

Abstract
The chronic liver disease primary biliary cirrhosis (PBC) is characterised by autoreactive B-cell and T-cell responses directed against mitochondrial antigens. In recent years these responses have been extensively characterised and the principal PBC associated autoantigen identified as pyruvate dehydrogenase complex (PDC). The identification of anti-PDC responses (present in over 95% of PDC patients) has given rise to important questions pertinent to our understanding of the pathogenesis of PBC. What specific role to anti-PDC responses play in target cell damage? How and why does immune tolerance break down to as highly conserved and ubiquitously expressed self-antigen as PDC? Why does breakdown in tolerance to an antigen present in all nucleated cells result in damage restricted to the intra-hepatic bile ducts? In attempting to answer these key questions we have, in this review, proposed a unifying hypothesis for the pathogenesis of PBC.
Keywords: animal models of disease, apoptosis, autoimmune disease, liver cirrhosis, biliary, molecular mimicry, xenobiotic
INTRODUCTION
Primary biliary cirrhosis (PBC) is a chronic liver disease with autoimmune features, characterised by destruction of the biliary epithelial cells (BEC) lining small intrahepatic bile ducts, and the progressive development of fibrotic chronic liver disease culminating in biliary cirrhosis [1]. It is a relatively common condition affecting up to 1 in 700 women over the age of 40 in the UK (the most commonly affected demographic group) [2]. A decade ago PBC was described as representing both a paradox and a paradigm for autoimmunity [3]. Over the last decade, significant progress has been made in our understanding and characterization of the autoimmune responses seen in PBC. Important questions remain, however, regarding the mechanism of tolerance breakdown and the link between the resulting autoreactive responses and target cell damage. In this review we will outline recent progress in our understanding of the immunological basis of PBC, and examine whether the equivocal description of 10years ago holds up to modern scrutiny. We will also propose a unifying hypothesis for disease aetiology which goes some way to reconciling the current, apparently contradictory, experimental data and disease pathogenesis models. It is our contention that whilst it may not yet be the beginning of the end for PBC, we may at least have reached the end of the beginning.
HUMORAL IMMUNE RESPONSES IN PBC
It has long been established that PBC is characterised by the presence of autoantibodies that are reactive with both mitochondrial and nuclear antigens. Anti-mitochondrial autoantibodies (AMA) reactive with the E2 components of the 2-oxo acid dehydrogenase complexes (in particular pyruvate dehydrogenase complex (PDC)), which are present in the serum of in excess of 95% of PBC patients and which represent a key diagnostic finding, have been extensively characterised in recent years. The characterization of the anti-E2 responses in PBC has been extensively reviewed elsewhere [4–6]. More recent studies have helped to clarify some of the outstanding issues regarding antibody responses to the non-PDC-E2 mitochondrial antigens implicated in PBC.
A significant proportion (>50%) of patients sera contain AMA that are reactive with the E1α component of PDC [7]. Although the antibody titres seen are an order of magnitude lower than those directed towards PDC-E2, it is perhaps surprising that little work has, to date, addressed the possible role of these autoantibodies in the immuno-pathogenesis of PBC. The over-expression of full length recombinant human E1α and several truncated internal polypeptides, and their use in epitope mapping studies using PBC sera, has revealed that reactivity is directed to the C-terminus of the molecule [8]. This is intriguing as the C-terminus contains the active site of the enzyme and anti-E1α antibodies have been shown to be inhibitory of PDC activity [9], a situation which is analogous to that seen with PDC-E2 [10]. These observations are consistent with findings from several other autoimmune diseases where autoantibodies have been shown to be specific for the functional sites of key enzymes [11]. Further work is required to study the binding of antibodies to epitopes around the active site, including the regulatory phosphorylation sites and cofactor binding motifs.
Antibodies reactive with branched chain 2-oxo acid dehydrogenase complex (BCOADC) E1α have recently been identified using highly purified human BCOADC as the antigen source [12]. The inhibitory capacity of this antibody appears not to have been tested as yet. It will be of interest to see if the auto-epitope of this enzyme also proves to be located in the catalytic domain, which also contains regulatory phosphorylation sites. The lack of detectable autoantibody reactivity to the E1 subunit of the 2-oxoglutarate dehydrogenase complex (OGDC) may be a consequence of this enzyme not being regulated by reversible phosphorylation [13].
Secretory IgA anti-PDC is present in saliva [14–16], bile [17] and urine [18] mimicking the distribution of tissue damage in PBC and suggesting at least some mucosal targeting of the autoantibody response. These antibodies retain the PDC inhibitory activity characteristic of serum anti-PDC [10,19–21].
Anti-nuclear autoantibodies (ANA) are seen at a lower frequency (only being present in about a third of PBC patients) and are seen more frequently and at much higher titres in the small subgroup anti-PDC negative PBC patients [22–24]. Autoantibodies specific for the proteins of the nuclear pore complex (gp210, p62) may be associated with more active or severe disease [25]. This may also be true for the autoantibodies that react with proteins (Sp100 and PML) forming the antigenic targets that produce the multiple nuclear dot (MND) pattern revealed by direct immunofluorescence [23,26]. Another subset of auto antibodies previously reported to be reactive with carbonic anhydrase II [27] have more recently been described as a nonspecific marker of autoimmunity rather than being associated with AMA-negative PBC [28].
CELLULAR IMMUNE RESPONSES IN PBC
BEC damage in PBC is seen in the context of a mixed portal tract inflammatory response. T-cells predominate, with CD8+ cells particularly prominent in the peri-ductular areas [29,30]. The cellular infiltrate includes significant numbers of eosinophils (especially in early disease), with RANTES expression by biliary epithelium being implicated in their accumulation [31]. Heterogeneous expression of ‘Th1’ and ‘Th2’ cytokine patterns is reported in liver [32–35]. Limited studies have described increased serum IL-18 levels particularly in advanced PBC and declining numbers of peripheral blood IL-4 producing CD4 cells [36,37].
CD4+ and CD8+ T-cells reactive with PDC are present in the peripheral blood and liver infiltrating T-cell populations in the majority of PBC patients and absent from controls [34,38–41]. Such responses are seen against native human PDC derived from heart muscle [42] confirming that T-cell responses to PDC in PBC are truly autoreactive in nature [43]. PDC reactivity is universal in PBC patient derived peripheral blood T-cells (and absent from controls) when cocultured with PDC pulsed autologous dendritic cells [44] suggesting that apparent absence of response to PDC in primary peripheral blood mononuclear cell (PBMC) culture in some PBC patients simply reflects culture artefact. T-cell responses appear to be principally directed against PDC-E2 [34,40,45] although studies of T-cell responses to the other component subunits of PDC have, to date, been limited [46]. An HLA DRB4*0101 restricted epitope spanning PDC-E2163–176 (the lipoic acid binding site in the inner lipoyl domain) has been identified [45] and extensively characterised [47]. Whether this epitope is unique, or even, for that matter, dominant, is at present unclear [48]. Recent work has characterised peripheral blood derived HLA-A2 restricted CD8+ T-cell lines reactive with PDC-E2159–167 [49]. At present no data regarding the antigen specificity of liver infiltrating CD8+ cells and the cytotoxic activity of the PBMC derived lines are available.
TARGET CELL BIOLOGY IN PBC
Both in situ and in vitro studies of human BEC, the target cell in PBC, have demonstrated expression of a number of important T cell ligands. On resting cells these include class I MHC antigens and adhesion receptors such as ICAM-1 [50]. Additionally, these epithelial cells express E-cadherin and potentially interact with the αEβ7-integrin (CD103) on T cells with an intraepithelial phenotype; such T cells have been observed in the liver [5]. Following stimulation by pro-inflammatory cytokines such as IFN-γ the cells also express high levels of class II MHC antigens [51]. Despite expression of these ligands, studies have failed to identify expression of the costimulatory ligands B7-1 (CD80) or B7-2 (CD86) on resting or activated cells [52]; this is consistent with the failure of BEC to present antigen to and directly activate resting T cells [53].
The capacity for cytokine-stimulated human BEC to form high-affinity adhesive bonds with T lymphocytes has been demonstrated by application of a sensitive flow cytometric assay [54]. Combination of this system with antibody blockade of specific adhesion molecules has allowed demonstration of the major contributions made by ICAM-1 and, to a lesser extent, LFA-3 to the adhesion of T cells to cultured BEC. These adhesive interactions are essential for effective induction of BEC cytolysis by activated lymphocytes [54].
The PBC autoantigen PDC is located on the inner surface of the inner mitochondrial membrane and is therefore normally separated from the extra-cellular immune system by three membranes. However, it has been reported that PDC-like epitopes are present on the surface of BEC within or freshly cultured from PBC liver samples [55]. Clearly this observation has significance for the aetiology of PBC. It is known that several apoptogenic proteins, including cytochrome c, are released from the mitochondrial intermembrane space at an early stage during the induction of apoptosis [56]. Studies from our group have shown that PDC is released from apoptotic mitochondria to the cytoplasm within 6 h of the induction of apoptosis, and that autoreactive epitopes are present on the still-intact cell surface at later time points [57]. It has been argued that BEC are particularly susceptible to this process, as other cell types efficiently ‘delete’ cytoplasmic PDC by glutathiolation, which eliminates the autoreactive epitope [58].
OUTSTANDING QUESTIONS
Key questions remain to be answered, however, if we are to fully understand the immuno-pathogenesis of PBC. Recent observations have allowed us to at least attempt to answer these questions.
What is the role (if any) of anti-PDC immune responses in BEC damage?
Until recently there have been few data to directly implicate PDC specific autoreactive immune responses in target cell damage. The available data suggest that anti-PDC antibody responses play little if any role in target cell damage. IgG anti-PDC responses are seen in patients with some bacterial infections in the apparent absence of the clinical features of PBC [59,60]. Moreover, the induction of high titre anti-PDC responses in mice by sensitization [61,62], and passive transfer of anti-PDC into naïve mice [63] are not associated, in isolation, with disease induction. The intriguing hypothesis that the secretory IgA anti-PDC identified in the secretions of PBC patients [15,16] causes BEC damage as a result of intra–cellular interaction with PDC [64] during transcytosis [5,65] appears not to have been born out [66].
In the absence of direct studies of BEC-directed cytotoxicity all data regarding the role played by autoreactive T-cells in BEC damage remain circumstantial. The body of such evidence is, however, strong. CD4+ and CD8+ T-cells reactive with self-PDC are present in peripheral blood. Affected portal tracts contain both CD4+ and CD8+ cells, the former showing specificity for self-PDC (the specificity of the latter not having been addressed yet), with a higher precursor frequency than for PBMC [41]. Apoptosis of the BEC in affected portal tracts is seen [67–69] in the context of localised Granzyme B transcription [70]. Recent observations suggesting that the induction of autoreactive T-cell responses to PDC is temporally associated with the development of bile duct lesions in an SJL/J mouse model is the strongest evidence to date to implicate self-PDC specific T-cell responses in bile duct damage [71,72] (although the precise relationship of such damage to that seen in humans in PBC remains unclear and is the source of some debate [73]).
What is the mechanism of breakdown of T-cell tolerance to PDC, a highly conserved and ubiquitously distributed self-antigen?
Evidence from murine modelling studies suggests that breakdown of tolerance to self-PDC at the B-cell level is relatively easy to achieve but is not, in isolation, associated with development of pathology [61]. This mirrors the observations made in human infectious disease models. Tolerance breakdown in such models results, we would suggest, entirely from cross-reactivity at the B-cell level. The development of breakdown of T-cell self-tolerance to PDC is, in contrast, a much more highly restricted phenomenon and, we further suggest, the key step in disease pathogenesis. Several models have been proposed for how such breakdown in T-cell tolerance to self-PDC might occurs in humans.
(i) The molecular mimicry model. Several studies have demonstrated cross-reactivity, at both the B-cell [74,75] and T-cell [76] level, between PDC and polypeptides derived from the sequences of potential pathogens. It is suggested that molecular mimicry between pathogen and self-antigen results in tolerance breakdown. Three conceptual problems arise with such models. The first is that the most prevalent ‘mimic’ of self-PDC is obviously bacterial PDC which is immunogenic but not, it would appear, pathogenic. The second problem is that, although studies demonstrating reactivity with epitopes derived from pathogen genetic sequences are tantalising they fail to show whether such potentially cross-reactive epitopes are generated in vivo during natural infection. Finally, a recent study addressing changes in specificity of human anti-PDC antibodies during affinity maturation argues directly against such molecular mimicry models [77].
Despite the lack of evidence to support simple molecular mimicry models, several findings do suggest some role for bacterial infection, with increased prevalence of bacterial infection in PBC patients [78,79], serological evidence of specific previous infection [80] and bacterial products being present in the mononuclear cells surrounding damaged interlobular bile ducts [81]. Features of the host-response seen in PBC such as the presence of MCP-2 and MCP-3 expressing mononuclear cells in the portal tract infiltrate and around the periphery of the archetypal epithelioid granulomata have also been interpreted as suggesting a role for localised bacterial infection [82]. Intriguingly, a role for bacterial DNA (rich in CpG dinucleotide repeats which are ligands for TLR9) in the induction of autoimmune responses has been proposed in both PBC and other autoimmune disease [83–85].
(ii) The ‘altered-self’ model. An alternative suggestion for the mechanism of breakdown of tolerance to self-PDC in PBC is that reactivity arises in response to a modified form of self, with subsequent reactivity to intact self-PDC. Two scenarios have been suggested. In the first, reactivity arises to self-PDC in the BEC following modification by xenobiotics excreted in the bile. This model is supported by the observation that AMA from PBC patients show greater reactivity to some synthetic structures designed to mimic xenobiotically modified lipoyl haptens than to native lipoylated PDC [86]. The second scenario is that PDC-E2 undergoes modification within cells undergoing apoptosis, generating novel or cryptic epitopes, leading to cross-priming of autoreactive T-cells by dendritic cells (DC) [57] and tolerance breakdown through epitope spreading. There is certainly evidence to suggest that BEC apoptosis occurs in PBC although this has to date been interpreted as representing the consequences of effector cell function [67–69,87]. As outlined above, BEC, in contra-distinction to other cell types, have been demonstrated to retain immunogenic PDC-E2 whilst undergoing apoptosis [58]. Caspase cleavage of PDC-E2 in vitro has been shown to generate potentially immunogenic protein fragments [88]. In this model, a primary aetiological factor would induce apoptosis of BEC, triggering tolerance breakdown through the liberation (during cleavage of PDC by caspases and other apoptotic mediators) of cryptic epitopes. As BEC killing would be, to a significant degree, mediated through the medium of apoptosis a cycle of ongoing damage would be established. A strength of this model is that it might help to explain the surface expression of PDC derived epitopes on BEC as cells undergoing apoptosis have previously been demonstrated to express other highly conserved autoantigens [89]. DC presentation of epitopes derived from phagocytosed apoptotic cells, generating productive immunity, has been demonstrated [90]. The outcome of such presentation is conventionally believed to be tolerance (and, indeed to be an important mechanism of induction and maintenance of peripheral tolerance). We would have to hypothesise therefore that for auto-reactivity to be induced, presentation of apoptotic BEC derived cryptic PDC derived epitopes would have to occur in an inflammatory (and therefore DC activating) environment. Perhaps this is another role for the mucosal bacterial infections identified to occur at increased frequency in PBC patients?
(iii) The endogenous retrovirus model. A study, to date published in abstract form only, has described the isolation of retroviral sequences from the BEC of PBC patients [91]. This has led to the suggestion that PBC is triggered through the actions of an endogenous retrovirus [92]. The potential for such an agent to generate the apoptosis necessary for altered-self models of tolerance breakdown is clear. The question of the role of endogenous retroviruses in the aetiology of autoimmunity is, however, deeply controversial [93] and further work is badly needed in this area in PBC.
How does breakdown of tolerance to self-pdc, a ubiquitous protein complex, result in target cell damage with such a restricted distribution?
There is a growing consensus that the specificity of tissue damage in PBC is a result of the unique microenvironment of the biliary tree. BEC are exposed to a highly unusual and potentially toxic environment containing both agents excreted in the bile (e.g. xenobiotics able to modify PDC-E2 or heavy metals able induce apoptosis [94]) and ascending infectious agents (bacterial cofactors or, conceivably, retroviral agents).
A UNIFYING HYPOTHESIS FOR THE PATHOGENESIS OF PBC
Emerging data from murine modelling studies allow us to reconcile these conflicting models. SJL/J mice are unresponsive to sensitization with self-PDC. Sensitization with foreign-PDC induces, over the short-term, antibody cross-reactive with self-PDC but no breakdown of T-cell self-tolerance [72]. Co-sensitization with self and foreign-PDC, in contrast, induces rapid breakdown of T-cell tolerance to self-PDC in the context of autoreactive antibodies (the immunological motif of PBC). The induction of B-cell responses cross-reactive with self-PDC does not, in isolation therefore lead to T-cell tolerance breakdown but acts as a cofactor for such tolerance breakdown if it occurs in the context of immunological exposure to self-PDC (in this context, antigen in the presence of adjuvant). Possible mechanisms whereby antiself antibody responses might promote T-cell tolerance breakdown are through presentation of self-PDC by activated cross-reactive B-cells [95–98] or uptake, processing and presentation of complexes of self-PDC and anti-PDC by dendritic cells [99]. In our murine model, foreign PDC (of bovine origin) shows a sufficient sequence difference to mouse PDC to allow a significant immune response, but sufficient similarity for the resulting immune response to be cross-reactive at the B-cell level (Fig. 1a). In a human model, either foreign but cross-reactive PDC of bacterial origin, cleaved self-PDC from apoptotic cells, or xenobiotic altered self-PDC could act to induce B-cell responses cross-reactive with native self-PDC and similarly able to promote T-cell tolerance breakdown in the correct immunogenetically susceptible individual and inflammatory context (Fig. 1b). The implication of this model, if correct, is that all the suggested aetiological pathways would converge in a final common pathway of cross-reactive B-cell promoted tolerance breakdown in an inflammatory environment containing self-PDC released from damaged cells.
Fig. 1.
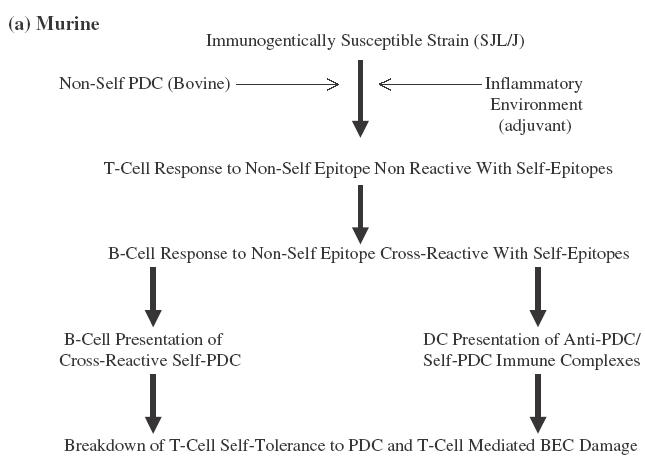
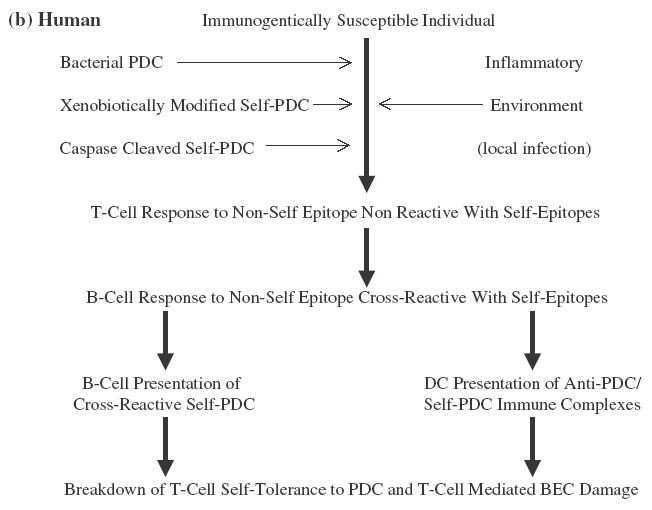
Suggested model for the breakdown of self-tolerance in (a) a murine model and (b) humans with PBC. In both cases the model hinges on the initial development of an immune response to a nonself form of PDC (bovine in the mouse model, bacterial or xenobiotically modified or caspase cleaved self in the suggested human model). Sufficient sequence diversity exists in each case between nonself and the equivalent self-PDC to allow the development, in the correct inflammatory environment (adjuvant driven in the murine model, secondary to local infection in the human model) of non-cross-reactive T-cell responses. There is, in each case, however, sufficient similarity between the priming nonself-PDC and self-PDC to allow full B-cell cross-reactivity. Cross-reactive B-cell responses then promote epitope spreading within the priming nonself-PDC variant from nonconserved to conserved epitopes resulting in the breakdown of T-cell tolerance to self-PDC characteristic of both the murine model and human PBC. Alternative suggested mechanisms for this B-cell effect are direct presentation of self-PDC by cross-reactive activated B-cells and uptake of complexes of self-PDC and cross-reactive antinon-self-PDC by professional antigen presenting cells. Self-PDC is present in the murine model as a result of deliberate cosensitization. It would be suggested to be present in the local microenvironment in the human model as a result of release by necrotic PDC rich cells.
This model may help us to understand some of the unusual, and as yet unexplained, features of PBC. These include the apparent restriction of the disease to the biliary tree (and, to a lesser extent the salivary gland) despite the universal expression of the autoantigen PDC, the apparent restriction of the disease to adults, and the recurrence of the disease following liver transplantation. One of the key aspects of the model is the prominent role played by environmental factors in the early disease stages. The tissue tropism of the disease could result from restricted exposure to the environmental trigger (through biliary excretion of PDC modifying xenobiotics or agents such as heavy metals able to induce apoptosis or, in the case of pathogens, agents ascending from the GI tract). Similarly, age related exposure patterns (e.g. the encountering of bioactive compounds such as drugs not typically given to children or work-place toxins) or cumulative exposure resulting in toxic levels being achieved only after many years might explain the age of onset of PBC. The prominent role played by apoptosis in the model may explain disease recurrence in MHC mismatched liver transplant recipients (a simple recurrence of autoreactivity being likely in MHC matched recipients). In this regard it is intriguing to note the increased rate of disease recurrence reported in patients receiving primary immuno-suppressive therapy in the form of tacrolimus compared with patients receiving cyclosporin [100]. Cyclosporin has the additonal property, not shared with tacrolimus of blocking the mitochondrial permeability transition (MPT) rendering cells, to some extent at least, resistant to apoptosis [101].
REFERENCES
- 1.Neuberger JM. Primary biliary cirrhosis. Lancet. 1997;350:875–9. doi: 10.1016/S0140-6736(97)05419-6. [DOI] [PubMed] [Google Scholar]
- 2.James OFW, Bhopal R, Howel D, Gray J, Burt AD, Metcalf JV. Primary biliary cirrhosis once rare, now common in the UK? Hepatology. 1999;30:390–4. doi: 10.1002/hep.510300213. [DOI] [PubMed] [Google Scholar]
- 3.Gershwin ME, Mackay IR. Primary biliary cirrhosis: paradigm or paradox for autoimmunity. Gastroenterology. 1991;100:822–3. doi: 10.1016/0016-5085(91)80033-6. [DOI] [PubMed] [Google Scholar]
- 4.Berg PA, Klein R, Lindenborn-Fotinos J. Antimitochondrial antibodies in primary biliary cirrhosis. J Hepatol. 1986;2:123–31. doi: 10.1016/s0168-8278(86)80015-0. [DOI] [PubMed] [Google Scholar]
- 5.Yeaman SJ, Kirby JA, Jones DEJ. Autoreactive responses to pyruvate dehydrogenase complex in the pathogenesis of primary biliary cirrhosis. Immunol Rev. 2000;174:238–49. doi: 10.1034/j.1600-0528.2002.00021h.x. [DOI] [PubMed] [Google Scholar]
- 6.Jones DEJ. Autoantigens in primary biliary cirrhosis. J Clin Path. 2000;53:1–8. doi: 10.1136/jcp.53.11.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fussey SPM, Bassendine MF, Fittes D, Turner IB, James OFW, Yeaman SJ. The E1α and β subunits of the pyruvate dehydrogenase complex are M2′d'and M2′e′ autoantigens in primary biliary cirrhosis. Clin Sci. 1989;77:365–8. doi: 10.1042/cs0770365. [DOI] [PubMed] [Google Scholar]
- 8.Palmer J, Yeaman S, Jones D. Epitope Specificity of Anti-PDC E1alpha Antibodies in Primary Biliary Cirrhosis. J. Hepatol. 2001;34:214. 10.1016/S0168-8278(01)80793-5. [Google Scholar]
- 9.Fregeau DR, Prindiville T, Coppel RL, Kaplan M, Dickinson RE, Gerswin ME. Inhibition of α-ketoglutarate dehydrogenase activity by a distinct population of autoantibodies recognising dehydrolipamide succinyltranferase in primary biliary cirrhosis. Hepatology. 1990;11:975–80. doi: 10.1002/hep.1840110611. [DOI] [PubMed] [Google Scholar]
- 10.Van de Water J, Fregeau D, Davis P, Ansari A, Danner D, Leung P, Coppel R, Gershwin ME. Autoantibodies of primary biliary cirrhosis recognise dihydrolipoamide acetyltransferase and inhibit enzyme function. J Immunol. 1988;141:2321–4. [PubMed] [Google Scholar]
- 11.Leung P, Gershwin M. The Molecular Structure of Autoantigens. Curr Opin Immunol. 1990;2:567–75. doi: 10.1016/0952-7915(90)90012-6. [DOI] [PubMed] [Google Scholar]
- 12.Mori T, Ono K, Hakozaki M, Kasukawa R, Kochi H. Autoantibodies of sera from patients with primary biliary cirrhosis recognize the alpha subunit of the decarboxylase component of human branched-chain 2-oxox acid dehydrogenase complex. J Hepatol. 2001;34:799–804. doi: 10.1016/s0168-8278(01)00027-7. [DOI] [PubMed] [Google Scholar]
- 13.Reed LJ, Yeaman SJ. Pyruvate Dehydrogenase. The Enzymes. 1987;XVIII:77–95. [Google Scholar]
- 14.Palmer JM, Jones DEJ, Doshi M, Yeaman SJ, Bassendine MF, Kirby JA. Secretory IgA anti-PDC in primary biliary cirrhosis: a novel mechanism for tissue damage? Hepatology. 1998;28:541A. [Google Scholar]
- 15.Palmer JM, Doshi M, Kirby J, Yeaman SJ, Bassendine MF, Jones DEJ. Secretory autoantibodies in primary biliary cirrhosis. Clin Exp Immunol. 2000;122:1–7. doi: 10.1046/j.1365-2249.2000.01403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynoso-Paz S, Leung PSC, Van de Water J, et al. Evidence for a locally driven mucosal response and the presence of mitochondrial antigens in saliva in primary biliary cirrhosis. Hepatology. 2000;31:24–9. doi: 10.1002/hep.510310106. [DOI] [PubMed] [Google Scholar]
- 17.Nishio A, Van de Water J, Leung PS, et al. Comparative studies of antimitochondrial autoantibodies in sera and bile in primary biliary cirrhosis. Hepatology. 1997;25:1085–9. doi: 10.1002/hep.510250506. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka A, Nalbandian G, Leung PSC, et al. Mucosal immunity and primary biliary cirrhosis: Presence of anti-mitochondrial antibodies in urine. Hepatology. 2000;32:910–5. doi: 10.1053/jhep.2000.19254. [DOI] [PubMed] [Google Scholar]
- 19.Rowley MJ, McNeilage JL, Armstrong JM, Mackay IR. Inhibitory autoantibody to a conformational epitope of the pyruvate dehydrogenase complex. Clin Immunol Immunopathol. 1991;60:356–70. doi: 10.1016/0090-1229(91)90093-p. [DOI] [PubMed] [Google Scholar]
- 20.Teoh KL, Mackay IR, Rowley MJ, Fussey SPM. Enzyme inhibitory autoantibodies to pyruvate dehydrogenase complex in primary biliary cirrhosis differ for mammalian, yeast and bacterial enzymes: implications for molecular mimicry. Hepatology. 1994;19:1029–33. [PubMed] [Google Scholar]
- 21.Ikuno N, Mackay IR, Jois J, Omagari K, Rowley MJ. Antimitochondrial autoantibodies in saliva and sera from patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2001;16:1390–4. doi: 10.1046/j.1440-1746.2001.02624.x. [DOI] [PubMed] [Google Scholar]
- 22.Michieletti P, Wanless I, Katz A, Scheuer P, Yeaman S, Bassendine M, Palmer J, Heathcote E. Antimitochondrial antibody negative primary biliary cirrhosis. A distinct syndrome of autoimmune cholangitis. Gut. 1994;35:260–5. doi: 10.1136/gut.35.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szostecki C, Guldner HH, Will H. Autoantibodies against ‘nuclear dots’ in primary biliary cirrhosis. Semin Liver Dis. 1997;17:71–8. doi: 10.1055/s-2007-1007184. [DOI] [PubMed] [Google Scholar]
- 24.Courvalin J-C, Worman HJ. Nuclear envelope protein autoantibodies in primary biliary cirrhosis. Semin Liver Dis. 1997;17:79–90. doi: 10.1055/s-2007-1007185. [DOI] [PubMed] [Google Scholar]
- 25.Invernizzi P, Podda M, Battezzati PM, et al. Autoantibodies against nuclear pore complexes are associated with more active and severe liver disease in primary biliary cirrhosis. J Hepatol. 2001;34:366–72. doi: 10.1016/s0168-8278(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 26.Zuchner D, Sternsdorf T, Szostecki C, Heathcote E, Cauch-Dudek K, Will H. Prevalence, kinetics and therapeutic modulation of autoantibodies against Sp100 and promyelocytic leukaemia protein in a large cohort of patients with primary biliary cirrhosis. Hepatology. 1997;26:1123–30. doi: 10.1002/hep.510260506. [DOI] [PubMed] [Google Scholar]
- 27.Gordon S, Quattrociocchi-Longe T, Khan B, Kodali V, Chen J, Silverman A, Kiechle F. Antibodies to carbonic anhydrase in patients with immune cholangiopathies. Gastroenterology. 1995;108:1802–9. doi: 10.1016/0016-5085(95)90143-4. [DOI] [PubMed] [Google Scholar]
- 28.Comay D, Cauch-Dudek K, Hemphill D, Diamandis E, Wanless I, Heathcote EJ. Are antibodies to carbonic anhydrase II specific for anti-mitochondrial antibody-negative primary biliary cirrhosis. Dig Dis Sci. 2000;45:2018–21. doi: 10.1023/a:1005548126211. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkland A, Loof L, Mendel-Hartvig I, Totterman TH. Primary biliary cirrhosis. High proportion of B-cells in blood and liver tissue produce anti-mitochondrial antibodies of several Ig classes. J Immunol. 1994;153:2750–7. [PubMed] [Google Scholar]
- 30.Leon MP, Spickett G, Jones DEJ, Bassendine MF. CD4+ T-cell subsets defined by isoforms of CD45 in primary biliary cirrhosis. Clin Exp Immunol. 1995;99:233–9. doi: 10.1111/j.1365-2249.1995.tb05538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuneyama K, Yasoshima M, Hiramatsu K, Harada K, Gershwin ME, Nakanuma Y. A putative role for eotaxin and RANTES in primary biliary cirrhosis: Eosinophilic infiltration and damaged bile ducts. Hepatol Res. 1999;16:68–77. [Google Scholar]
- 32.Harada K, Van de Water J, Leung PS, Coppel RL, Ansari AA, Nakanuma Y, Gershwin ME. In situ nucleic acid hybridisation of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25:791–6. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 33.Berg PA, Klein R, Rocken M. Cytokines in primary biliary cirrhosis. Semin Liver Dis. 1997;17:115–23. doi: 10.1055/s-2007-1007189. [DOI] [PubMed] [Google Scholar]
- 34.Van de Water J, Ansari A, Prindiville T, et al. Heterogeneity of autoreactive T-cell clones specific for the E2 component of the pyruvate dehydrogenase complex in primary biliary cirrhosis. J Exp Med. 1995;181:723–33. doi: 10.1084/jem.181.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–76. doi: 10.1136/gut.49.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekiya H, Komatsu T, Isono E, Furukuwa M, Matsushima S, Yamaguchi N, Yamauchi K, Hayashi N. Decrease in the prevalence of IL-4 producing CD4+ T cells in patients with advanced stage primary biliary cirrhosis. Am J Gastroenterol. 1999;94:3589–94. doi: 10.1111/j.1572-0241.1999.01547.x. [DOI] [PubMed] [Google Scholar]
- 37.Yamano T, Higashi T, Nouso K, et al. Serum interferon-gamma-inducing factor/IL-18 levels in primary biliary cirrhosis. Clin Exp Immunol. 2000;122:227–31. doi: 10.1046/j.1365-2249.2000.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van de Water J, Ansari AA, Surh CD, Coppel R, Roche T, Donkovsky HB, Kaplan M, Gershwin ME. Evidence for the targeting by 2-oxo-dehydrogenase enzymes in the T-cell response of primary biliary cirrhosis. J Immunol. 1991;146:89–94. [PubMed] [Google Scholar]
- 39.Lohr H, Fleischer B, Gerken G, Yeaman SJ, Meyer-zum-Buschenfelds KH, Manns M. Autoreactive liver-infiltrating T cells in primary biliary cirrhosis recognise inner mitochondrial epitopes and the pyruvate dehydrogenase complex. J Hepatol. 1993;18:322–7. doi: 10.1016/s0168-8278(05)80276-4. [DOI] [PubMed] [Google Scholar]
- 40.Jones DEJ, Palmer JM, Yeaman SJ, James OFW, Bassendine MF, Diamond AG. T-cell responses to the components of pyruvate dehydrogenase complex in primary biliary cirrhosis. Hepatology. 1995;21:995–1002. [PubMed] [Google Scholar]
- 41.Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T-cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–40. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer JM, Bassendine MF, James OFW, Yeaman SJ. Human pyruvate dehydrogenase complex as an autoantigen in primary biliary cirrhosis. Clin Sci. 1993;85:289–93. doi: 10.1042/cs0850289. [DOI] [PubMed] [Google Scholar]
- 43.Jones DEJ, Palmer JM, Yeaman SJ, Bassendine MF, Diamond AG. T-cell responses to natural human proteins in primary biliary cirrhosis. Clin Exp Immunol. 1997;116:562–8. doi: 10.1046/j.1365-2249.1997.3101202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akbar SMF, Yamamoto K, Ninomiya T, Abe M, Hiasa Y, Masumoto T, Horiike N, Onji M. Peripheral blood T-cell responses to pyruvate dehydrogenase complex in primary biliary cirrhosis. Role of antigen-presenting cells. Eur J Clin Invest. 2001;31:639–46. doi: 10.1046/j.1365-2362.2001.00847.x. [DOI] [PubMed] [Google Scholar]
- 45.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-resticted immunodominant T-cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune disease. J Exp Med. 1995;181:1835–45. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robe AJ, Palmer JM, Yeaman SJ, Jones DEJ. The E3 BP (protein X) component of pyruvate dehydrogenase complex (PDC) is not a T-cell autoantigen in primary biliary cirrhosis (PBC) J Hepatol. 2000;32:128. [Google Scholar]
- 47.Shigematsu H, Shimoda S, Nakamura M, et al. Fine specificity of T cells reactive to human PDC-E2, 163–176 peptide, the immunodominant autoantigen in primary biliary cirrhosis: implications for molecular mimicry and cross–recognition among mitochondrial autoantigens. Hepatology. 2000;32:901–909. doi: 10.1053/jhep.2000.18714. [DOI] [PubMed] [Google Scholar]
- 48.Palmer JM, Diamond AG, Yeaman SJ, Bassendine MF, Jones DEJ. T-cell responses to the putative autoepitope in primary biliary cirrhosis. Clin Exp Immunol. 1999;116:133–9. doi: 10.1046/j.1365-2249.1999.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kita H, Lian Z, Van de Water J, et al. Identification of HLA-A2-restricted CD8+ cytotoxic T-cell responses in primary biliary cirrhosis: T-cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–23. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leon MP, Bassendine MF, Thick M, Gibbs P, Burt AD, Kirby JA. Hepatic allograft rejection. regulation of the immunogenicity of human intrahepatic biliary epithelial cells. J Liver Transp Surg. 1996;2:37–45. doi: 10.1002/lt.500020107. [DOI] [PubMed] [Google Scholar]
- 51.Ayres RCS, Neuberger JM, Shaw J, Joplin R, Adams DH. Intercellular-adhesion molecule-1 and MHC antigens on human intra-hepatic bile duct cells-effects of pro-inflammatory cytokines. Gut. 1993;34:1245–9. doi: 10.1136/gut.34.9.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leon MP, Kirby JA, Gibbs P, Burt AD, Bassendine MF. Immunogenicity of biliary epithelial cells: study of the expression of B7 molecules. J Hepatol. 1995;22:591–5. doi: 10.1016/0168-8278(95)80456-0. [DOI] [PubMed] [Google Scholar]
- 53.Leon MP, Bassendine MF, Wilson JL, Ali S, Thick M, Kirby JA. Immunogenicity of biliary epithelium: investigation of antigen presentation to CD4+ T cells. Hepatology. 1996;24:561–7. doi: 10.1002/hep.510240317. [DOI] [PubMed] [Google Scholar]
- 54.Leon MP, Bassendine MF, Gibbs P, Thick M, Kirby JA. Immunogenicity of biliary epithelium: Study of the adhesive interaction with lymphocytes. Gastroenterology. 1997;112:968–77. doi: 10.1053/gast.1997.v112.pm9041260. [DOI] [PubMed] [Google Scholar]
- 55.Joplin R, Johnson GD, Matthews JB, Hamburger J, Lindsay JG, Hubscher SG, Strain AJ, Neuberger JM. Distribution of pyruvate dehydrogenase dihydrolipoamide acetyltransferase (PDC-E2) and another mitochondrial marker in salivary gland and biliary epithelium from patients with primary biliary cirrhosis. Hepatology. 1994;19:1375–80. [PubMed] [Google Scholar]
- 56.Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta. 1998;1366:139–49. doi: 10.1016/s0005-2728(98)00109-1. [DOI] [PubMed] [Google Scholar]
- 57.Macdonald P, Kirby J, Jones DEJ. Primary biliary cirrhosis (PBC): does apoptosis contribute to altered autoantigen cleavage and targeting? Immunology 2001. 2001;104:16. [Google Scholar]
- 58.Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. 2001;108:223–32. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein R, Wiebel M, Engelhart S, Berg PA. Sera from patients with tuberculosis recognise the M2a-epitope (E2-subunit of pyruvate dehydrogenase complex) specific for primary biliary cirrhosis. Clin Exp Immunol. 1993;92:308–16. doi: 10.1111/j.1365-2249.1993.tb03397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler P, Hamilton-Miller J, Baum H, Burroughs AK. Detection of M2 antibodies in patients with recurrent urinary tract infection using an ELISA and purified PBC specific antigens. Evidence for a molecular mimicry mechanism in the pathogenesis of primary biliary cirrhosis. Biochem Mol Biol Int. 1995;35:473–85. [PubMed] [Google Scholar]
- 61.Krams SM, Surh CD, Coppel RL, Ansari AA, Reubner B, Gershwin ME. Immunization of experimental animals with dihydrolipoamide acetyltransferase, as a purified recombinant polypeptide, generates mitochondrial antibodies but not primary biliary cirrhosis. Hepatology. 1989;9:411–6. doi: 10.1002/hep.1840090311. [DOI] [PubMed] [Google Scholar]
- 62.Jones DEJ, Palmer JM, Kirby JA, et al. Experimental autoimmune cholangitis. a mouse model of immune-mediated cholangiopathy. Liver. 2000;20:351–6. doi: 10.1034/j.1600-0676.2000.020005351.x. [DOI] [PubMed] [Google Scholar]
- 63.Robe AJ, Palmer JM, Kirby J, Jones DEJ. No role for autoreactive antibody in the breakdown of T-cell tolerance to pyruvate dehydrogenase complex, the autoantigen in primary biliary cirrhosis. Hepatology. 2001;34:121A. [Google Scholar]
- 64.Mazanec MB, Coudret CL, McCool T, Baldwin P, Fletcher DR. Intracellular interaction between IgA antibody and viral nucleoprotein. Clin Immunol Immunopathol. 1995;28:541A. [Google Scholar]
- 65.Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, Coppel RL. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–25. doi: 10.1034/j.1600-0528.2002.017402.x. 10.1034/j.1600-065X.2000.174001210.x. [DOI] [PubMed] [Google Scholar]
- 66.Duner E, Pinton P, Spirli C, Neuberger JM, Keogh A, Joplin R, Rizzuto R, Strazzabosco M. Cholangiocyte mitochondrial function is normal is normal in primary biliary cirrhosis and is not altered by long-term incubation with IgA from PBC serum. Hepatology. 2000;32:169A. [Google Scholar]
- 67.Harada K, Ozaki S, Gershwin ME, Nakanuma Y. Enhanced apoptosis relates to bile duct loss in primary biliary cirrhosis. Hepatology. 1997;26:1399–405. doi: 10.1002/hep.510260604. [DOI] [PubMed] [Google Scholar]
- 68.Graham AM, Dollinger MM, Howie SE, Harrison DJ. Bile duct cells in primary biliary cirrhosis are ‘primed’ for apoptosis. Eur J Gastroenterol Hepatol. 1998;10:540–1. doi: 10.1097/00042737-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 69.Harada K, Furubo S, Ozaki S, Hiramatsu K, Sudo Y, Nakanuma Y. Increased expression of WAF1 in intrahepatic bile ducts in primary biliary cirrhosis relates to apoptosis. J Hepatol. 2001;34:500–6. doi: 10.1016/s0168-8278(00)00075-1. [DOI] [PubMed] [Google Scholar]
- 70.Fox CK, Furtwaengler A, Nepomuceno RR, Martinez OM, Krams SM. Apoptotic pathways in primary biliary cirrhosis and autoimmune hepatitis. Liver. 2001;21:272–9. doi: 10.1034/j.1600-0676.2001.021004272.x. [DOI] [PubMed] [Google Scholar]
- 71.Jones DEJ, Palmer JM, Yeaman SJ, Kirby JA, Bassendine MF. Breakdown of tolerance to pyruvate dehydrogenase complex in experimental autoimmune cholangitis a murine model of primary biliary cirrhosis. Hepatology. 1999;30:65–70. doi: 10.1002/hep.510300123. [DOI] [PubMed] [Google Scholar]
- 72.Jones DEJ, Palmer JM, Bennett K, et al. Investigation of a mechanism for accelerated breakdown of immune-tolerance to the primary biliary cirrhosis associated autoantigen, pyruvate dehydrogenase complex. Laboratory Invest. 2002;82:211–9. doi: 10.1038/labinvest.3780413. [DOI] [PubMed] [Google Scholar]
- 73.Sasaki M, Long SA, Van de Water J, et al. The SJL/J mouse is not a model for PBC. Hepatology. 2002;35:1284. doi: 10.1053/jhep.2002.32540. [DOI] [PubMed] [Google Scholar]
- 74.Bogdanos D-P, Okamoto M, Butler P, Williams R, Baum H, Burroughs AK, Vergani D. Cross-reactivity to Lactobacillus Delbrueckii/human pyruvate dehydrogenase complex-E2 characterises primary biliary cirrhosis. Hepatology. 2000;32:300A. [Google Scholar]
- 75.Vilagut L, Vila J, Vias O, Pares A, Gines A, Jiminez de Anta MT, Rodes J. Cross-reactivity of anti-Mycobacterium gordonae antibodies with the major mitochondrial autoantigens in primary biliary cirrhosis. J Hepatol. 1994;21:673–7. doi: 10.1016/s0168-8278(94)80117-7. [DOI] [PubMed] [Google Scholar]
- 76.Shimoda S, Nakamura M, Shigematsu H, Tanimoto H, Gushima T, Gershwin ME, Ishibashi H. Mimicry peptides of human PDC-E2, 163–176 peptide, the immunodominant T–cell epitope of primary biliary cirrhosis. Hepatology. 2000;31:1212–1216. doi: 10.1053/jhep.2000.8090. [DOI] [PubMed] [Google Scholar]
- 77.Potter KN, Thomson RK, Hamblin A, Richards SD, Lindsay JG, Stevenson FK. Immunogenetic analysis reveals that epitope shifting occurs during B-cell affinity maturation in parimary biliary cirrhosis. J Molec Biol. 2001;306:37–46. doi: 10.1006/jmbi.2000.4210. [DOI] [PubMed] [Google Scholar]
- 78.Burroughs AK, Rosenstein IJ, Epstein O, Hamilton-Miller JM, Brumfitt W, Sherlock S. Bacteriuria and primary biliary cirrhosis. Gut. 1984;25:133–7. doi: 10.1136/gut.25.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butler P, Valle F, Hamilton-Miller JM, Brumfitt W, Baum H, Burroughs AK. M2 mitochondrial antbodies and urinary rough mutant bacteris in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J Hepatol. 1993;17:408–14. doi: 10.1016/s0168-8278(05)80225-9. [DOI] [PubMed] [Google Scholar]
- 80.Mayo I, Arizti P, Pares A, Oliva J, Doforno RA, De Sagarra MR, Rodes J, Castano JG. Antibodies against the COOH-terminal region of E. coli C1pP protease in patients with primary biliary cirrhosis. J Hepatol. 2000;33:528–36. doi: 10.1034/j.1600-0641.2000.033004528.x. 10.1016/S0168-8278(00)80003-3. [DOI] [PubMed] [Google Scholar]
- 81.Tsuneyama K, Harada K, Kono N, et al. Scavenger cells with gram-positive bacterial lipoteichoic acid infiltrate around the damaged interlobular bile ducts of primary biliary cirrhosis. J Hepatol. 2001;35:156–63. doi: 10.1016/s0168-8278(01)00084-8. 10.1016/S0168-8278(01)00084-8. [DOI] [PubMed] [Google Scholar]
- 82.Tsuneyama K, Harada K, Yasoshima M, Hiramatsu K, Mackay CR, Mackay IR, Gershwin ME, Nakanuma Y. Monocyte chemotactic protein-1, -2, and -3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: Implications foe pathogenesis. J Pathol. 2001;193:102–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH725>3.0.CO;2-P. 10.1002/1096-9896(2000)9999:9999::AID-PATH7253.3.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 83.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature. 2002;416:603–7. doi: 10.1038/416603a. 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 84.Vinuesa CG, Goodnow CC. DNA drives autoimmunity. Nature. 2002;416:595–8. doi: 10.1038/416595a. [DOI] [PubMed] [Google Scholar]
- 85.Jones DEJ, Palmer JM, Burt AD, Walker C, Robe AJ, Kirby J. Bacterial motif DNA as an adjuvant for the breakdown of immune-self-tolerance to pyruvate dehydrogenase complex. Hepatology. doi: 10.1053/jhep.2002.35067. in press. [DOI] [PubMed] [Google Scholar]
- 86.Long SA, Quan C, Van de Water J, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956–63. doi: 10.4049/jimmunol.167.5.2956. [DOI] [PubMed] [Google Scholar]
- 87.Iwata M, Harada K, Kono N, Kaneko S, Kobayashi K, Nakanuma Y. Expression of Bcl-2 familial proteins is reduced in small bile duct lesions of primary biliary cirrhosis. Hum Pathol. 2000;31:179–84. doi: 10.1016/s0046-8177(00)80217-8. [DOI] [PubMed] [Google Scholar]
- 88.Matsumura S, Van de Water J, Kita H, Coppel RL, Tsuji T, Yamamoto K, Ansari AA. Contribution to antimitochondrial antibody production. cleavage of pyruvate dehydroganse complex-E2 by apoptosis-related proteases. Hepatology. 2002;35:14–22. doi: 10.1053/jhep.2002.30280. 10.1053/jhep.2002.30280. [DOI] [PubMed] [Google Scholar]
- 89.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface blebs on cultured keratinocytes. J Exp Med. 1994;179:1317–30. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I restricted CTLs. Nature. 1998;392:86–9. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 91.Xu L, Guo L, Keogh A, Joplin R, Shen Z, Peng X, Neuberger J, Mason A. Isolation and characterization of a novel virus associated with primary biliary cirrhosis. Hepatology. 2000;32:87A. [Google Scholar]
- 92.Mason AL, Xu L, Guo L, Garry RF. Retroviruses in autoimmune liver disease: genetic or environmental agents. Arch Immunol Therap Experiment. 1999;47:289–97. [PubMed] [Google Scholar]
- 93.Obermayer-Straub P, Manns MP. Hepatitis C and D, retroviruses and autoimmune manifestations. J Autoimmun. 2001;16:275–85. doi: 10.1006/jaut.2000.0488. 10.1006/jaut.2000.0488. [DOI] [PubMed] [Google Scholar]
- 94.Kim MS, Kim BJ, Woo HN, Kim KW, Kim KB, Kim IK, Jung YK. Cadmium induces caspase-mediated cell-death: suppression by Bcl-2. Toxicology. 2000;145:27–37. doi: 10.1016/s0300-483x(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 95.Mamula MJ, Lin RH, Jameway CA, Hardin JA. Breaking T cell tolerance with foreign and self co-immunogens: a study of B and T cell epitopes of cytochrome c. J Immunol. 1992;149:789–95. [PubMed] [Google Scholar]
- 96.Mamula MJ, Janeway CA. Do B cells drive the diversification of immune responses? Immunol Today. 1993;14:151–2. doi: 10.1016/0167-5699(93)90274-O. [DOI] [PubMed] [Google Scholar]
- 97.Mamula MJ. Epitope spreading. the role of self peptides and autoantigen processing by B lymphocytes. Immunol Rev. 1998;164:231–9. doi: 10.1111/j.1600-065x.1998.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 98.Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T-cell priming in vivo. J Immunol. 1999;162:5696–703. [PubMed] [Google Scholar]
- 99.Fuchs E, Matzinger P. B cells drive diversification but not directly. Immunol Today. 1993;14:153. doi: 10.1016/0167-5699(93)90274-O. [DOI] [PubMed] [Google Scholar]
- 100.Liermann Garcia RF, Evangalista Garcia C, McMaster P, Neuberger J. Transplantation for primary biliary cirrhosis. retrospective analysis of 400 patients in a single centre. Hepatology. 2001;33:22–7. doi: 10.1053/jhep.2001.20894. [DOI] [PubMed] [Google Scholar]
- 101.Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cycosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci. 1998;18:5151–9. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]