

Abstract
The use of NK cells in adoptive therapy for malignant disease is an area of great potential. Currently the only NK cell line in clinical trials is NK-92, an activated NK cell line with a broad range of cytotoxicity against malignant cells. The activity of NK-92 against pre-B acute lymphoblastic leukaemias, however, is highly variable. In this study we compare the cytotoxic mechanisms and signalling pathways utilized by NK-92 ci and IL-2 activated NK cells to mediate killing of pre-B acute lymphoblastic leukaemia cell lines. Deficiencies in TNF family mediated apoptosis, phosphoinositide-3 kinase dependent and phosphoinositide-3 kinase independent killing limit the efficiency of NK-92 ci against pre-B acute lymphoblastic leukaemia cells. Importantly, treatment of the poorly killed leukaemia cells with TNF-α augmented both phosphoinositide-dependent and -independent cytolysis.
Keywords: NK-92, acute lymphoblastic leukaemia, cytotoxicity, PI-3 kinase, TNF
INTRODUCTION
The use of ex vivo expanded cytotoxic cells is an area of extensive investigation for the treatment of malignant disease. While the goal of routine expansion of tumour-specific T cells for adoptive therapy may still be some way off, an alternative source of potentially therapeutic cytotoxic cells are the natural killer (NK) cells. In contrast to T cells, NK cells are not antigen specific but rather their activation appears to be determined by the balance of inhibitory and activating signals received by the NK cell upon conjugation with target cells [1]. Once activated, NK cells can kill their targets either by the granule exocytosis pathway or via the tumour necrosis factor (TNF) family of molecules [2,3]. One significant advantage to the use of NK cells for therapy is the lack of MHC restriction of their cytotoxic activity. This allows an NK cell line to be used in the treatment of a number of individuals, with limitations only imposed by the expression of inhibitory receptors for specific MHC class I molecules.
The NK-92 cell line, derived from a non- Hodgkin's lymphoma patient, has a phenotype resembling an activated NK cell [4]. This cell line has been shown to exert strong cytotoxic activity against a wide range of tumour cell types, including leukaemias and melanomas [5,6]. The specificity of the NK-92 mediated killing has been shown by its ability to purge normal bone marrow of seeded K562 leukaemia cells [7] and to kill leukaemia and melanoma cells transferred into SCID mice [6]. Importantly, preclinical data suggest that the NK-92 cell line will have a low tumorigenic risk in immunocomprimized individuals [5,6]. This risk can be further diminished by irradiation that does not reduce cytotoxic activity [7]. Based on the high cytotoxic activity and specificity for malignant cells possessed by NK-92, clinical trails have opened to evaluate the feasibility of using this line for adoptive transfer therapy [8]. Preliminary indications are that the intravenous administration of NK-92 is safe and that the cells are not rejected by the patient's immune system. The prolonged cytotoxic activity of NK-92 cells requires the presence of IL-2 [5]. For this reason stable IL-2 producing derivatives of NK-92, NK-92 ci and NK-92 mi, were established [9]. The cytotoxic activity of these derivative lines is similar to that of the parental NK-92 cells when measured using standard NK cell targets. Importantly for clinical utility, the level of local IL-2 produced by the transfected NK-92 lines does not cause toxicity.
While NK-92 is an efficient killer of a wide variety of leukaemia cell types, its level of cytotoxicity is lowest against B-lineage acute lymphoblastic leukaemia (ALL) [5]. ALL is the single most common malignancy in children, with an incidence rate of 34 per million children less than 15 years of age [10]. While current chemotherapy regimes result in excellent long-term event free survivals, relapsed ALL continues to be a significant clinical challenge and novel treatment strategies for this disease are needed. In order to design rational strategies to improve NK-92 mediated killing of paediatric pre-B ALL cells we compared the cytotoxic mechanisms and activation pathways utilized by NK-92 ci and IL-2-activated primary NK (ANK) cells against pre-B ALL cell lines. Our results indicate that the level of NK-92 ci killing of pre-B ALL cells is determined by three distinct mechanisms and demonstrate strategies to enhance the potential use of the line as a therapeutic agent for this disease.
MATERIALS AND METHODS
Cell culture
All cell lines, except NK-92 and NK-92 ci, were maintained in RPMI medium (Biofluids, Rockville, MD, USA), supplemented with 10% fetal bovine serum (GibcoBRL, Grand Island, NY, USA), 20 mm HEPES and 2 mml-glutamine. NK-92 cells were cultured in Myelocult medium (StemCell Technologies, Vancouver, BC, Canada). The NK-92 and NK-92 ci cell lines were provided by Dr H.G. Klingemann (Chicago, USA). The precursor B ALL cell lines used were ALL1 (BCR-ABL), REH (ETV6-AML1) and RS4; 11 (MLL-AF-4). The ALL1 cell line was a gift from Dr F. Uckun (St. Paul, MN, USA). The remaining cell lines (REH, RS4: 11, K562 and Jurkat) were obtained from ATCC. Where indicated, cell lines were treated with 20 ng/ml human recombinant TNF-α (Pharmingen, Torrey Pines, CA, USA) for 18 h. IL-2 activated NK cells were prepared by selection of CD56 positive PBMCs from a healthy donor using StemSep (StemCell Technologies, Vancouver, Canada). The CD56 positive cells were then cultured for 3 days in the presence of 100 U/ml IL-2 at which time the cultured cells were greater than 95% CD56 positive (data not shown). LFA-1 expression was evaluated using the CD11a specific antibody clone Hl111 (Pharmingen).
Annexin V assay
Apoptotic cells were detected using Annexin V-FITC (Pharmingen). Briefly, unlabelled target cells were incubated with CMA treated NK-92 ci cells at an E : T ratio of 10 : 1 for 4 h at 37°C. Following washing in PBS and Annexin V binding buffer, the cells were resuspended in 100 μl of binding buffer and incubated on ice with 5 μl Annexin V-FITC and 10 μg of PE conjugated anti-CD56 mAb (clone B159, Pharmingen) for 15 min The cells were then analysed by flow cytometry performed on a FACScalibur cell analyser using CellQuest (Becton Dickinson, Mountain View, CA, USA). CD56 staining was used to gate out NK-92 cells from the analysed population.
Chromium release assays
Cytotoxicity of NK-92 ci was measured using standard 51Cr release assays (CRA). 5 × 105 target cells were labelled with 100 μCi for 2 h prior to assay and incubated with effector cells for the time indicated in figure legends. Unless stated otherwise NK-92 ci CRAs were carried out at an E:T ratio of 15 : 1. Perforin degradation was achieved by incubating NK-92 ci cells with 400 nm Concanamycin A (CMA) (Sigma, St Louis, MO, USA) for 2 h prior to assay. CMA was then present at 200 nm throughout the CRA. To inhibit Ca2+ mediated events 2 mm EGTA (Sigma) was present throughout the CRA. Where indicated, NK-92 ci cells were incubated with the PI-3 kinase inhibitor, Wortmannin (Sigma), at 100 nm for 30 min prior to the assay. Wortmannin was present at 50 nm throughout the CRA. Percent specific lysis was determined by the following formula:
[(experimental release − spontaneous release)/(total release − spontaneous release)] × 100.
RESULTS
Comparison of pre-B ALL killing by NK-92, NK-92 ci and IL-2 activated NK cells
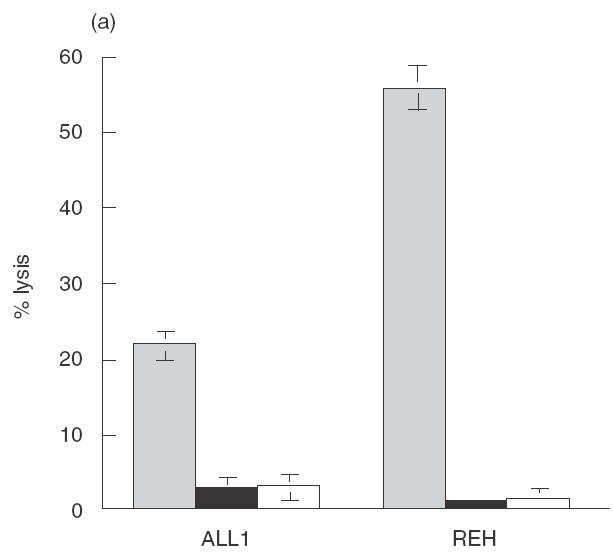
It has been reported previously that as a group pre-B ALL cells are less sensitive to killing by NK-92 than other leukaemias [5]. As pre-B cells have been reported to be less sensitive than other B cell stages to NK cell cytotoxicity [11], we sought to determine if the reduced killing by NK-92 was a reflection of a general insensitivity of pre-B ALL cells to NK killing or rather the result of a deficiency in NK-92 cytotoxicity. While killing of the standard NK targets, K562 and Jurkat, by NK-92 and NK-92 ci was similar, significant differences in the level of killing obtained against pre-B ALL cells was observed, most noticeably against REH (Fig. 1a). For 2 of the 3 pre-B ALL lines tested, ALL1 and REH, more killing was observed with ANK cells compared to NK-92 ci (Fig. 1a). This was most pronounced for ALL1 cells, with differences of over 60% in the amount of killing observed. In contrast, RS4; 11 was killed to the same degree by both effector cells. The killing of the established NK cell target lines was similar with NK-92 ci or ANK effector cells. The level of LFA-1 expression by each of the pre-B ALL cell lines is shown in Fig. 1(b). It is clear from these data that in this system the level of killing does not correlate with expression of this adhesion molecule. Expression of LFA-1 was not altered by treatment with TNF-α (data not shown).
Fig. 1.
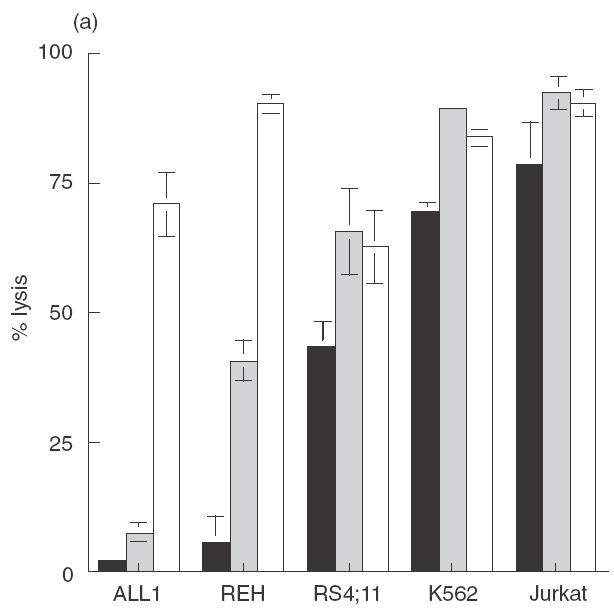
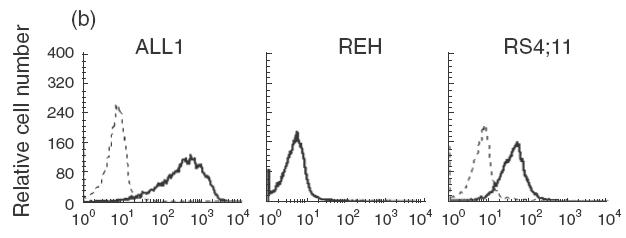
Cytotoxic activity against pre-B ALL cell lines by NK-92, NK-92 ci and IL-2 activated NK cells. (a) The 3 pre-B ALL lines (ALL1, REH and RS4; 11), K562 and Jurkat were incubated with either NK-92 (▪), NK-92 ci (▪) or ANK (□) in a 6-h CRA. The results presented were obtained with an E : T ratio of 15 : 1. The results shown are the averages ± SE of 3 experiments. (b) The level LFA-1 expression by the 3 pre-B ALL cell lines as detected by flow cytometry. The isotype control is shown by a dashed line and the specific antibody signal shown by a solid line.
Mechanisms of NK-92 ci mediated cytotoxicity
NK-92 cells have been reported to express high levels of the granule contents, perforin and granzyme B, and several members of the TNF family [12]. These distinct killing mechanisms can be distinguished on the basis of the effect of various inhibitors on target cell death. The drug concanamycin A (CMA), an inhibitor of vacuolar type H+-ATPase, specifically inhibits perforin-mediated cytotoxicity by causing degradation of the protein in the lytic granules [13]. TNF family mediated killing is unaffected by the presence of CMA. The calcium chelator EGTA inhibits the Ca2+ dependent release and multimerization of perforin. The effect of CMA and EGTA on NK-92 ci killing of the well described NK cell target cell lines K562 and Jurkat are shown in Fig. 2(a). In the case of the K562 cell line both CMA and EGTA treatment inhibited all killing, whereas for the Jurkat cell line the treatments resulted in only partial inhibition of killing. These results indicate that NK-92 ci cells can kill Jurkat cells via the perforin-dependent and independent mechanisms.
Fig. 2.
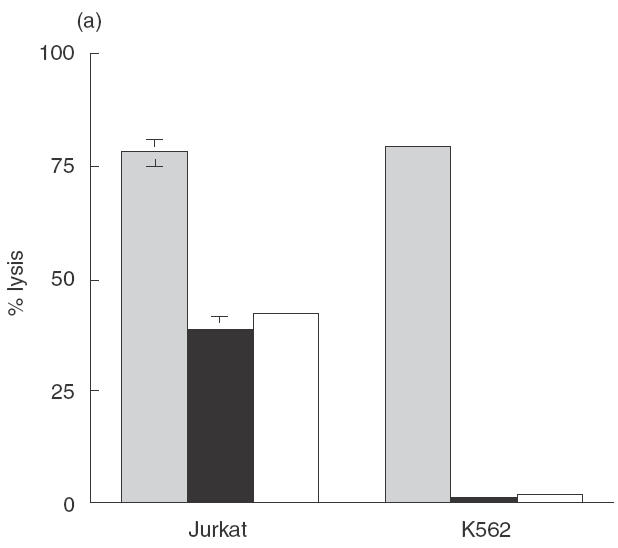
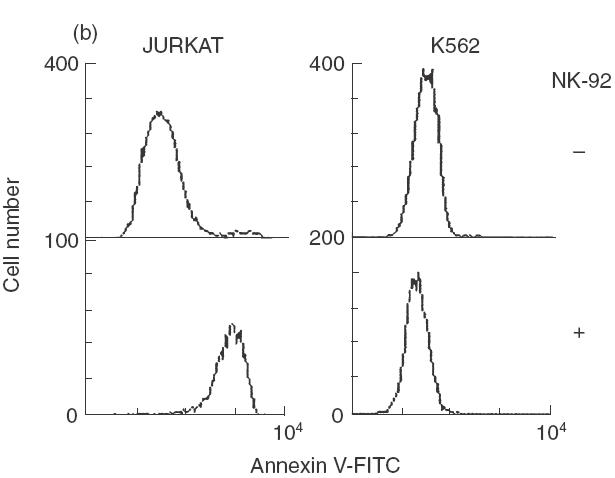
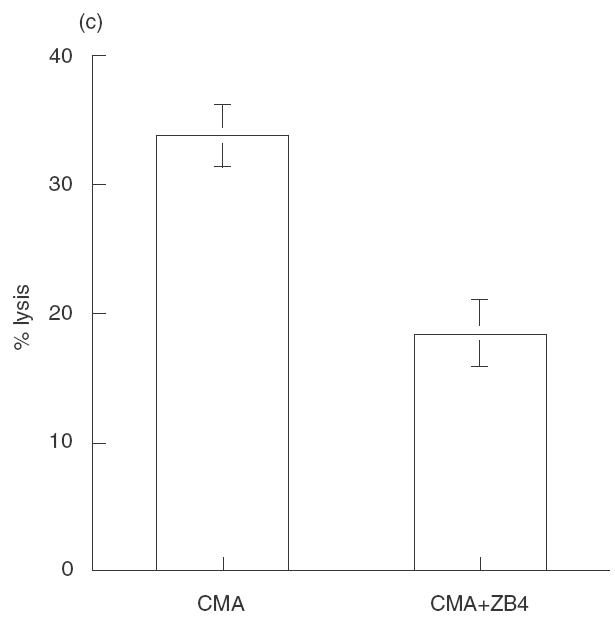
NK-92 ci utilizes both perforin dependent and independent killing mechanisms. (a) Target cells, Jurkat and K562, were incubated with either untreated NK-92 ci (▪), CMA treated NK-92 ci (▪) or NK-92 ci in the presence of EGTA (□), in a 5-h CRA at an E:T ratio of 10:1. (b) NK-92 ci killing induces Annexin V binding to target cells. Jurkat and K562 cells were incubated either alone (upper panels) or with CMA treated NK-92 ci cells (lower panels) for 4 h prior to labelling with Annexin V-FITC. NK-92 ci cells were gated out on the basis of CD56 staining. (c) CD95 blocking antibody partially inhibits apoptotic killing. Jurkat cells were incubated with CMA treated NK-92 in the presence or absence or 2 μg/ml ZB4 antibody. Chromium release was measured after a 6-h CRA. The results shown are the averages ± SE of 6 experiments.
Members of the TNF family induce apoptosis in the target cell. To confirm that the perforin-independent chromium release from Jurkat cells was the result of the apoptosis, Annexin V binding assays were performed to detect the presence of phosphotidylserine on the outer leaflet of the cell membrane, a characteristic of apoptotic death. Figure 2(b) shows that Jurkat cells incubated in the presence of CMA-treated NK-92 ci cells bind a significant amount of Annexin V. In contrast, K562 cells incubated under identical conditions revealed no such binding activity. These results demonstrate that the nonperforin mediated killing activity of NK-92 ci cells induces apoptotic cell death of the susceptible Jurkat cells.
Several members of the TNF family of molecules have been reported to mediate apoptotic killing by NK cells [3,14]. It has been reported that the K562 cell line is insensitive to killing mediated by CD95 ligand whereas Jurkat cells are sensitive to the action of this molecule [15]. The antibody ZB4 has been shown to inhibit killing induced by the interaction between CD95 and CD95 ligand [16]. We found that perforin-independent killing of Jurkat cells is inhibited by an average of 50% in the presence of ZB4, indicating that the CD95 ligand apoptotic pathway is involved in, but is not completely responsible for, NK-92 ci mediated apoptosis of Jurkat cells (Fig. 2c).
Sensitivity of paediatric pre-B ALL cell lines to NK-92 ci killing mechanisms
Having demonstrated the functional activity of both perforin dependent and independent cytotoxic mechanisms in NK-92 ci cells, we analysed the sensitivity of the paediatric pre-B cell lines REH and ALL1 to these mechanisms. The results in Fig. 3(a) show that all killing of the cell lines by NK-92 ci was inhibited by the presence of CMA or EGTA, indicating that TNF family mediated cytotoxic mechanisms do not result in detectable killing of the pre-B cell lines. CD95 is expressed on 12% and 18% of REH and ALL1 cells, respectively (data not shown). As at least some apoptotic killing induced by NK-92 ci is mediated by CD95 ligation (Fig. 2c), the insensitivity of the pre-B ALL lines to CD95 mediated apoptosis was confirmed using an anti-CD95 antibody cross-linking approach [17]. Despite significant killing of Jurkat cells in this assay, no death of the pre-B ALL lines was detected, even after 12 h incubation (data not shown).
Fig. 3.
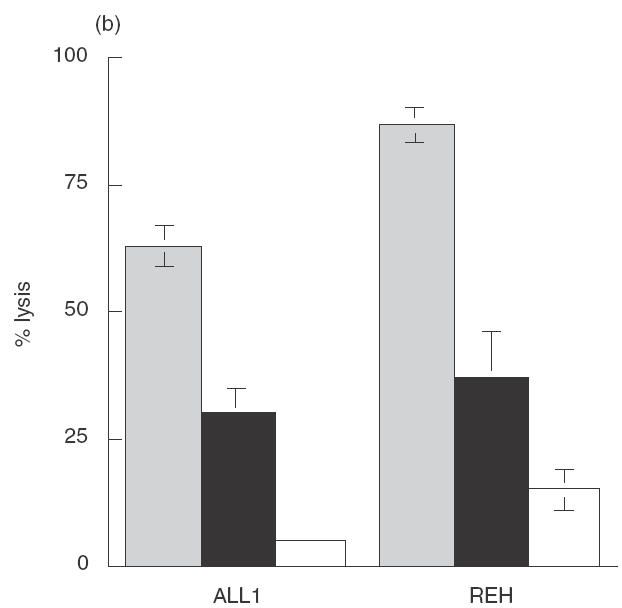
Perforin dependent and independent killing of pre-B ALL cell lines by NK-92 ci and ANK. Target cells were incubated with either untreated (▪), CMA treated (▪) or EGTA treated (□) effector cells, in a 6-h CRA at an E:T ratio of 10:1. (a) NK-92 effectors. (b) IL-2 activated NK cell effectors. The results shown are the averages ± SE of 5 experiments.
The insensitivity of pre-B ALL lines to apoptosis induction by NK-92 ci suggested that the greater killing achieved with primary ANK cells (Fig. 1a) may reflect the ability of the latter cytotoxic cells to induce apoptosis. To address this possibility, we measured the level of pre-B leukaemia cell killing achieved by primary ANK cells in the presence of CMA and EGTA. Figure 3(b) shows clearly that ANK cells are capable of inducing perforin-independent killing of the leukaemic cells.
Comparison of signalling pathways used by NK-92 ci and ANK cells
Activation of NK cells can be mediated by a number of cell surface receptors that mediate signalling either via an ITAM motif or a Phosphoinositide-3 (PI-3) kinase binding motif [18] and it has been proposed that PI-3 kinase plays a key role in the signal transduction leading to NK cell granule exocytosis and cytotoxicity [19]. To determine whether the difference in killing of pre-B cells by NK-92 ci compared to ANK cells was the result of different triggering of activation pathways, we examined the effect of the specific PI-3 kinase inhibitor, wortmannin, on cytotoxicity. In the case of ALL1 cells, Fig. 4 shows that while the level of PI-3 kinase independent cytotoxicity is similar for both NK-92 ci and ANK (8% versus 22%), the contribution of PI-3 kinase dependent killing was far greater for ANK cells (49%) than for NK-92 ci (5%). For REH cells, however, the contribution of PI-3 kinase dependent killing is 19% for NK-92 ci and 18% for activated NK cells, indicating a similar level of involvement for the enzyme in both cytotoxic cell types. In this case, the increase in killing seen with ANK effectors is the result of enhanced wortmannin insensitive cytotoxicity (22% versus 72%). For RS4; 11 cells, the relative contribution of the two pathways to the overall killing was similar for NK-92 ci and ANK cells.
Fig. 4.
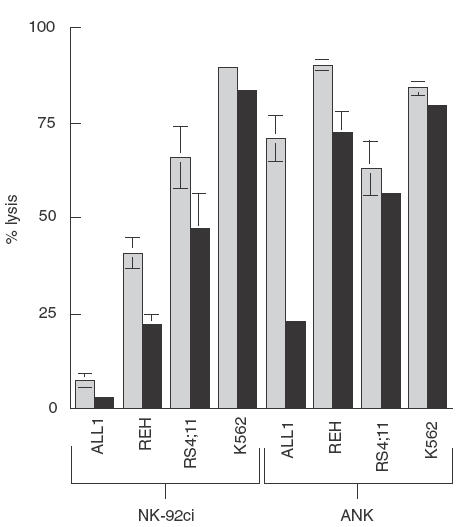
Inhibition of PI-3 kinase-dependent killing of pre-B ALL cells. Effector cells were either untreated (▪) or treated with 100nm wortmannin for 30 min (▪) prior to a 5-h CRA at an E:T ratio of 15:1. The results shown are the averages ± SE of 3 experiments.
Both PI-3 kinase dependent and independent killing of ALL1 cells are augmented by TNF-α
It has been previously reported that treatment of resistant leukaemia cells with TNF-α up-regulates their sensitivity to NK-92 cytotoxicity [20]. To determine if this approach was relevant to the killing of pre-B ALL cells by NK-92 ci, we treated the leukaemia cell lines with TNF-α for 18 h prior to incubation with NK-92 ci. This treatment resulted in significantly increased ALL1 cell death (Fig. 5). Inhibition with wortmannin revealed that increases in both the PI-3 kinase dependent and independent killing contributed to the overall improvement in killing efficiency. All killing of the treated ALL1 cells was inhibited by CMA indicating that there is no increase in sensitivity to perforin independent cytotoxic mechanisms (data not shown). TNF-α treatment did not affect the level of killing obtained against REH or RS4; 11 cells.
Fig. 5.
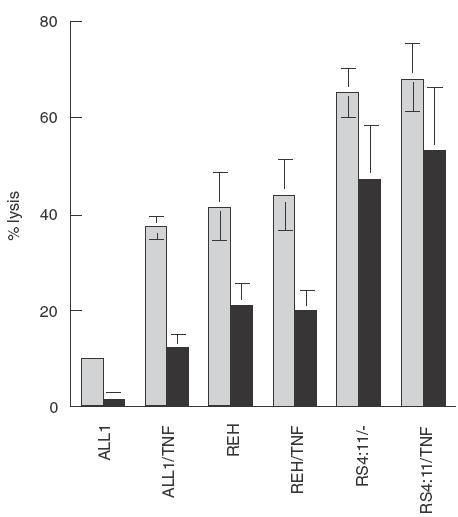
Augmentation of ALL1 cell killing by TNF-α. Pre-B ALL cells were untreated or incubated for 18h with 20ng/ml TNF-α. Cells were washed several times prior to incubation with effectors. The effector cells were either untreated (▪) or incubated with wortmannin for 30min (▪) prior to CRA. The results presented were obtained in a 5-h CRA at an E:T ratio of 15:1. The results shown are the averages ± SE of 3 experiments.
DISCUSSION
Adoptive transfer with the NK cell line NK-92 represents an area of significant clinical potential in the fight against malignant disease. Understanding the mechanisms controling the cytotoxicity of this line will be critical for realizing that potential. The poor killing of pre-B ALL cells by NK-92 [5] will limit the application of the cell line for treatment of this disease. In this study we have compared the mechanisms involved in the killing of pre-B ALL cells by NK-92 ci and IL-2 activated NK cells. Our results show that for each of the pre-B ALL lines tested, greater killing was obtained using the NK-92 ci cell line compared to the parental NK-92 line. This finding suggests that for treatment of pre-B ALL, use of the NK-92 ci line may represent a more effective strategy. Although it has been reported that susceptibility of B-ALL cells to NK killing requires the expression of LFA-1 by the leukaemia cell [21], the data presented here demonstrate that the killing of the pre-B ALL cell lines by NK-92 ci and ANK cells does not correlate with LFA-1 expression. Interestingly, this result suggests that the use of activated NK effectors may be beneficial in the treatment of LFA-1 negative B-ALL. For two of the pre-B ALL lines tested, ALL1 and REH, significantly more killing was obtained using ANK effectors than with NK-92 ci. Our results demonstrate that the ability of NK-92 ci cells to deliver perforin-independent killing signals, activate PI-3 kinase-dependent signalling pathways and exert strong PI-3 kinase-independent cytotoxicity are all limiting in the killing of pre-B ALL cells. The ability of TNF-α treatment to alleviate the deficiencies in PI-3 dependent and independent, but not TNF family mediated, killing of ALL1 cells indicates that the activity of NK-92 against poorly killed pre-B ALL cells can be manipulated to achieve greater killing efficiency.
It has been reported that NK-92 cells contain transcripts for several members of the TNF family of molecules, including CD95 ligand, TRAIL, TWEAK and TNF-α [12]. However, the functionality of these molecules on NK-92 cells has not been tested. Our results indicate that NK-92 ci cells are indeed capable of delivering an apoptotic signal to sensitive target cells, and that this is in part mediated by CD95 ligand. However, when used against pre-B ALL cells, the proteins involved in perforin independent killing of Jurkat cells are ineffective against the leukaemia cells. In contrast, IL-2 activated NK cells deliver a significant apoptotic signal to pre-B ALL cells. In the case of REH cells, the difference in killing obtained by NK-92 ci and ANK can be attributed to the CMA resistant cytotoxicity exerted by ANK cells. The identity of the protein(s) involved in delivering the apoptotic signal to the leukaemia cells is currently under investigation. Identification of this protein will provide a strategy to increase NK-92 ci mediated killing of pre-B ALL cells via transfection of the gene into the NK cell line. It is of interest that the apoptotic killing was inhibited to a greater degree by EGTA than by CMA. While this may simply reflect a more significant inhibition of perforin mediated killing by EGTA, we favour an alternate explanation for two reasons; increasing the CMA treatment to 4 h at 800 nm failed to increase inhibition (data not shown), and under the same conditions killing by NK-92 ci cells was completely inhibited. These results imply that the apoptotic signal delivered by IL-2 activated NK cells is, at least in part, calcium dependent.
Comparison of the inhibitory effect of wortmannin on NK-92 ci and ANK cells revealed a lack of PI-3 kinase involvement in the killing of K562 and Jurkat target cells. The results presented here are in contrast to a recent report that wortmannin inhibited virtually all killing of targets, including K562, by NK-92 cells [19] but are in agreement with findings reported with other NK effectors [22,23]. A 10-fold higher concentration of wortmannin had no additional effect, suggesting that it was not the result of inefficient inhibition (data not shown). Similar patterns of killing were seen with the parental NK-92 line (data not shown), indicating that the results are not simply the result of using the IL-2 transfected NK-92 ci line. Our results favour the model in which PI-3 kinase plays a contributory, but not pivotal, role in killing by activated NK cells.
The difference in killing of the p190 BCR-ABL positive ALL1 cells by NK-92 ci and ANK cells is dramatic. The delivery of either inhibitory signals to NK-92 ci or activating signals to ANK cells may account for this difference. Extensive analysis of NK-92 has revealed a very limited expression of NK inhibitory receptors and no influence of target cell HLA expression has been observed [12,24–26]. NK-92 does express several activating receptors, including NKp30, NKp46, and NKG2D (detected by RT-PCR) that have been reported to signal through PI-3 kinase [12,27,28], the pathway responsible for most of the ALL1 killing. As the density of activating receptors on the surface of NK cells influences killing [29], higher levels of expression of one or more of these receptors on ANK may explain the difference in killing. Alternatively, NK-92 ci may lack a PI-3 kinase dependent activating receptor present on ANK cells. Although NK-92 cells do not express NKp44 [12], signalling via this receptor is mediated through an ITAM motif and thus is unlikely to be responsible for the killing reported here [30]. Analysis of the cell surface expression of the known activating receptors on NK-92 ci is needed to resolve this issue. The killing of pre-B ALL cell lines described here supports the suggestion that malignant cells with a common origin may differ in their expression of ligands for NK activating receptors [29].
In order to improve the therapeutic potential of NK-92 for pre-B ALL cells we attempted to increase PI-3 kinase mediated killing using TNF-α. We chose this approach for two reasons. Firstly, it has been reported that TNF-α induced NK-92 killing of a previously resistant leukaemia cell line and that this corresponded with the activation of adhesion molecules, including CD44, on the leukaemia cell surface [20]. CD44 can interact with CD44 molecules on other cells and CD44 directed NK killing is PI-3 kinase dependent [23,31]. Secondly, previous results have suggested that TNF-α may up-regulate expression of ligands for NKp30 and NKp46 [25]. We found that TNF-α significantly increased killing of ALL1 cells and that this was mediated by increased activation of both PI-3 kinase-dependent and -independent pathways.
In summary, we have used the recent increases in understanding of NK cell mediated cytotoxicity to specifically address the issue of poor pre-B ALL killing by the NK-92 ci cell line. We find that deficiencies in apoptosis induction, PI-3 kinase activation and PI-3 kinase-independent killing all contribute to the level of cytotoxicity directed against pre-B ALL cells. The extent of these deficiencies varies between pre-B ALL lines. We have demonstrated that, in the case of a poorly pre-B ALL target, two of these deficiencies can be addressed by TNF-α induced changes in the leukaemia cell.
Acknowledgments
Excellent technical assistance was provided by Sharon Bader, Greg Doho and Luke Terrett. G.R. is the recipient of a Candlelighters Canada/MRC postdoctoral fellowship.
REFERENCES
- 1.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 2.Shresta S, Pham CTN, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets? Curr Opin In Immunol. 1998;10:581–7. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- 3.Kashii Y, Giorda R, Herberman RB, Whiteside TL, Vujanovic NL. Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol. 1999;163:5358–5366. [PubMed] [Google Scholar]
- 4.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92CI) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8. [PubMed] [Google Scholar]
- 5.Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, McGuirk J, O'Reilly RJ. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res. 1998;4:2859–68. [PubMed] [Google Scholar]
- 6.Tam YK, Miyagawa B, Ho VC, Klingemann HG. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line ‘NK-92CI’. J Hematother. 1999;8:281–90. doi: 10.1089/106161299320316. [DOI] [PubMed] [Google Scholar]
- 7.Klingemann H-G, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 8.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 9.Tam YK, Maki G, Miyagawa B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum General Ther. 1999;10:1359–73. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 10.Reis LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. United States SEER Program 1975–95. Bethesda: NCI; 1999. Cancer Incidence and Survival Among Children and Adolescents; pp. 99–4649. NIH pub. no. [Google Scholar]
- 11.Storkus WJ, Dawson JRB. Cell sensitivity to natural killing. correlation with target cell stage of differentiation and state of activation. J Immunol. 1986;136:1542–7. [PubMed] [Google Scholar]
- 12.Maki G, Klingemann H-G, Martinson JA, Tam YT. Factors regulating the cytotxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res. 2001;10:369–83. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- 13.Kataoka T, Shinohara S, Takayama H, Takaku K, Kondo S, Yonehara S, Nagai K. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156:3678–86. [PubMed] [Google Scholar]
- 14.Johnsen AC, Haux J, Steinkjer B, Nonstad U, Egeberg K, Sundan A, Ashkenazi A, Espevik T. Regulation of APO-2 ligand/TRAIL expression in NK cells-involvement in NK cell-mediated cytotoxicity. Cytokine. 1999;11:664–72. doi: 10.1006/cyto.1999.0489. 10.1006/cyto.1999.0489. [DOI] [PubMed] [Google Scholar]
- 15.Alderson M, Tough TW, Braddy S, Davis-Smith T, Roux E, Schooley K, Miller RE, Lynch DH. Regulation of apoptosis and T cell activation by Fas-specific mAb. Int Immunol. 1994;6:1799–806. doi: 10.1093/intimm/6.11.1799. [DOI] [PubMed] [Google Scholar]
- 16.Oka M, Hirazawa K, Yamamoto K, Iizuka N, Hazama S, Suzuki T, Kobayashi N. Induction of Fas-mediated apoptosis on circulating lymphocytes by surgical stress. Ann Surg. 1996;223:434–40. doi: 10.1097/00000658-199604000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cifone MG, De Maria R, Roncaioli P, Rippo MR, Azuma M, Lanier LL, Santoni A, Testi R. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J Exp Med. 1994;180:1547–52. doi: 10.1084/jem.180.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanier LL. On guard – activating NK cell receptors. Nat Immunol. 2001;2:23–7. doi: 10.1038/83130. [DOI] [PubMed] [Google Scholar]
- 19.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, Djeu JY. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cell. Nat Immunol. 2001;1:419–25. doi: 10.1038/80859. [DOI] [PubMed] [Google Scholar]
- 20.Maki G, Krystal G, Dougherty G, Takei F, Klingemann HG. Induction of sensitivity to NK-mediated cytotoxicity by TNF-alpha treatment: possible role of ICAM-3 and CD44. Leukemia. 1998;12:1565–72. doi: 10.1038/sj.leu.2401145. [DOI] [PubMed] [Google Scholar]
- 21.Ruggeri L, Capanni M Casucci, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94:333–9. [PubMed] [Google Scholar]
- 22.Colucci F, Schweighoffer E, Tomasello E, Turner M, Ortaldo JR, Vivier E, Tybulewicz VLJ, Di Santo JP. Natural cytotoxicity uncoupled from the Syk and ZAP-70 intracellular kinases. Nat Immunol. 2001;3:288–94. doi: 10.1038/ni764. [DOI] [PubMed] [Google Scholar]
- 23.Sconocchia G, Titus JA, Segal DM. Signaling pathways regulating CD44-dependent cytolysis in natyral killer cells. Blood. 1997;90:716–25. [PubMed] [Google Scholar]
- 24.Burshtyn DN, Biassoni R, Cantoni C, et al. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intra-cellular domains. Immunity. :77–85. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 25.Peruzzi M, Wagtmann N, Long EO. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J Exp Med. 1996;184:1585–90. doi: 10.1084/jem.184.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopalan S, Long EO. A major histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med. 1999;189:1093–9. doi: 10.1084/jem.189.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, Phillips JH. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285:730–2. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 28.Spaggiari GM, Carosio R, Pende D, Marcenaro S, Rivera P, Zocchi MR, Moretta L, Poggi A. NK cell-mediated lysis of autologous antigen-presenting cells is triggered by the engagement of the phosphotidylinositol 3 kinase upon ligation of the natural cytotoxicity receptors NKp30 and NKp44. Eur J Immunol. 2001;31:1656–65. doi: 10.1002/1521-4141(200106)31:6<1656::aid-immu1656>3.0.co;2-v. 10.1002/1521-4141(200106)31:61656::AID-IMMU16563.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 29.Pende D, Cantoni C, Rivera P, et al. Role of NKG2D in tumor cell lysis mediated by human NK cells: cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur J Immunol. 2001;31:1076–86. doi: 10.1002/1521-4141(200104)31:4<1076::aid-immu1076>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 30.Vitale M, Bottino C, Sivori S, et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med. 1998;187:2065–72. doi: 10.1084/jem.187.12.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Droll A, Dougherty ST. Chiu RK, Dirks JF, McBride WH, Cooper DL, Dougherty GJ. Adhesive interactions between alternatively spliced CD44 isoforms. J Bio Chem. 1995;270:11567–73. doi: 10.1074/jbc.270.19.11567. [DOI] [PubMed] [Google Scholar]