though the anti-FasL fragment 1 antibody reacted with FasL in immunoblotting experiments (data not shown). Similarly, sFasL-mediated apoptosis was also inhibited by anti-FasL fragments 2, 3, 4 and 5 antibodies, but not by anti-FasL fragment 1 antibody (data not shown). These results suggested that a region contained within FasL fragments 2, 3, 4 and 5, but not FasL fragment 1, was involved in sFasL- as well as membrane FasL-mediated apoptosis.
Fig. 3.
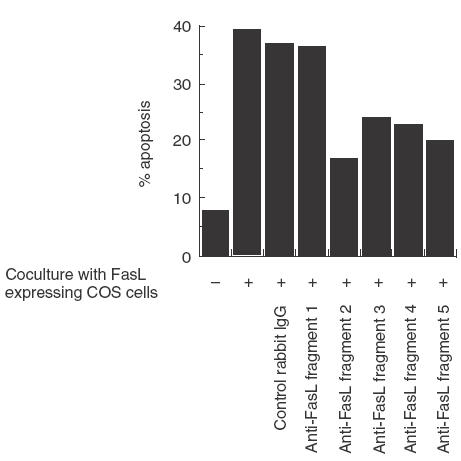
Inhibition by anti-FasL fragment antibodies of membrane FasL-induced apoptotic cell death of Fas-expressing Jurkat cells. Wild-type FasL cDNA transfected COS cells were incubated with PBS, control rabbit IgG (1 μg/ml), or affinity-purified rabbit anti-FasL fragment antibodies (1 μg/ml). Jurkat-NU cells were then co-cultured with the COS cells. Cell death was estimated by two-colour fluorescence analysis employing TUNEL staining with FITC and OKT3-PE antibodies. Data shown are representative of three independent experiments. Addition of rabbit anti-FasL fragment antibodies into the Jurkat-NU cell culture without FasL exerted no effects on cell death of the target cells (data not shown).
Schneider et al. have already shown that FasL aa 206 and 218 are important for Fas/FasL molecular interaction [52], and are located within our FasL fragment 3 construct. Our results indicated that a region within fragment 2 (aa 103–179) but not fragment 1 (aa103–146) was also important. This finding was confirmed by results obtained using rabbit anti-FasL fragment antibodies 1 (Fig. 2).
Precise epitope mapping of anti-FasL autoantibodies in SLE patients by immunoblotting analysis
To characterize the epitope(s) recognized by anti-FasL autoantibodies more precisely, we synthesized FasL deletion-mutant proteins fragment 1·5 (aa 103–156) and fragment 1·8 (aa 103–163). We then performed immunoblotting on these proteins using anti-FasL autoantibodies that potently inhibited Fas/FasL-mediated apoptosis. We found that the autoantibodies recognized fragment 2, but not 1·8 or 1·5 (Fig. 4). These results suggested that aa 163–179 of human FasL constituted one of the major autoantibody epitopes.
Fig. 4.

Precise epitope mapping of anti-FasL autoantibody from SLE patients by immunoblotting analysis. Recombinant human FasL fragments 1·5, 1·8 and 2 were produced and purified using Ni-NTA resin. The proteins were blotted onto polyvinylidene difluoride membranes and the parallel gels stained with Quick-CBB. The membranes were reacted with anti-FasL autoantibodies from SLE patients. Seven patients were positive for the anti-FasL autoantibodies out of 21 patients studied. The autoantibodies from seven patients showed the same reaction pattern and inhibited the Fas/FasL-mediated apoptosis.
Molecular modelling of the Fas/FasL complex
We then investigated whether the epitope was located on the outer surface of the FasL molecule to ascertain the access of the autoantibodies to the epitope. To this end, we generated a molecular model of the FasL-Fas trimolecular complex using a knowledge-based protein modelling method and the known tridimensional structures of the 55-kDa tumour necrosis factor receptor and lymphotoxin-α (TNF-α) [53]. The computer-predicted tertiary structure of the complex revealed that the aa 162–169 domain was located on the outermost side of FasL facing the Fas receptor (Fig. 5). Thus, it appears that this region of FasL could be recognized easily by the antibodies.
Fig. 5.
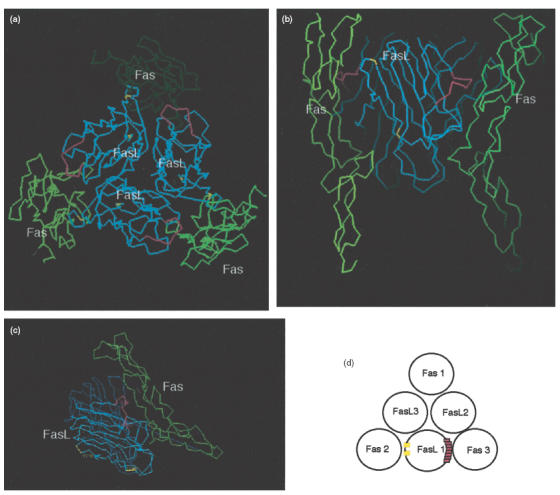
Molecular modelling of Fas/FasL trimolecular complex. (a–c) A molecular model of the FasL/Fas complex was generated by a knowledge-based protein modelling method and the known tridimensional structures of lymphotoxin α (TNF-α) and TNF receptor. The computer-mediated structure model predicted the tertiary structure of the complex. Regions coloured red indicate FasL aa 162–169. Regions coloured yellow show aa 206 and aa 218, both of which were reported to be involved in Fas/FasL molecular interaction. FasL aa 162–169 is located on the outermost side of FasL facing towards its receptor, Fas. (d) Schematic representation of Fas/FasL trimolecular complex. FasL aa 162–169 (FasL 1) faces towards the Fas molecule (Fas 3), as opposed to Fas (Fas 2), to which aa 206 and aa 218 of the same FasL molecule (FasL 1) faces.
The aa 163–179 epitope recognized by the anti-FasL autoantibodies with apoptosis-inhibiting potential clearly overlapped the aa 162–169 region predicted to be involved in Fas/FasL interaction by the molecular modelling. This result suggested that anti-FasL autoantibodies directed against aa 163–179 could potentially inhibit efficient molecular interaction between Fas and FasL, resulting in the inhibition of Fas/FasL-induced apoptosis of lymphocytes.
Apoptosis inducing activities of mutant FasL secreted by COS cells transiently transfected with FasL mutant cDNAs
The availability of an efficient system for the production of recombinant FasL allowed us to test the validity of our structural model for FasL by generating a number of mutants [52]. We performed the same experiments as described in Schneider et al. [52] and confirmed their findings that FasL aa P206 and Y218 were involved in Fas/FasL molecular interaction (Fig. 6). Due to the predicted accessibility of the aa 162–169 region of FasL to Fas, and the epitope determined using anti-FasL autoantibodies, the region aa 161–179 was chosen for mutagenesis. COS cells were transfected with empty pME18S vector, vector carrying full-length wild-type human FasL cDNA or one of a panel of mutant FasL cDNAs. After 52 h, the supernatants were recovered and assayed for cytotoxic activity using Jurkat-NU cells. We assessed sFasL concentrations by ELISA using antibodies that recognized the N-terminal region of sFasL. We also confirmed that all the mutant FasL proteins retained the proper conformation by immunoblotting using rabbit anti-FasL fragment 5 antibody, and by flow cytometric analysis of the cDNA-transfected COS cells using conformation-sensitive anti-FasL MoAbs 4A5 and NOK-2 (data not shown).
Fig. 6.
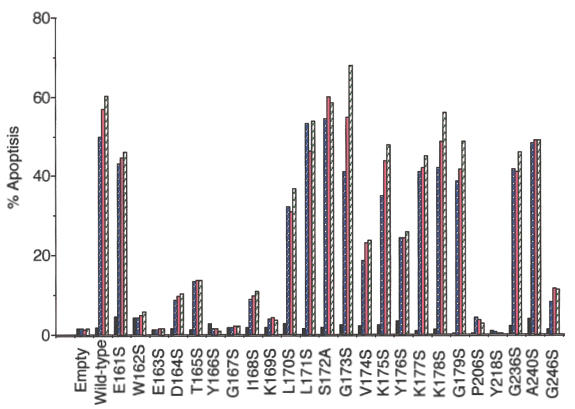
Apoptosis-inducing activities of recombinant wild-type and mutant FasL proteins secreted by COS cells. COS cells were transfected with pME18S empty vector, pME18S carrying the full-length human FasL cDNA, or FasL constructs carrying point mutants. The culture supernatants were recovered 52 h after transfection and assayed for sFasL by ELISA. Cytotoxic activity of the mutant sFasL was determined per ng of protein. Fas-expressing Jurkat-NU cells were cultured with the supernatants containing wild-type and mutant sFasL proteins for 24 h. Cell death was then estimated by DNA staining with PI. Data shown are representative of three independent experiments. ▪, Soluble FasL 0 ng/ml;  , 0·8 ng/ml;
, 0·8 ng/ml;  , 1·6 ng/ml;
, 1·6 ng/ml;  , 3·2 ng/ml.
, 3·2 ng/ml.
As shown in Fig. 6, supernatants from COS cells transfected with the wild-type human FasL expression plasmid contained potent apoptosis-inducing activity, which increased in a dose-dependent manner. Supernatants from COS cells transfected with most of the pME18S-FasL point mutant vectors (aa 162–169) showed no apoptosis-inducing activity (Fig. 6). However, the FasL point mutants (aa161, aa 170–179) showed almost comparable levels of apoptosis-inducing potential to wild-type FasL. These results suggested that human FasL mutations at positions aa 162–169 lost apoptosis-inducing potential, whereas mutations at positions aa161 and aa 170–179 retained apoptosis-inducing potential, indicating involvement of aa 162–169 in Fas/FasL molecular interaction.
Fine epitope mapping of anti-FasL antibodies with immunoblotting
To gain insights into the possible mechanisms involved in the development of the autoantibodies, we performed an aa sequence homology search of the epitope sequence using the blast search program at NCBI. We found highly homologous peptide sequences from various infections agents, including human immunodeficiency virus (HIV), human herpes viruses, Epstein–Barr virus (EBV), Staphylococcus and E. coli. Table 3 shows some of the sequences of potential importance.
Table 3.
Homologous peptide sequences with the epitope of anti-FasLautoantibodyA.A. s
| A.A. sequence | Description |
|---|---|
| 161 EWEDTYGIVL 170 | Human FasL (the epitope) |
| 130 TWNDTYGSNN 139 | (U35946) envelope glycoprotein (humanimmunodeficiency virus type 1) |
| 335 LWDQTYGVPD 344 | (P03189) V120 EBV capsid assemblyprotein BOLF1 human herpesvirus 4>(strain B95-8) |
| 114 EWGDA YVPER 123 | (P06492)HSV11 alpha trans-inducingprotein (VMW65)human herpesvirus 1(strain 17) |
| 297 EWES TYS ILG 306 | (U94706) phospho-N-muramic acid-pentapeptide translocase(Staphylococcus aureus) |
In order to define the epitope more precisely, we conducted a competitive inhibition study using several 10 aa peptides. The chemically synthesized peptides were: FasL aa 161–170 (EWEDTYGIVL), FasL aa 165–174 (TYGIVLLSGV), FasL aa 171–180 (LSGVKYKKGG), HIV type 1 derived peptide (TWNDTYGSNN), EBV-derived peptide (LWDQTYGVPD), and Staphylococcus-derived peptide (EWESTYSILG) (Table 3). While immunoblotting showed that the FasL peptide aa161–170 efficiently inhibited anti-FasL autoantibodies binding to recombinant FasL fragment 2, the FasL aa171–180 peptide did not inhibit binding and the FasL aa 165–174 peptide inhibited binding only when used at high concentrations (Fig. 7). These results showed clearly that one of the major epitopes of anti-FasL autoantibodies is located within the FasL aa 161–170 peptide.
Fig. 7.
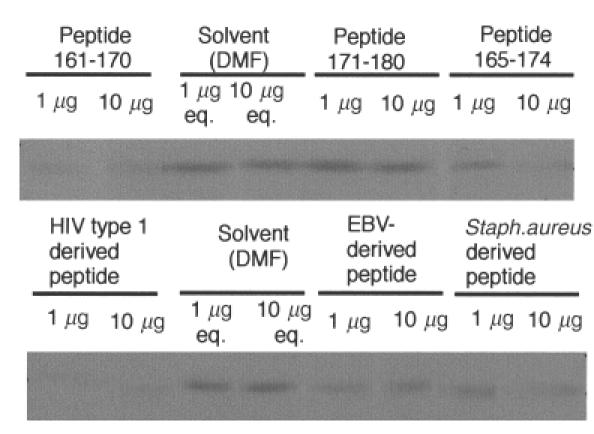
FasL-derived and infectious agent-derived synthetic peptides inhibit the binding of SLE patient anti-FasL autoantibodies to FasL fragment 2 by immunoblot analysis. An equal amount of recombinant FasL fragment 2 was run on a SDS-PAGE gel, transferred onto a membrane and reacted with anti-FasL autoantibodies in the presence of synthetic peptide. FasL peptide aa 161–170 and the HIV-derived peptide efficiently blocked autoantibody binding to FasL fragment 2, while FasL peptide aa 165–174 and the Staphylococcus-derived peptide moderately inhibited binding; eq. denotes equivalent amounts of solvent. We found that the peptide 161–170 inhibited the binding to the fragment 2 of the anti-FasL autoantibodies in four of four patients tested.
We next tested the effects of the highly homologous infectious agent-derived peptides on autoantibody binding. We found that the HIV-derived homologous peptide inhibited the binding efficiently, as did the Staphylococcus-derived peptide. However, autoantibody binding-inhibition by the EBV-derived peptide was less evident [54]. None the less, our results indicated that the anti-FasL autoantibodies could cross-react with peptides from several other organisms.
Aberrant expression of FasL on lymphocytes in patients with SLE
Immunoblot analysis was carried out to detect FasL protein expression on SLE PBMC using rabbit affinity-purified antihuman FasL fragment 5 antibody. While unstimulated PBMC from normal individuals did not express FasL protein, stimulation with PHA rapidly induced FasL protein expression, which suggested tight regulation of FasL expression upon activation in normal lymphocytes. However, in the majority of SLE patients, freshly isolated PBMC spontaneously expressed FasL protein (Fig. 8). PHA activation of PBMC from SLE patients failed to induce up-regulation of FasL expression. Furthermore, in some, but not all SLE patients (e.g. patients 1 and 2, Fig. 8), the extent of spontaneous FasL expression by PBMC far exceeded that of mitogen-induced FasL expression on PBMC from normal individuals. Thus, the autoantigen for the anti-FasL autoantibodies was aberrantly expressed on PBMC from SLE patients, such that FasL could become a stimulating (or ‘boosting’) antigen for the autoantibody secreting B lymphocytes.
Fig. 8.
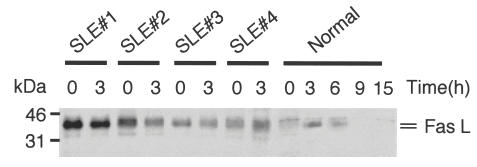
Aberrant FasL protein expression on peripheral blood lymphocytes from SLE patients. Freshly isolated PBMC from four SLE patients were used either immediately (0) or stimulated by 3 h culture with PHA (3). As a control, normal PBMC were stimulated with PHA for up to 15 h. An equal amount of proteins from the cell lysates was analysed by immunoblotting using affinity-purified antihuman FasL fragment 5 antibody. Results shown are representative of six independent experiments.
DISCUSSION
SLE is a disease characterized by the production of autoantibodies directed against various self-components. Autoantibody profiles in SLE suggest that tolerance to multiple unrelated antigens may be lost [16]. The FasL/Fas interaction induces apoptosis in Fas-expressing cells and is associated with the establishment and maintenance of peripheral tolerance [24–31].
In this study, we first conducted epitope mapping of the anti-FasL autoantibodies from SLE patients that inhibited Fas/FasL mediated apoptosis using deletion-mutant FasL proteins. The autoantibodies recognized a region that may play an important role in Fas/FasL molecular interactions.
We next developed rabbit antibodies against the FasL deletion-mutant proteins, which were then characterized functionally by measuring the ability of each antibody to block FasL-mediated apoptosis. Although this approach could not delineate precisely the regions containing the epitopes, we found that anti-FasL fragments 2, 3, 4 and 5 antibodies potently inhibited FasL mediated apoptosis, whereas the anti-FasL fragment 1 antibody did not. Thus, it appeared that fragment 2, but not fragment 1, contained the region important for FasL-mediated apoptosis. The antibody binding sites that blocked FasL-mediated apoptosis would be expected to overlap at least partially with the Fas-interacting region, while the epitopes of non-blocking antibodies (such as anti-FasL fragment 1 antibody) would most probably lie in regions distinct from the Fas/FasL interacting domains.
Our study suggested that the FasL aa 200–220 region was also involved in Fas/FasL interaction, as anti-FasL autoantibodies in SLE patients recognized FasL fragments 3 and II−. However, we did not characterize this region further, as Schneider et al. have already reported the importance of this region. Our study confirmed the importance of this region in Fas/FasL molecular interactions by both computer modelling and by the creation of point mutations in FasL at positions 206 and 218 (Fig. 6) [51].
Using knowledge-based computer modelling, we calculated the tertiary structure of the Fas/FasL trimolecular complex. The model also suggested that the FasL aa 162–169 region was important for interaction with Fas, rather than failure of FasL trimerization. To clarify further the functionally pivotal epitope for the anti-FasL autoantibodies, we introduced site-directed mutations into human FasL cDNA. The mutant FasL proteins retained the wild-type tertiary conformation because the conformation sensitive anti-FasL MoAbs 4A5 and NOK-2 recognized the mutant FasL proteins to the same extent as wild-type FasL. Also, the mutant FasL proteins expressed on the cell surface of the transfected COS cells were still able to be cleaved by MMP to become sFasL. We found that the FasL aa 162–169 region was important for apoptosis induction, as the FasL point mutants within this region failed to induce apoptosis. This finding confirmed the prediction of the computer-aided molecular modelling experiments that the region was involved in Fas/FasL molecular interaction. Thus, anti-FasL autoantibodies with apoptosis-inhibitory potential appeared to recognize an epitope at aa 162–169.
Immunoblotting analysis using synthetic peptides revealed that the binding of anti-FasL autoantibodies to FasL fragment 2 was inhibited by the addition of the peptide 161–170, but not peptide171–180, while peptide 165–174 only partially blocked binding. These results suggested further that a major epitope of anti-FasL autoantibodies was located within the FasL aa 161–170 region. Similarly, HIV- and Staphylococcus-derived peptides cross-reacted with the autoantibodies, which suggested that cross-reactivity or molecular mimicry may be involved in the production of autoantibody in SLE. Thus, the observation that the FasL aa 161–170 epitope may be shared with infectious agent-derived proteins is a major finding, as it suggests that pathogen infection could provoke the SLE autoimmune response.
We also found excessive FasL autoantigen expression on PBMC from SLE patients, but not from normal individuals or patients with RA. Lymphocytes from the majority of SLE patients showed much higher FasL expression levels than normal lymphocytes fully stimulated with PHA.
It has been suggested that molecular mimicry is a major cause of autoantibody production. One important question is why antibodies against nuclear proteins are detected only in patients with autoimmune disease, even though the nuclear protein autoantigens are ubiquitously present in cells of all individuals. More recent studies have suggested that the excessive apoptosis in patients with autoimmune disease provokes dissemination of nuclear proteins, and subsequently triggers autoantibody production in patients, but not in normal individuals [55]. Our results suggested that the aberrant expression of FasL on lymphocytes may be involved in the autoantigen-specific stimulation of autoantibody-secreting B lymphocytes, at least in some SLE patients.
Based on our findings, we propose the following hypothesis. Individuals who are genetically predisposed to developing SLE are infected first by particular pathogens that express a protein containing a peptide sequence homologous to human FasL aa 161–170. B lymphocytes then produce antibodies against the human FasL-like peptide. In predisposed individuals, the FasL aberrantly expressed on lymphocytes would then become a cross-reactive stimulating antigen for the B cells producing the antihuman FasL-like peptide antibody. Autoantigen-specific stimulation of the cross-reactive B cells would lead to the introduction of somatic mutations into the B cell IgV genes, and the subsequent selection by the autoantigen would lead to the development of an authentic autoantigen-specific immune response. The anti-FasL autoantibodies developed in these individuals would interfere with the appropriate induction of apoptosis in the self-reactive lymphocytes, and lead to manifestation of the disease.
While the infectious agents would also induce antihuman FasL-like peptide antibodies in normal individuals, the lack of lymphocytes expressing FasL would mean that the B cells would not be stimulated. Therefore, after removal of the infectious agents by the immune system, the human FasL-like peptide antibody response would subside, and the authentic autoantibody response would never develop.
Acknowledgments
This work was supported in part by the 1999–2001 Grant for the promotion of the advancement of education and research in graduate schools sponsored by the Promotion and Mutual Aid Corporation for Private Schools of Japan, the 2000–2001 SRF Foundation Grant for Biomedical Research (Tokyo, Japan) and the Grant from Comprehensive Research on Ageing and Health, Ministry of Health and Welfare of Japan and Japan Foundation for Ageing and Health.
REFERENCES
- 1.Hahn BH. Antibodies to DNA. N Engl J Med. 1998;338:1359–68. doi: 10.1056/NEJM199805073381906. [DOI] [PubMed] [Google Scholar]
- 2.Madaio MP. B cells and autoantibodies in the pathogenesis of lupus nephritis. Immunol Res. 1998;17:123–32. doi: 10.1007/BF02786437. [DOI] [PubMed] [Google Scholar]
- 3.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–6. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 4.Radic MZ, Weigert M. Origins of anti-DNA antibodies and their implications for B-cell tolerance. Ann NY Acad Sci. 1995;764:384–96. doi: 10.1111/j.1749-6632.1995.tb55853.x. [DOI] [PubMed] [Google Scholar]
- 5.Canbral AR, Alarcon-Segovia D. Autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 1998;10:409–16. doi: 10.1097/00002281-199809000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Elkon K. Autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 1995;7:384–8. doi: 10.1097/00002281-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Lefkowith JB, Gilkeson GS. Nephritogenic autoantibodies in lupus: current concepts and continuing controversies. Arthritis Rheum. 1996;39:894–903. doi: 10.1002/art.1780390605. [DOI] [PubMed] [Google Scholar]
- 8.Tsokos GC. Lymphocytes, cytokines, inflammation, and immune trafficking. Curr Opin Rheumatol. 1995;7:376–83. doi: 10.1097/00002281-199509000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Vyse TJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Annu Rev Immunol. 1998;16:261–92. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- 10.Tsao BP, Cantor RM, Kalunian KC, Wallace DJ, Hahn BH, Rotter JI. The genetic basis of systemic lupus erythematosus. Proc Assoc Am Physicians. 1998;110:113–7. [PubMed] [Google Scholar]
- 11.Elkon KB. Apoptosis in SLE – too little or too much? Clin Exp Rheumatol. 1994;12:553–9. [PubMed] [Google Scholar]
- 12.Mountz JD, Wu J, Cheng J, Zhou T. Autoimmune disease. A problem of defective apoptosis. Arthritis Rheum. 1994;37:1415–20. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- 13.Rose LM, Latchman DS, Isenberg DA. Bcl-2 and Fas, molecules which influence apoptosis. Autoimmunity. 1994;17:271–8. doi: 10.3109/08916939409010667. [DOI] [PubMed] [Google Scholar]
- 14.Carson DA, Ribeiro JM. Apoptosis and disease. Lancet. 1993;341:1251–4. doi: 10.1016/0140-6736(93)91154-e. [DOI] [PubMed] [Google Scholar]
- 15.Tax WJ, Kramers C, van Bruggen MC, Berden JH. Apoptosis, nucleosomes, and nephritis in systemic lupus erythematosus. Kidney Int. 1995;48:666–73. doi: 10.1038/ki.1995.336. [DOI] [PubMed] [Google Scholar]
- 16.Elkon K. Autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 1995;7:384–8. doi: 10.1097/00002281-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Takeno M, Nagafuchi H, Kaneko S, et al. Autoreactive T cell clones from patients with SLE support polyclonal autoantibody production. J Immunol. 1997;158:3529–38. [PubMed] [Google Scholar]
- 18.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–56. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 19.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–78. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 20.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi T, Tanaka M, Brannan CI, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–76. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins JA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 23.Suda T, Nagata S. Why do defects in the Fas-Fas ligand system cause autoimmunity? J Allergy Clin Immunol. 1997;100:S97–101. doi: 10.1016/s0091-6749(97)70013-7. [DOI] [PubMed] [Google Scholar]
- 24.Alderson MR, Tough TW, Davis-Smith T, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–7. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabelitz D, Pohl T, Pechhold K. Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes. Immunol Today. 1993;14:338–9. doi: 10.1016/0167-5699(93)90231-9. [DOI] [PubMed] [Google Scholar]
- 26.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/ (Fas/CD95) Nature. 1995;373:438–41. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 27.Brunner T, Mogil RJ, LaFace D, et al. Cell-autonomous Fas (CD95)/Fas–ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–4. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 28.Ju S-T, Panka DJ, Cui H, et al. Fas (CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–8. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 29.Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–71. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 30.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–69. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 31.Rathmell JC, Cooke MP, Ho WY, et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–4. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 32.Cheng J, Zhou T, Liu C, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–62. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 33.Suda T, Hashimoto H, Tanaka M, Ochi T, Nagata S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J Exp Med. 1997;186:2045–50. doi: 10.1084/jem.186.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courtney PA, Crockard AD, Williamson K, McConnell J, Kennedy RJ, Bell AL. Lymphocyte apoptosis in systemic lupus erythematosus: relationships with Fas expression, serum soluble Fas and disease activity. Lupus. 1999;8:508–13. doi: 10.1191/096120399678840765. [DOI] [PubMed] [Google Scholar]
- 35.Nozawa K, Kayagaki N, Tokano Y, Yagita H, Okumura K, Hasimoto H. Soluble Fas (APO-1, CD95) and soluble Fas ligand in rheumatic diseases. Arthritis Rheum. 1997;40:1126–9. doi: 10.1002/art.1780400617. [DOI] [PubMed] [Google Scholar]
- 36.Georgescu L, Vakkalanka RK, Elkon KB, Crow MK. Interleukin-10 promotes activation-induced cell death of SLE lymphocytes mediated by Fas ligand. J Clin Invest. 1997;100:2622–33. doi: 10.1172/JCI119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNally J, Yoo DH, Drappa J, et al. Fas ligand expression and function in systemic lupus erythematosus. J Immunol. 1997;159:4628–36. [PubMed] [Google Scholar]
- 38.Kovacs B, Liossis SN, Dennis GJ, Tsokos GC. Increased expression of functional Fas-ligand in activated T cells from patients with systemic lupus erythematosus. Autoimmunity. 1997;25:213–21. doi: 10.3109/08916939708994730. [DOI] [PubMed] [Google Scholar]
- 39.Lorenz HM, Grunke M, Hieronymus T, et al. In vitro apoptosis and expression of apoptosis-related molecules in lymphocytes from patients with systemic lupus erythematosus and other autoimmune diseases. Arthritis Rheum. 1997;40:306–17. doi: 10.1002/art.1780400216. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;98:1107–13. doi: 10.1172/JCI118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaishnaw AK, Toubi E, Ohsako S, et al. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum. 1999;42:1833–42. doi: 10.1002/1529-0131(199909)42:9<1833::AID-ANR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 42.Mysler E, Bini P, Drappa J, et al. The apoptosis-1/Fas protein in human systemic lupus erythematosus. J Clin Invest. 1994;93:1029–34. doi: 10.1172/JCI117051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakata K, Sakata A, Vela-Roch N, et al. Fas (CD95)-transduced signal preferentially stimulates lupus peripheral T lymphocytes. Eur J Immunol. 1998;28:2648–60. doi: 10.1002/(SICI)1521-4141(199809)28:09<2648::AID-IMMU2648>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki N, Ichino M, Mihara S, Kaneko S, Sakane T. Inhibition of Fas/Fas ligand-mediated apoptotic cell death of lymphocytes in vitro by circulating anti-Fas ligand autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:344–53. doi: 10.1002/1529-0131(199802)41:2<344::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 45.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 46.Brenner CA, Tam AW, Nelson PA, et al. Message amplification phenotyping (MAPPing): a technique to simultaneously measure multiple mRNAs from small numbers of cells. Biotechniques. 1989;7:1096–103. [PubMed] [Google Scholar]
- 47.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Meth. 1991;139:271–9. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 48.Kaneko S, Suzuki N, Koizumi H, Yamamoto S, Sakane T. Rescue by cytokines of apoptotic cell death induced by IL-2 deprivation of human antigen-specific T cell clones. Clin Exp Immunol. 1997;109:185–93. doi: 10.1046/j.1365-2249.1997.4191324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harlow ED, Lane D. Antibodies, a laboratory manual. New York: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 50.Kayagaki N, Yamaguchi N, Nagao F, et al. Polymorphism of murine Fas ligand that affects the biological activity. Proc Natl Acad Sci USA. 1997;94:3914–9. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanda Y, Tanaka Y, Shirakawa K, et al. Increased soluble Fas-ligand in sera of bone marrow transplant recipients with acute graft-versus-host disease. Bone Marrow Transplant. 1998;22:751–4. doi: 10.1038/sj.bmt.1701427. [DOI] [PubMed] [Google Scholar]
- 52.Schneider P, Bodmer JL, Holler N, et al. Characterization of Fas (Apo-1, CD95)–Fas ligand interaction. J Biol Chem. 1997;272:18827–33. doi: 10.1074/jbc.272.30.18827. [DOI] [PubMed] [Google Scholar]
- 53.Banner DW, D'Arcy A, Janes W, et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993;73:431–45. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 54.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein–Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997;100:3019–26. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shoenfeld Y, Alarcon-Segovia D, Buskila D, et al. Frontiers of SLE: review of the 5th International Congress of Systemic Lupus Erythematosus, Cancun, Mexico, April 20–25, 1998. Semin Arthritis Rheum. 1999;29:112–30. doi: 10.1016/s0049-0172(99)80042-0. [DOI] [PubMed] [Google Scholar]